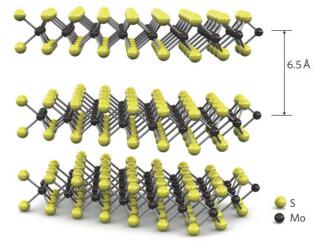
 图1
MoS2三维结构示意图[14]
Figure1.
Three-dimensional representation of the structure of MoS2[14]
图1
MoS2三维结构示意图[14]
Figure1.
Three-dimensional representation of the structure of MoS2[14]
引用本文:
林潇羽, 王璟. 二维过渡金属硫族化合物纳米材料的制备与应用研究进展[J]. 化学学报,
2017, 75(10): 979-990.
doi:
10.6023/A17060282

Citation: Lin Xiaoyu, Wang Jing. Research Progress on Preparation and Application of Two-Dimensional Transition Metal Dichalcogenides Nanomaterials[J]. Acta Chimica Sinica, 2017, 75(10): 979-990. doi: 10.6023/A17060282
Citation: Lin Xiaoyu, Wang Jing. Research Progress on Preparation and Application of Two-Dimensional Transition Metal Dichalcogenides Nanomaterials[J]. Acta Chimica Sinica, 2017, 75(10): 979-990. doi: 10.6023/A17060282
二维过渡金属硫族化合物纳米材料的制备与应用研究进展
English
Research Progress on Preparation and Application of Two-Dimensional Transition Metal Dichalcogenides Nanomaterials
Abstract:
Two-dimensional (2D) materials have received great attentions in recent years, including BN, transition metal dichalcogenides, transition metal oxides and black phosphorus. Among them, graphene-like transition metal dichalcogenides (TMDCs), such as MoS2, WS2, MoSe2, TiS2, are emerging as key materials in electronics and chemical industry because of their excellent physical and chemical properties. Because of the quantum confinement and surface effects, the 2D nanomaterials exhibit completely different properties from their bulk, leading to a new field in material science and technology. The ability to prepare high quality and large scale TMDCs is the foundation for their practical applications. Until now, many methods have been employed to prepare various morphologies of TMDCs, including mechanical cleavage, intercalation-exfoliation, ultrasonic-assisted liquid-phase exfoliation, chemical vapor deposition and hydrothermal synthesis. In this paper, the authors introduce the crystal structures and electronic properties of TMDCs briefly. The dimension from bulk to single or few layers leads to changes of these nanomaterials, showing novel properties in electronic transfer rate, catalytic activity, etc. Then the top-down and bottom-up preparation methods are summarized, and the advantages and disadvantages of these methods are discussed. At present, the challenge is that there are no proper ways to prepare TMDCs in large scale with controlled thickness and general application. As every single material has its performance limitation, the hotpot in preparation lies in the hybridization with other materials to create functional composites, aiming to improve their electronic and optical properties for special devices, and the most commonly used components are graphene and other 2D materials. And the authors also introduce the research progress in applications systematically, with emphasis on electronic devices, optoelectronic devices, sensing platforms, energy storage devices and catalyst, showing a wide range of applications. In addition, the authors also give some perspectives on the challenges and prospects in this field.
-
Key words:
- transition metal dichalcogenides
- / nanomaterials
- / preparation
- / application
-
1 引言
在过去的十几年里, 人们在二维材料方面的研究取得了巨大的进展. 2004年, 英国曼切斯特大学的Novoselov等[1]成功剥离出石墨烯, 开辟了材料研究的新领域.随后, 与石墨烯具有类似结构和性能的一种新材料——类石墨烯, 引起了物理、化学、材料和电子等众多领域研究人员的广泛关注.目前研究较多并取得一定成果的类石墨烯二维材料有六方氮化硼[2, 3]、过渡金属硫族化合物[4~6]、过渡金属氧化物[7]、过渡金属碳化物或碳氮化物[8, 9]、黑磷[10~12]等.其中, 过渡金属硫族化合物(Transition Metal Dichalcogenides, TMDCs)作为一种典型的层状材料, 具有类似甚至优于石墨烯的特性, 例如TMDCs的带隙可调, 因此可用于场效应晶体管和逻辑电路.除此以外, TMDCs在润滑、光电探测、传感、储能、催化等方面都有广泛的应用.
本文综述了TMDCs的晶体结构和电子特性, 常用制备方法及其在电子器件、光电器件、传感、微波吸收、储能、催化等方面的应用, 以期为后续的研究理清思路.
2 过渡金属硫族化合物的晶体结构和电子特性
TMDCs有40多种类型[13], 基本化学式为MX2, 其中M为过渡金属元素, X为硫族元素, 例如MoS2、WS2、MoSe2、TiSe2等.与石墨类似, TMDCs也是片层结构, 六方对称的晶胞在(001) 晶面方向进行堆叠, 层与层之间靠范德华力相互作用, 平均层间距为6~7 Å; 与石墨不同的是, TMDCs片层呈三明治形状, 由两个六边形结构的X原子层夹着一层M原子层形成一个MX2分子层, 可用“X-M-X”表示, 如图 1所示[14].
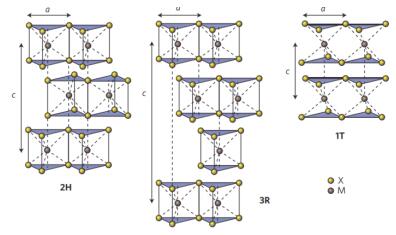
根据原子堆积方式和金属原子配位方式的不同, TMDCs通常有三种晶体结构[15]: 1T、2H和3R, 如图 2所示, 不同晶体结构之间性质存在较大差异.以MoS2为例, 在自然状态下1T-MoS2是热力学非稳定相, 2H-MoS2是热稳定相, 两者之间可以相互转化.
