Received Date:
04 July 2017 Available Online:
15 November 2017
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21335001 and 21575126) and the Zhejiang Provincial Natural Science Foundation (No. LR14B050001)
Abstract:
Optical sensors are devices that transform the interaction between medium and analyte to optical signal. Optical interference is a technique that has been widely applied in optical sensors, which is label-free, fast and non-invasive. Light reflected from the top and bottom surfaces of single layer film, or each interfaces of multilayer film in optical sensors leads to constructive and destructive fringes of the optical interference pattern. Nanoporous films with large surface-to-volume ratio are beneficial to improve the sensitivity and lower the limit of detection of the sensors, which is typically used in the form of single layer, double layer or multilayer (usually served as photonic crystal). In this article, we introduce and review the applications of nanoporous films of silicon, anodic aluminum oxide, titanium dioxide and metal-organic framework in optical sensors based on the optical interference. A perspective of developments in this research field is also provided.
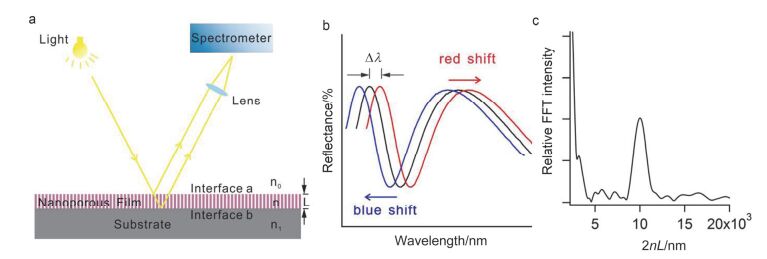
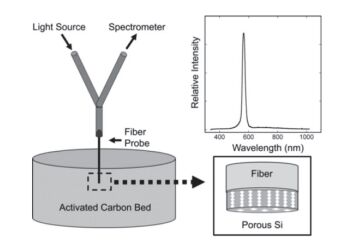
Figure 1.
(a) Schematic illustration of optical interference of nanoporous film. (b) Reflectometric interference spectrum of nanoporous film. (c) Fourier transforms of reflectometric interference spectrum of nanoporous film.
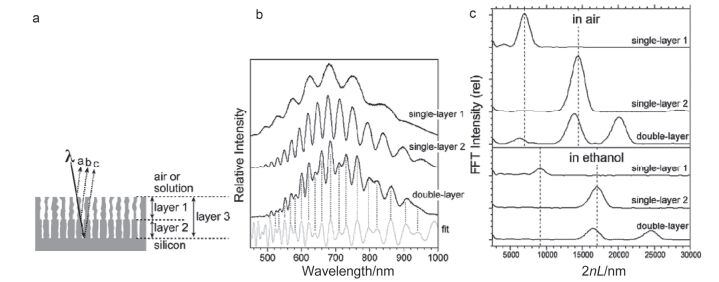
Figure 2.
(a) Schematic of the porous Si double-layer biosensor consisting of a top layer with large pores and a bottom layer with smaller pores. (b) Relative reflectance spectra of thermally oxidized porous Si single-and double-layers. (c) Fourier transforms of thermally oxidized p-type porous Si single-and double-layers in air and immersed in ethanol[45].
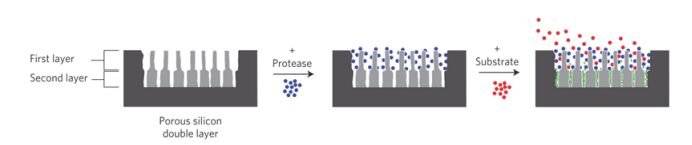
Figure 3.
Schematic illustration of real-time monitoring of enzyme activity in a mesoporous silicon double layer. Active protease (pepsin, blue) is absorbed into the first layer. The assay is then carried out by addition of substrate (α-casein, red). The pores of the second layer are too small to admit the protease or its substrate. However, digestion of the substrate by pepsin produces fragments (green) sufficiently small to infiltrate the second layer.[49]
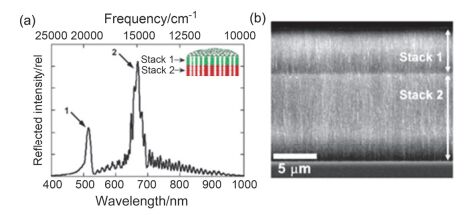
Figure 4.