石墨烯是一种半金属材料, 而块体TMDCs展现出多种电子特性.根据金属原子的配位和氧化状态, 可以分为金属、半金属、半导体和超导体, 块体MoS2和WS2属于半导体材料, WTe2和TiSe2属于半金属, NbS2和VSe2具有金属性质, NbSe2和TaS2则表现出低温超导特性[16].
与石墨烯相比, TMDCs家族中半导体类材料最显著的特点在于带隙可调.以MoS2为例, 块体MoS2为间接带隙半导体, 带隙宽度为1.29 eV, 随着层数的减少, MoS2带隙逐渐变大, 最后单层MoS2发生间接带隙到直接带隙的转变, 带隙宽度为1.8 eV[14]. WS2和MoSe2也表现出相同的带隙特征[17, 18].在MoS2的拉曼光谱中可以观测到强烈的E2g1和Ag1振动模式, 层数对频率、强度以及两峰之间的宽度有着强烈的影响, 随着层数的增加, Ag1和E2g1分别出现蓝移和红移的现象, 因此利用拉曼光谱可以对MoS2的层数进行分析[19].由于量子限域效应和表面效应, 单层和少层TMDCs表现出不同于块体材料的性质, 例如单层MoS2光致发光效应和产氢催化能力的增强, 这些特性使其在电子器件、催化等领域具有广阔的应用前景.
3 过渡金属硫族化合物纳米材料的制备
材料的高质量可控制备是发挥其性能的重要基础, 目前制备TMDCs的方法可以分为“自上而下”的剥离法和“自下而上”的合成法.
3.1 机械剥离法
机械剥离法(Mechanical cleavage)是一种传统的制备手段, 以其相对简便的操作和剥离程度高的特点成为制备二维材料的成熟方法. 2004年, Novoselov等[20]就利用该法剥离出单层石墨烯.随后, 又相继剥离出单层2D材料BN、MoS2、NbSe2和Bi2Sr2CaCu2Ox. Li等[21]也利用该方法得到了平均厚度为0.8 nm的单层以及少层MoS2.通过拉曼光谱分析, 单层MoS2在384和400 cm-1处有强烈的拉曼峰, 对应模E2g1和Ag1.机械剥离法获得的样品结晶质量高、缺陷少、性质稳定, 并且与块体材料具有相同的晶体结构, 非常适合于后续的性质研究及单一电子器件的装配, 但是样品产量低, 一般用于实验室制备, 难以大规模应用.
3.2 超声液相剥离法
超声液相剥离法(Ultrasonic-assisted liquid-phase exfoliation)是2008年由Coleman研究组报道的一种分离石墨烯的方法[22], 随后他们利用该方法陆续剥离得到MoS2、NbSe2、TaSe2、MoTe2、b-BN和Bi2Te3等[23].这种利用超声打破层间范德华力, 实现材料从本体剥离的过程类似于聚合物在特殊溶液中的溶解[24], 可以用热力学的混合焓理论[22]以及分子层与溶剂分子之间的电子传输作用来解释.因此, 选择表面能与分子层间界面能相匹配的溶剂, 可以有效提高剥离效率.
常用的溶剂有三类:有机溶剂、离子溶剂和水-表面活性剂混合溶液. Coleman等[23]考察了多种溶剂对MoS2和WS2剥离效率的影响, 发现N-2-甲基-吡咯烷酮(NMP)的匹配效果最好, 可获得0.3 mg/mL的MoS2和0.15 mg/mL的WS2.而后, Wang等[25]利用N-甲基吡咯烷酮(NMP)、N-乙烯基吡咯烷酮(NVP)和环己基吡咯烷酮(CHP)作为超声剥离介质, 其中NMP为介质, 所得产物中大约39%的MoS2纳米片厚度小于5 nm.此外, 在有机溶剂中加入辅助剂可以改善剥离的效果.例如在NMP或者环己酮中添加NaOH, 可以通过增大材料的层间距来提高剥离效果.
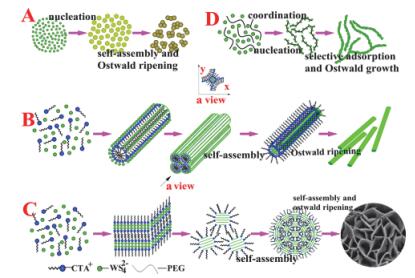
另一方面, Smith等[26]直接采用水作为溶剂, 同时加入胆酸钠作为表面活性剂以抑制产物团聚, 构成水-表面活性剂体系得到了多种TMDCs和BN纳米材料(图 3).样品产量与表面活性剂浓度、初始TMDCs浓度以及超声时间有关, 这为TMDCs纳米材料的批量制备提供了参考.
超声液相剥离法工艺简单, 可以大批量工业化生产, 获得的产物可应用于纳米复合热电材料、超级电容器和锂离子电池等方面, 尤其是最近有关在水相条件下成功剥离MoS2的文献报道更彰显了这一制备方法的工业化应用前景.但是该方法的剥离效果受到超声功率、超声时间以及剥离介质等多种因素的影响, 并且产品厚度难以控制、形貌单一.
3.3 插层剥离法
插层剥离法(Intercalation-Exfoliation)是通过在层间插入小分子或者在片层上加上非共价连接的分子或聚合物以获得层间化合物, 达到削减层间作用力和剥离层状材料的目的.插入分子的方法以电化学插入、气相插入和液相插入为主, 插层试剂主要包括热稳定性差的硫酸、硝酸、溴素和化学稳定性差的碱金属Li、K、Cs、Rb等.
1986年, Joensen等[27]首次采用锂离子插层法获得了MoS2.其基本原理是先利用锂离子插层剂嵌入到MoS2粉末中, 形成LixMoS2 (x≥1) 插层化合物, 再通过插层化合物与质子性溶剂剧烈反应所产生出的大量H2增大MoS2的层间距, 进而得到多层甚至单层MoS2. Matte等[28]采用相同方法, 将块体材料浸入丁基锂溶液中100 ℃处理72 h, 得到单层及少层MoS2和WS2. Eda等[29]进一步对MoS2产物的晶体结构进行研究, 结果发现:室温下获得的产物为1T相和2H相的混合物, 二者各占约50%.随着退火处理温度的提高, 热力学非稳定相1T逐渐消失, 300 ℃下热力学稳定相2H可达到95%.
为了改进化学锂离子插层法反应温度高、时间长等缺点, Zeng等[30, 31]运用电化学锂离子插层剥离法, 将层状块体材料作为阴极, 锂箔作为阳极, 充电过程锂离子插入层状材料层隙中, 放电过程锂与水反应生成H2以达到剥离目的.利用该思路还可以制备多种TMDCs (MoS2, WS2, TiS2, WSe2, NbSe2等)、BN和石墨烯纳米片.这种方法可以在室温下进行, 反应时间较化学锂离子插层法更短, 并且制备过程可通过放电曲线调控.
锂离子插层法剥离效率高、应用范围广, 几乎可以将所有的层状材料剥离至单层或者少层, 但是该过程的操作比较复杂, 可能会改变样品的电学性质, 限制了其在电子器件方面的应用.
3.4 水(溶剂)热合成法
水热合成法(Hydrothermal synthesis)和溶剂热合成法(Solvothermal synthesis)是采用水或者溶剂作为反应介质在高温、高压环境中使通常难溶或不溶的物质溶解并进行重结晶反应的方法.反应温度范围一般在100~1000 ℃、压力范围1 MPa~1 GPa, 具有操作简单、污染小、成本低廉以及纯度高的特点, 是一种非常具有潜力的TMDCs合成方法.
研究人员很早就利用水热法合成出TMDCs[32, 33]. He等[34]以硫酸钛和硫化钠为反应物, 在200 ℃下水热反应4 h, 洗涤干燥后获得了TiS2粉末. Zhang等[35]将硒粉和钼酸钠溶解在水合肼中, 而后转入密闭的反应釜中200 ℃反应48 h, 获得了纳米花状的MoSe2. Chakravarty等[36]采用同样的思路, 将钨酸铵和单质硒溶解在水合肼中, 150~170 ℃水热反应48 h后得到微米棒状WSe2, 而采用钨酸为钨源时, 在相同的条件下获得的产物为纳米颗粒状.
随着制备手段和制备条件的不断丰富, 研究人员发现各种反应条件都会影响合成过程, 其中表面活性剂的加入可以有效地控制产物的形貌和结构. Wu等[37]采用硫代钨酸铵为原料, 吡啶为溶剂, 三辛胺(TOA)为表面活性剂, 制备出了空心球状的WS2.将空心球在氩气气氛下退火, 随着退火温度的升高(873~1123 K), WS2晶化程度加强, 但空心球结构被破坏.随后, Cao等[38]利用钨酸钠和硫脲为原料, 以十六烷基三甲基溴化铵(CTAB)为表面活性剂制备出纳米花状的WS2, 该样品表现出良好的可见光吸收性能.作者进一步研究发现, 反应时间对产物形态的影响远不如表面活性剂的种类和用量, 并提出了表面活性剂作用机理[39](图 4).
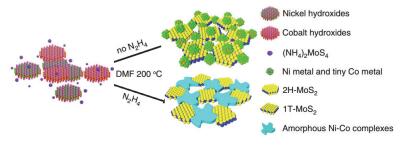
水(溶剂)热法不仅可以合成单组分的纳米材料, 还可用于复合材料的制备. Huang等[40]开发了一种阳离子表面活性剂辅助的水热合成法, 制备出类石墨烯MoS2/石墨烯(GL-MoS2/G)复合材料. Shelke等[41]采用水热法制备出WS2/rGO (reduced graphene oxide)纳米杂化材料用于紫外光探测. Li等[42]利用超薄镍钴氢氧化物纳米片和四硫代钼酸铵作为前驱体, 通过在N, N-二甲基甲酰胺体系中引入水合肼, 实现了MoS2从2H和1T的混合相到纯1T相的完全转变, 并且调节了镍钴部分的结晶状态, 最终形成了非晶态镍钴配合物和1T相MoS2的复合材料(图 5), 非晶态镍钴配合物起到稳定1T相的作用.与传统的制备方法不同, 水热法制备的复合材料实现了分子层面的复合, 增强相与相、相与基体之间的能量和物质流动, 在催化、吸波等方面具有广泛的应用前景.
总的说来, 水热法可以通过控制反应条件对产物进行有效调控, 但受到反应温度所限, 产物晶体结构不够完整, 并且无法精确控制产物的厚度.
3.5 化学气相沉积法
化学气相沉积法(Chemical Vapor Deposition, CVD)是工业中常用的制备无机纳米材料的方法, 主要包含两种思路, 以MoS2为例: (1) 钼源先在衬底表面沉积, 而后在高温下硫化或发生钼源热分解; (2) 钼源在高温下升华后和硫蒸气反应生成MoS2, 沉积到衬底表面成核生长. CVD可以大量制备厚度可控、大小可调和电学特性良好的2D纳米薄膜[43].
基于思路(1), Terrones研究组[44]以WO3和S为前驱体, 通过预沉积步骤, 将WOx沉积在Si/SiO2基底上, 再利用S蒸气直接硫化处理, 可以合成面积为1 cm2的单层和少层WS2薄膜.同样的思路, 可以用MoO3或Mo和S为前驱体, 制备MoS2.并且MoS2薄膜的大小和厚度分别由衬底的大小和Mo薄膜的厚度决定, 使得其合成过程高度可控[45, 46].但该方法获得的MoS2薄膜单晶畴区极小(大约20 nm), 薄膜的电子迁移率只有0.004~0.04 cm2/(V·s), 比机械剥离的MoS2纳米片[0.1~10 cm2/(V·s)]差了几个数量级, 缺陷多导致的低电子迁移率和低电流通断比将限制材料的后续研究和应用. Laskar等[47]用蓝宝石衬底代替Si/SiO2基底, 先用电子束蒸发沉积5 nm厚的Mo, 然后高温硫化处理, 获得了(0001) 晶向的高质量单晶MoS2薄膜.并且随着生长温度的提高, 薄膜的电学特性越来越接近于天然MoS2, 基于所合成的MoS2制备的场效应晶体管的电子迁移率可以达到(12±2) cm2/(V·s).