(a) Reflectivity spectrum of a sample containing two photonic crystals, one on top of the other (double stack). A cross-sectional schematic of the structure is shown in the inset. (b) Cross-sectional secondary electron SEM of the double-stack porous silicon photonic crystal[61].
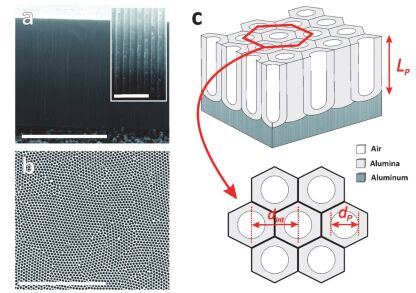
Figure 6.
(a) Cross-section SEM image of AAO (scale bar=5 μm) and inset showing detail of the cylindrical nanopores (scale bar=500 nm). (b) Top-view SEM image of AAO (scale bar=3 μm). (c) Schematic illustration of the structure of AAO[73].
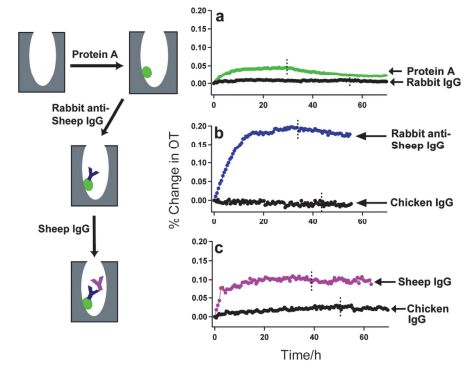
Figure 7.
Experiments demonstrating specific binding affinity and controls for the AAO immunosensor. (a) Protein A and rabbit IgG separately dosed on bare AAO sample, demonstrating the greater relative affinity of the surface for protein A. (b) Rabbit anti-sheep IgG and chicken IgG introduced to a protein-A-modified sample. (c) Sheep IgG and chicken IgG introduced to a protein-A-modified sample that contains the protein A+rabbit anti-sheep IgG assembly[76].
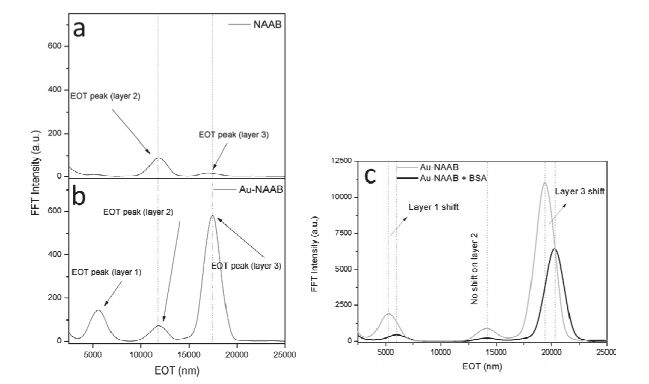
Figure 8.
Comparison of the Fast Fourier Transform plots of NAAB and Au-NAAB with the same layer 1 and layer 2 thicknesses. (a) RIFTS of NAAB. (b) RIFTS of Au-NAAB. (c) The optical response of the Au-NAAB before and after the introduction of the BSA protein[79].
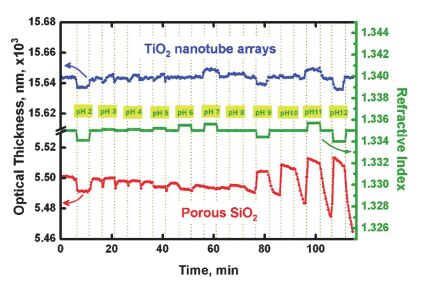
Figure 9.
Optical thickness response curves of TiO2 nanotube arrays and porous SiO2 upon exposure to buffer solutions in the range pH=2~12. The cell was flushed with pH=7.4 PBS buffer solution between each buffer exposure[91].