基于思路(2), Najmaei等[48]报道了MoS2的可控气相合成方法, 制备出大面积、高结晶度的单层和少层MoS2薄膜.在该方法中, MX2的成核位点控制至关重要.他们发现二维MoS2偏向于在基底边缘、污点或者粗糙的基底表面生长, 因此通过基底表面图案化设计可以控制成核位点, 不同位点的单晶相互连接得到厘米尺寸的薄膜. Lee等[49]采用APCVD (Ambient Pressure Chemical Vapor Deposition)的方法, 利用芳香族化合物二萘嵌苯-3, 4, 9, 10-四羧酸四钾盐(PTAS)作为“种子诱导剂”促进MoS2核的生长, 合成了结晶度高、缺陷少、形态均匀的MoS2, 并且实现了衬底和MX2薄膜的简易分离.随后, 为了获得畴区尺寸可调的MoS2, Shi等[50]以金箔为生长基底, 通过基底温度和载气组分的控制, 实现了MoS2畴区尺寸从纳米到微米级的调控, 并获得边缘长度为81 μm的三角形单晶MoS2.将金箔上合成的纳米MoS2直接用于电催化析氢, 具有高效的产氢效率.
CVD除了可以制备大面积薄膜, 还可以合成各种精细结构, 该过程受到多种因素的影响. Wang等[51]利用MoO3和Se粉末为前驱体制备了MoSe2纳米片, 并研究了Se/Mo比对MoSe2纳米片形态的影响.研究发现, 当Se/Mo比为20、10和5时, 产物形态为片状, 厚度分别为32、10和8 nm; 当Se/Mo比减小至2时, 产物为纳米颗粒和纳米短棒的混合形态.
就目前而言, CVD法可以用于合成高质量的TMDCs, 但制备条件比较苛刻, 并且适用性相对较窄, 除了MoS2、WS2、MoSe2等少数外[52], 一般难以采用该方法.如何拓展该法的普适性还有待进一步研究.
3.6 胶体合成法
胶体合成法(Colloidal synthesis)是在高沸点溶剂中加入还原剂、表面修饰剂等, 将前驱物热解, 以此得到尺寸均一的无机纳米晶. Mahler等[53]通过胶体合成法制备了1T相WS2纳米片, 当在合成过程中加入六甲基二硅胺(HMDS)后, 导致WS2形态和晶体结构剧烈变化, 实现1T-WS2到2H-WS2的转变.
与前几种方法相比, 有关TMDCs胶体合成法的研究较少, 但是该方法可以精确控制反应条件, 获得高结晶度的纳米材料, 可运用于光催化、储能等方面[54, 55].
4 过渡金属硫族化合物的应用研究
4.1 电子器件
石墨烯以及类石墨烯材料由于其出众的电学和力学性质, 一直是人们研究的热点[56]. 2004年, Novoselov等[1]就报道了第一个以石墨烯为基础的场效应晶体管(Field Effect Transistor, FET), 表现出突出的双极性电场效应.但是石墨烯本身固有的“0”带隙效应, 严重限制了FET的效率以及在逻辑电路方面的应用.随后, 许多研究致力于打开石墨烯的带隙, 但都以牺牲导电性[57]或者消耗大量能量[58]为代价.寻求具备合适带隙的类石墨烯材料成为新的解决方法.在TMDCs中, 以WS2和MoS2为代表的半导体类材料, 具有合适的可调带隙.这个特性使得TMDCs能够取代石墨烯, 被用于场效应晶体管、逻辑电路等电子器件领域.
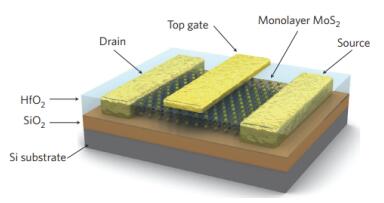
尽管MoS2具有合适的带隙宽度, 但是单层MoS2制成的背栅晶体管的电子迁移率仅有0.5~3 cm2/ (V·s)[20], 难以实际应用.随后Chen等[59]和Konar等[60]的研究表明, 引入高介电常数的栅介质层, 可有效地提升器件中载流子迁移率, 其原因在于高电介质可以通过介电屏蔽作用抑制MoS2的库伦杂质散射. Radisavljevic等[14]采用原子沉积技术制备了30 nm厚的高介电常数HfO2作为栅极介质, 采用微机械剥离法制备了6.5 Å厚的MoS2作为导电通道(图 6), 使得MoS2电子器件的电子迁移率从10 cm2/(V·s)提高至200 cm2/(V·s), 接近改性后的石墨烯纳米带[电子迁移率为200 cm2/(V·s)[61]]以及2 nm厚的应变硅[电子迁移率为250 cm2/(V·s)[62]], 同时电流开关比高达108.除了HfO2外, 利用Al2O3代替SiO2作为介质绝缘层[63], 或者利用水溶性聚乙烯醇(PVA)作为封盖层[64], 都可以提升MoS2晶体管的电子迁移率.
Yoon等[65]采用非平衡格林函数对MoS2晶体管的性能极限进行了研究.结果表明, MoS2晶体管的电流通断比可以达到1010, 且不受短沟道效应的影响, 适用于构建低功率器件.除了MoS2晶体管外, 其他TMDCs晶体管的研究也取得了一定的进展. Ovchinnikov等[66]制备了n型WS2场效应晶体管, 其电子迁移率随温度降低而增大, 当温度降低至83 K左右时, 单层WS2的迁移率最高可以达到140 cm2/(V·s), 而双层WS2可以达到300 cm2/(V·s)以上.
无论何种材料都存在性能极限, 随着二维材料的研究不断深入, 利用石墨烯等材料的高电子迁移特性以及TMDCs的带隙可调特性制备新型复合材料, 以期构建性能更加优越的电子器件成为必然趋势. Kwak等[67]研究了MoS2-石墨烯异质结的特性.该异质结在MoS2和石墨烯之间形成了肖特基势垒, 而由栅极电压控制的肖特基阻抗可以用来调节石墨烯上电子的传输特性.在不损耗石墨烯电子迁移能力的前提下, 该FET的电流开关比达到100, 可以初步实现逻辑开关功能.之后, Srivastava等[68]对各种FET材料进行了总结, 虽然复合材料能弥补单一组分的缺陷, 但是总体性能并没有达到预期值, TMDCs电子器件的性能还没有完全释放, 这个领域仍有许多问题亟待解决.
逻辑电路也是一类重要的电子器件, 通过将简单的单元集成在同一个芯片上, 可以实现更加复杂多样的功能.在电子电路小型化、功能化、集成化不断发展的趋势下, TMDCs的能带结构和平面特性使其非常适于成为纳米电子器件的基本构筑单元.
Radisavljevic等[69]制备了由两个MoS2晶体管组成的电子器件, 该电子器件可以实现信号放大和简单的逻辑操作.当输入逻辑信号“0”(低电压输入), 可以转变成逻辑信号“1”(高电压输出); 也可以实现NOR门, 当两个逻辑信号都为“off”时, 逻辑电路输出为“1”, 否则输出为“0”(输出电压为0), 由此可以推导出其他的逻辑运算(AND、OR等等).对于逻辑电路, 为了使上一个逻辑门的输出可以作为下一个逻辑门的输入, 其电压增益需要高于1. Zou等[70]利用Y2O3作为缓冲层, 研制出HfO2/Y2O3/MoS2顶栅晶体管.将其作为反相器, 电压增益可以达到16, 非常适合于逻辑器件集成.
4.2 光电器件
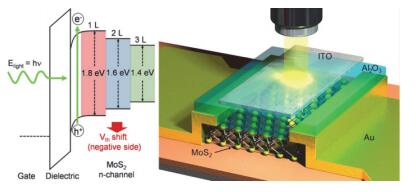
TMDCs的光学性质和其能带结构有关, 而能带结构又和材料的层数相关, 调控材料的层数即可改变其光电特性.并且, MoS2具有良好的光敏特性和响应速度, 因此可用于光电探测领域. Lee等[71]设计了一种顶栅结构分别为单层、双层和三层MoS2, 栅电极为透明导电玻璃(ITO), 栅介质层为Al2O3的光电晶体管.结果显示, 三层MoS2(带隙1.35 eV)光电晶体管适合于红光的探测, 单层(带隙1.82 eV)和双层MoS2(带隙1.65 eV)可以作为绿光探测器, 如图 7所示.
考虑到传统的光电探测器一般采用SiO2、HfO2和Al2O3作为栅介质, 如果要实现高灵敏度, 需要较高的源极偏压和施加额外的栅极偏压.为此Wang等[72]研制了铁电介质为栅极的MoS2光电探测器, 其中三层MoS2作为光敏半导体通道, 铁电材料聚偏二氟乙烯-三氟乙烯共聚物[P(VDF-TrFE)]作为顶栅介质, 利用P(VDF-TrFE)的剩余极化来降低MoS2半导体通道的暗电流.同时, 剩余极化能在局部产生109 V/m的强静电场, 远远大于传统场效应晶体管的栅极偏压, 使得铁电介质栅极MoS2光电探测器具有超灵敏的响应.该器件对1 nW的激光功率表现出2570 A/W响应度, 探测率高达2.2×1012 Jones.
对于光电探测器而言, 光响应波段的宽窄也是衡量其性能的重要指标.由于光电探测器是基于光电效应, 可探测的光波段取决于材料的带隙, 而MoS2等半导体固有的带隙宽度(1.29~1.8 eV)限制了其响应光谱区的范围.有研究发现, 在半导体的原子晶格上引入缺陷是调整带隙以实现宽光谱响应的重要方法. Xie等[73]通过控制MoS2原子晶格的缺陷程度, 主要是硫空位缺陷, 使得多层MoS2的带隙变窄, 实现了从445 nm(蓝光)到2717 nm(中红外)的超宽检测范围.随着波长的增加, 虽然响应度和探测率从50.7 mA/W和1.55×109 Jones降低至28.9 mA/W和0.88 Jones(栅极偏压为10 V), 但是与传统的单层MoS2探测器相比(入射光波长为550 nm, 栅极偏压为50 V时, 光响应度≤7.5 mA/W), 仍具有较高的光响应度.
尽管在MoS2上引入硫空位等缺陷能拓展光电探测器的响应范围, 但是缺陷的存在也影响了光电性能, 增加了光响应时间(4~30 s), 进而限制了其在光快速响应方面的应用. Chang等[74]采用CVD法获得了单层MoS2和MoSe2, 两者具有相近的电子迁移率[17~23 cm2/ (V·s)]和电流通断比(104~105), 但是TEM和光致发光谱显示MoSe2具有更少的缺陷, 而基于MoSe2的光电晶体管的光响应时间仅需25 ms, 使得MoSe2在光电子器件方面有着独特的应用前景.
太阳能电池是利用光伏效应将太阳能转化成电能的设备. TMDCs具有可控带隙宽度, 可响应多种波段的太阳光, 并且化学性质稳定, 是光伏电池的新型材料. Bernardi等[75]利用密度泛函理论(Density Functional Theory, DFT)等方法计算了MoS2、MoSe2和WS2对太阳光的吸收能力.理论计算表明, 厚度为1 nm的单层TMDCs, 其光吸收率可以达到5%~10%, 远远大于相同厚度Si和GaAs.同时研究人员构建了以MoS2/石墨烯为活性层材料的肖特基太阳能电池和基于MoS2/WS2异质结的激子太阳能电池, 发现在厚度为1 nm左右时, 能量转换效率(PCE)就可以达到1%, 虽然比GaAs材料小了30倍, 但所需厚度仅有GaAs材料的千分之一, 由此展现出TMDCs在超薄光伏器件方面的巨大优势.随后, 大量的研究集中于提升TMDCs的能量转换效率. Ma等[76]利用紫外臭氧处理化学剥离得到的WS2, 将其作为有机太阳能电池的空穴传输层, 电池的能量转换效率可以达到8.37%. Tsai等[77]将MoS2与p-Si组成II型异质结太阳能电池, 使能量转化效率达到5.23%.而后, Deng等[78]利用微电子和光子结构分析(AMPS-1D)软件模拟分析n-MoS2/p-Si异质结太阳能电池, 并从理论上进一步证明MoS2是理想的太阳能电池材料, 其极限转化效率可以达到19.0%, 而实现高效率的关键是有效控制Si和MoS2/Si界面上的缺陷水平.
除了光电探测器和太阳能电池, TMDCs还可以作为有机发光二极管(OLEDs)空穴注入材料和主体材料[79].另外, MoS2-聚乙烯吡咯烷酮(PVP)纳米复合材料作为闪存型存储器的电学双稳态材料也有报道[80].
4.3 传感器
传统的半导体氧化物传感器虽然具有高灵敏度的特点, 但是该类型传感器一般需要较高的工作温度(200~600 ℃)[81].因此, 开发一种室温下的高灵敏传感器成为研究的热点.二维TMDCs具有比表面积大、表面活性高以及高韧性等特性, 可用于构建超高灵敏、快速响应传感器. Li等[21]将机械剥离法制备得到的单层和少层n型半导体MoS2制成FET器件用于室温下NO气体的检测, 随着NO的吸附量增多, FET通道电阻增大、电流减小, 以此反映NO气体量, 检出下限为0.8 ppm.和机械剥离法不同, 用锂离子剥离法制备的纳米MoS2性质有所改变. Zeng等[30]采取锂离子剥离法制备的MoS2为p型半导体, 将其制成薄膜晶体管(Thin-film transistors, TFTs)用于NO气体的检测, 其对NO的响应与n型MoS2的FET完全相反, 随吸附量增多, 通道电阻减小、电流增大, 检出下限达到190 ppt.由此说明了制备方法会改变材料的特征, 进而影响器件的性能和工作原理.
除了NO气体, Late等[82]基于MoSe2纳米片构建了NH3气体传感器, 由于气体吸附导致的电子转移可以通过拉曼光谱的位移来反映, 以此检测环境中NH3量, 检测下限可以达到50 ppm. Li等[83]利用WS2纳米片构建了NH3气体传感器.对于5 ppm的NH3, 该传感器的响应时间和恢复时间仅需120和150 s, 并且改善了对甲醇、乙醇和甲醛等干扰物质的选择性.此外, 基于MoS2的NO2气体传感器[84]和基于WS2的湿度传感器[85]也有相关报道.这些传感器, 尤其是对有毒气体的检测, 在环境监测方面可以发挥重要的作用.
石墨烯、碳纳米管以及碳纳米粒子等在生物传感器方面表现出良好的性能[86~88].已有报道表明, TMDCs比石墨烯荧光淬灭能力更强, 并且对单链DNA和双链DNA表现出差异性的亲和能力, 有望获得更加灵敏的传感器. Zhu等[89]利用单层MoS2纳米片构建传感平台来检测核酸序列.染料标记的单链DNA探针(pDNA)可以通过核酸碱基和MoS2纳米片之间的范德华力而吸附在MoS2上, 然后染料的荧光淬灭, 而当pDNA与其互补的靶DNA(cDNA)相互识别后, 因为MoS2对单链DNA(ssDNA)和双链DNA(dsDNA)的亲和力不同, 染色标记的探针脱落, 体系展现出很强的荧光特征, 所以可以利用探针的荧光信号变化对DNA进行定量测定.利用Au修饰单层MoS2后, DNA与Au之间的Au-SH键可以进一步增强MoS2对DNA的吸附, 从而显著提高传感器的灵敏度和选择性[90].利用ssDNA和MoS2之间的高亲和力以及蛋白质标记技术, 可以将蛋白质检测转化为高灵敏度的DNA检测, Xiang等[91]以MoS2纳米片构建荧光生物传感平台来检测蛋白质.以链霉亲和素作为检测样品, 其检测下限可以达到0.67 ng/mL.对于一些重金属, 例如Hg2+和Pd2+, 利用Hg2+与DNA探针上T碱基的T-T错配反应形成T-Hg2+-T结构, 可以使荧光基团转化成游离态而展示很强的荧光.对于长度短、同源性高、低丰度的MicroRNAs, 利用WS2构建的传感器同样可以有效检测[92].
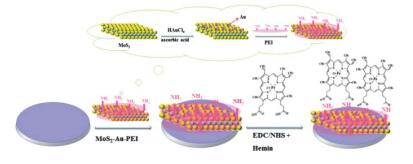
电化学传感器是化学、生物检测的另一个重要思路.前文介绍了一种染料标记的DNA检测方法, 鉴于其复杂性, Wang等[93]构建了无标记的电化学DNA传感器, 利用甲基蓝作为电化学活性指示剂, 采用微分脉冲伏安法测试整个DNA固定和杂交过程.研究人员以tlh基因片段作为目标DNA, 检测范围从1.0×10-16 mol/L到1.0×10-10 mol/L, 检测限为1.9×10-17 mol/L, 显著低于之前的报道.电化学生物传感器将电化学的高灵敏度和生物传感器的高特异性结合起来, 是一种更加优化的检测手段.盐酸克伦特罗(CLB)是一种人工合成的β-2-肾上腺素兴奋剂, 俗称“瘦肉精”, 对人体有破坏心血管和中枢神经的危害, 其检测工作一直受到人们的关注. Yang等[94]在玻璃碳电极上分层构建了MoS2-Au-PEI-hemin复合材料制成电化学传感器(图 8), 通过各组分协同作用使其对CLB具有优异的响应, 线性响应范围从10 ng/mL至2 μg/mL, 检测限为1.92 ng/mL, 并且可对真正猪肉样品进行检测.