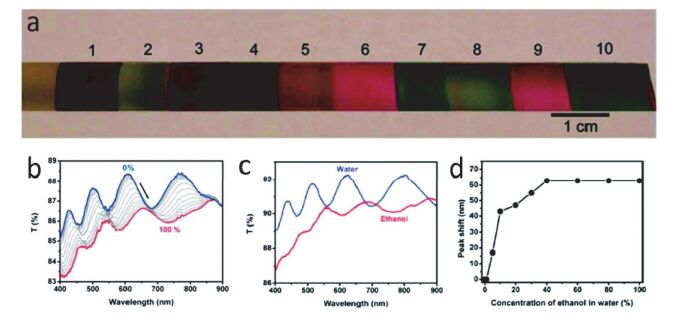
Figure 10.
(a) Photo of a series of ZIF-8 films of various thicknesses on Si substrates. UV-vis transmission spectra of ZIF-8 film on glass after exposure to (b) propane vapor of various concentrations from 0% (blue) to 100% (red) and (c) ethanol (red) or water (blue). (d) Interference peak (originally at 612 nm) shift versus ethanol concentration (V/V%) in ethanol/water solutions[102].
Sailor, M. J.; Link, J. R. Chem. Commun. 2005, 1375.
[27]
Janshoff, A.; Dancil, K.-P. S.; Steinem, C.; Greiner, D. P.; Lin, V. S. Y.; Gurtner, C.; Motesharei, K.; Sailor, M. J.; Ghadiri, M. R. J. Am. Chem. Soc. 1998, 120, 12108. doi: 10.1021/ja9826237
[28]
Meade, S. O.; Yoon, M. S.; Ahn, K. H.; Sailor, M. J. Adv. Mater. 2004, 16, 1811. doi: 10.1002/(ISSN)1521-4095
[29]
Berger, M. G.; Arens-Fischer, R.; Thönissen, M.; Krüger, M.; Billat, S.; Lüth, H.; Hilbrich, S.; Theiß, W.; Grosse, P. Thin Solid Films 1997, 297, 237. doi: 10.1016/S0040-6090(96)09361-3
Pacholski, C.; Sartor, M.; Sailor, M. J.; Cunin, F.; Miskelly, G. M. J. Am. Chem. Soc. 2005, 127, 11636. doi: 10.1021/ja0511671
[46]
Pacholski, C.; Yu, C.; Miskelly, G. M.; Godin, D.; Sailor, M. J. J. Am. Chem. Soc. 2006, 128, 4250. doi: 10.1021/ja056702b
[47]
Pacholski, C.; Perelman, L. A.; Vannieuwenhze, M. S.; Sailor, M. J. Phys. Status Solidi A 2009, 206, 1318. doi: 10.1002/pssa.v206:6
[48]
Kilian, K. A.; Boecking, T.; Gaus, K.; Gal, M.; Gooding, J. J. ACS Nano 2007, 1, 355. doi: 10.1021/nn700141n
[49]
Orosco, M. M.; Pacholski, C.; Sailor, M. J. Nat. Nanotechnol. 2009, 4, 255. doi: 10.1038/nnano.2009.11
[50]
Zhu, Y.; Soeriyadi, A. H.; Parker, S. G.; Reece, P. J.; Gooding, J. J. J. Mater. Chem. B 2014, 2, 3582. doi: 10.1039/C4TB00281D
[51]
Kilian, K. A.; Lai, L. M. H.; Magenau, A.; Cartland, S.; Bocking, T.; Di, G. N.; Gal, M.; Gaus, K.; Gooding, J. J. Nano Lett. 2009, 9, 2021. doi: 10.1021/nl900283j
[52]
Gupta, B.; Lowe, S. B.; Gooding, J. J.; Mai, K.; Wakefield, D.; Di, G. N.; Gaus, K.; Reece, P. J. Anal. Chem. 2015, 87, 9946. doi: 10.1021/acs.analchem.5b02529
[53]
Kelly, T. L.; Gao, T.; Sailor, M. J. Adv. Mater. 2011, 23, 1776. doi: 10.1002/adma.201004142
[54]
Tsang, C. K.; Kelly, T. L.; Sailor, M. J.; Li, Y. Y. ACS Nano 2012, 6, 10546. doi: 10.1021/nn304131d
Schwartz, M. P.; Yu, C.; Alvarez, S. D.; Migliori, B.; Godin, D.; Chao, L.; Sailor, M. J. Phys. Status Solidi A 2007, 204, 1444. doi: 10.1002/pssa.v204:5
[57]
Schwartz, M. P.; Alvarez, S. D.; Sailor, M. J. Anal. Chem. 2007, 79, 327. doi: 10.1021/ac061476p
[58]
Chen, M. Y.; Klunk, M. D.; Diep, V. M.; Sailor, M. J. Adv. Mater. 2011, 23, 4537. doi: 10.1002/adma.201102090
[59]
Chen, M. Y.; Sailor, M. J. Anal. Chem. 2011, 83, 7186. doi: 10.1021/ac201636n
Jessensky, O.; Muller, F.; Gosele, U. Appl. Phys. Lett. 1998, 72, 1173. doi: 10.1063/1.121004
[72]
Garcia-Vergara, S. J.; Habazaki, H.; Skeldon, P.; Thompson, G. E. Nanotechnology 2007, 18.