4.4 微波吸收
随着电子器件的普及, 电磁污染成为一种新的环境问题.二维TMDCs独特的电性能和丰富的结构类型, 使其具备应用于微波吸收领域的潜力. 2015年Ning等[95]首次考察了超声剥离MoS2纳米片的电磁参数, 并与MoS2块体进行对比, 发现前者的介电常数是后者的两倍.当吸收剂质量分数为60%, 拟合厚度为2.4 mm时, 纳米片的最大反射损耗为-38.42 dB, 是体相材料的4倍, 有效带宽为4.1 GHz (9.6~13.76 GHz), 与碳基纳米材料的吸波性能相当.分析认为由Mo和S的空位所引入的缺陷偶极极化以及纳米片的高比表面积是导致其高电磁损耗的主要原因.随后, Liang等[96]首次考察了水热合成MoS2纳米片的微波吸收特性, 当拟合厚度为2.2 mm时, 吸收剂的最大反射损耗为-47.8 dB, 对应12.8 GHz, 有效带宽4.5 GHz (11~15.5 GHz).值得注意的是, 当厚度仅有1.9 mm时, 有效带宽达5.2 GHz.高导电性、较大的界面极化和高介电损耗是导致MoS2纳米片展示优异电磁损耗特性的主要原因.作者还考察了水热反应温度对产物电磁参数的影响, 发现180 ℃制备的MoS2其介电常数的实部和虚部高于其他样品.
除了片状的单一组分, 研究人员也开始将工作拓展到其他形貌以及复合材料的微波吸收特性研究. Zhang等[97]利用水热法批量制备出由2D纳米片堆积而成的3D MoS2纳米微球, 并将其与聚偏氟乙烯(PVDF)复合, 研究其在2~40 GHz宽频率范围内的电磁波吸收性能, 结果发现:当吸收剂质量分数为25%, 拟合厚度为2.5 mm时, MoS2/PVDF的最大反射损耗为-26.11 dB, 峰值在11.36 GHz, 有效带宽3.44 GHz (9.92~13.36 GHz).改变填料比例, 可以有效调节吸波性能:在18~40 GHz波段, 吸收剂质量分数为30%, 拟合厚度为1.5 mm时, 最大反射损耗为-27.47 dB, 峰值对应18.47 GHz; 将吸收剂质量分数降为20%时, 匹配厚度将显著增大, 厚度为3.5 mm时, 最大反射损耗-32.67 dB, 峰值在28.93 GHz.最后作者还对材料的微波吸收机制进行了探讨, 认为各种偏振、破坏性干扰理论、多次反射均是材料吸收微波的主要机制. Ding等[98]首次制备并考察了rGO/MoS2 3D复合结构的吸波性能, 匹配厚度为2.5 mm时, 其有效带宽为5.92 GHz, 最大反射损耗为−31 dB.文中作者还将自己的工作与他人进行了对比, 结果表明rGO-MoS2复合材料在轻质吸波材料领域的潜在应用. Zhang等[99]先用溶剂热法制备了NiS2纳米微球, 并以此为核, 在其表面生长了MoS2纳米片, 构成NiS2@MoS2的核壳结构.当吸收剂质量分数为20%, 拟合厚度为2.2 mm时, NiS2@MoS2/PVDF的最大反射损耗为-41.05 dB, 峰值对应12.08 GHz, 有效带宽4.40 GHz (10.16~14.56 GHz).与单组分吸波材料NiS2/ PVDF (-12.47 dB)和MoS2/PVDF (-15.40 dB)相比, 吸波性能明显提升.研究发现, 虽然NiS2和MoS2都具有较高的复介电常数, 为电介质损耗型吸波材料, 但是当吸收剂质量分数为30%时, MoS2的加入反而可以降低NiS2@MoS2的复介电常数, 以趋向阻抗匹配.同时, 材料之间的协同作用、多次反射等均有可能影响复合材料的吸波特性.改变复合吸收剂组分, 比如选用磁损耗型吸收剂, 相信对改善阻抗匹配效果、提高材料的吸波性能非常有利.
4.5 储能器件
锂离子电池(Lithium-ion batteries, LIBs)是一种新型化学电源, 理论能量密度可以达到410 Wh/kg, 是镍镉电池的2~3倍, 输出电压能达到铅蓄电池的2倍左右[100].但是金属锂电极在电化学循环过程中, 由于电极表面不均匀沉淀, 易形成锂枝晶和不均匀溶出, 而形成失去电化学活性的“死锂”.迄今为止, 碳基、硅基、锡基以及合金基材料都被提出作为负极材料, 尤其是用嵌锂化合物代替金属锂负极促使了LIBs的广泛应用, 但是这些材料在脱嵌锂的过程中会产生较大的体积膨胀, 大大影响电池的循环性能. TMDCs的层状结构有利于锂离子在电极中快速扩散, 同时少层TMDCs因为堆积结构非常松散, 在嵌锂前后可以极大地适应结构变化, 体积膨胀率低, 被认为是理想的LIBs负极材料.
2005年, Guo等[101]利用水热法合成直径约为200 nm的MoS2纳米微球作为LIBs的负极材料, 首次放电比容量达到了1272 mA·h·g-1, 是石墨电极的3~4倍.对于直接用做负极材料的TMDCs, 通常难以同时获得高容量和良好的循环特性.所以在循环20次以后, 比容量仅剩下400 mA·h·g-1. Du等[102]利用锂离子化学剥离法得到了MoS2纳米片, 再通过水热处理对剥离的MoS2进行改性, 得到重新堆积的MoS2.与原始MoS2相比, 重堆积MoS2的晶胞沿c轴方向拓宽了0.235 Å, 为锂离子进入S原子层提供更大的空间.将重堆积的MoS2作为LIBs负极材料, 在50次循环后比容量仍能达到750 mA·h·g-1, 而原始MoS2已经降低至226 mA·h·g-1. Wang等[103]直接将单层MoS2组装成三维管状结构, 独特的空心和孔隙结构非常适合用于构建LIBs负极材料, 放电比容量和循环性能都优于未经处理的MoS2纳米片. Zhou等[104]利用(3-巯基丙基)三甲氧基硅烷(MPS)对水热法制备的WS2进行硫化处理, 以修复WS2的硫空位和去除残留的氧原子.在锂离子存储性能方面, 当电流密度为0.8 A·g-1时, 硫化处理和未处理WS2的初始比容量分别为857.7和746.9 mA·h·g-1, 50次循环后降为566.8和381.7 mA·h·g-1.硫化处理使得WS2的锂离子存储性能提升了48.4%.之后, 作者还试验了SnS2, 并得到相同的效果, 说明硫化处理可以成为一种通用改性方法.
但是, 无论是原始还是改性后的TMDCs, 导电性能差一直是制约其发展的重要因素.大量的研究表明, 在LIBs中添加少量的石墨烯就可以改善LIBs的电化学性能[105, 106].于是, 将TMDCs与高导电性材料石墨烯复合成为改善TMDCs电极的重要思路. Chang等[107]以半胱氨酸(L-cysteine)作为模板和硫源, 再加上钼酸钠和氧化石墨烯, 利用水热法制备了MoS2/石墨烯复合材料, 当Mo和C的物质的量比为1:2, 电流密度为100 mA· g-1时, 该体系的比容量可以达到1100 mA·h·g-1, 在循环100次后比容量基本不变.当电流密度达到1000 mA·h·g-1时, 比容量依旧可以高达900 mA·h·g-1, 体现了MoS2/石墨烯作为LIBs负极材料的巨大潜力. Luo等[108]制备了MoSe2/rGO杂化材料, 其中石墨烯不仅作为MoSe2成核生长的位点, 而且是LIBs的电子传输通道.当电流密度为100 mA·h·g-1时, 该体系的首次放电比容量可以达到1399 mA·h·g-1, 可逆比容量为1001 mA·h·g-1.循环100次后, 可逆比容量仍可以达到715 mA·h·g-1, 展现出优异的储能特性, 同时该体系在2000 mA·g-1高电流密度下依旧具有高容量和良好的恢复能力, 展现出优良的倍率性能. Wang等[109]在制备MoS2/ rGO时, 利用表面活性剂乙醇对MoS2的形态进行控制, 获得纳米片状MoS2.当电流密度为100 mA·h·g-1时, 该体系的首次充放电比容量分别为1454和1902 mA·h· g-1, 库伦效率可以达到76.45%.
超级电容器是一种功率密度高、循环寿命长的新型储能元件.根据电荷存储的机理不同, 超级电容器主要可以分为双电层电容器(Electrical Double Layer Capacitor)、法拉第赝电容器(Pseudocapacitor)和混合型电容器(Hybrid capacitor). MoS2纳米材料具有巨大的比表面积和层间空间, 并且Mo原子在氧化还原过程中拥有多种价态, 因此, 以MoS2作为电极材料兼具双电层和法拉第赝电容器的特点[110, 111].另有研究表明, MoS2作为电极材料时, 比氧化物具有更快的电子传输能力[112], 比石墨烯有更高的理论比电容[113], 展现出优异的性能.
Wang等[114]制备了MoS2/聚苯胺(PANI)纳米复合材料, 以此为电极材料制备的超级电容器, 其比电容可以达到390 F·g-1, 高于一般的碳材料电极.虽然低于RuO2、MnO2等金属氧化物, 但是制备成本低、循环稳定性高, MoS2/PANI-38 (MoS2质量分数为38%)在1000次循环后仍能保持80%的比电容.并且, MoS2和PANI的比例对电容器的储能机理也有影响:当MoS2质量分数为8%时, 电容器的储能机理以双电层为主, 当MoS2质量分数为24%和38%时, 由于MoS2的团聚, 电容器的储能机理为法拉第赝电容.目前, 影响双电层形成的关键问题是复合材料中MoS2的团聚.如果能够解决上述问题, 将有望进一步提升此类复合材料的储能性能.
4.6 催化
氢能被认为是21世纪最清洁的能源, 是代替化石燃料的最佳选择之一.目前, 制取氢气的方法包括热分解法、太阳能光解法以及水电解法.在水电解法中, 催化剂的加入能显著降低电化学反应的过电位, 使得电解水具有非常高的制氢效率.寻求高效、价格低廉的催化剂用于析氢反应(Hydrogen Evolution Reaction, HER)是该领域的研究重点.
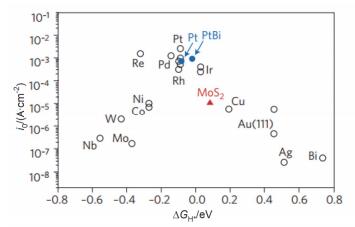
一直以来, 氢原子在催化剂表面的吉布斯自由能ΔGH*被用于反映HER电催化剂的本征活性, 交换电流密度和ΔGH*之间的关系可以用“火山模型”描述, 图 9中Pt、Rh、Re等贵金属位于火山峰处, 具有最高的交换电流密度和接近于0的氢原子吸附能.虽然Pt等贵金属作为HER催化剂具有最高的活性, 但是昂贵的价格限制了其