[73]
Law, C. S.; Sylvia, G. M.; Nemati, M.; Yu, J.; Losic, D.; Abell, A. D.; Santos, A. ACS Appl. Mater. Interfaces 2017, 9, 8929. doi: 10.1021/acsami.7b01116
Figure 1
(a) Schematic illustration of optical interference of nanoporous film. (b) Reflectometric interference spectrum of nanoporous film. (c) Fourier transforms of reflectometric interference spectrum of nanoporous film.
Figure 2
(a) Schematic of the porous Si double-layer biosensor consisting of a top layer with large pores and a bottom layer with smaller pores. (b) Relative reflectance spectra of thermally oxidized porous Si single-and double-layers. (c) Fourier transforms of thermally oxidized p-type porous Si single-and double-layers in air and immersed in ethanol[45].
Figure 3
Schematic illustration of real-time monitoring of enzyme activity in a mesoporous silicon double layer. Active protease (pepsin, blue) is absorbed into the first layer. The assay is then carried out by addition of substrate (α-casein, red). The pores of the second layer are too small to admit the protease or its substrate. However, digestion of the substrate by pepsin produces fragments (green) sufficiently small to infiltrate the second layer.[49]
Figure 4
(a) Reflectivity spectrum of a sample containing two photonic crystals, one on top of the other (double stack). A cross-sectional schematic of the structure is shown in the inset. (b) Cross-sectional secondary electron SEM of the double-stack porous silicon photonic crystal[61].
Figure 6
(a) Cross-section SEM image of AAO (scale bar=5 μm) and inset showing detail of the cylindrical nanopores (scale bar=500 nm). (b) Top-view SEM image of AAO (scale bar=3 μm). (c) Schematic illustration of the structure of AAO[73].
Figure 7
Experiments demonstrating specific binding affinity and controls for the AAO immunosensor. (a) Protein A and rabbit IgG separately dosed on bare AAO sample, demonstrating the greater relative affinity of the surface for protein A. (b) Rabbit anti-sheep IgG and chicken IgG introduced to a protein-A-modified sample. (c) Sheep IgG and chicken IgG introduced to a protein-A-modified sample that contains the protein A+rabbit anti-sheep IgG assembly[76].
Figure 8
Comparison of the Fast Fourier Transform plots of NAAB and Au-NAAB with the same layer 1 and layer 2 thicknesses. (a) RIFTS of NAAB. (b) RIFTS of Au-NAAB. (c) The optical response of the Au-NAAB before and after the introduction of the BSA protein[79].
Figure 9
Optical thickness response curves of TiO2 nanotube arrays and porous SiO2 upon exposure to buffer solutions in the range pH=2~12. The cell was flushed with pH=7.4 PBS buffer solution between each buffer exposure[91].
Figure 10
(a) Photo of a series of ZIF-8 films of various thicknesses on Si substrates. UV-vis transmission spectra of ZIF-8 film on glass after exposure to (b) propane vapor of various concentrations from 0% (blue) to 100% (red) and (c) ethanol (red) or water (blue). (d) Interference peak (originally at 612 nm) shift versus ethanol concentration (V/V%) in ethanol/water solutions[102].

 下载:
下载:

 下载:
下载:











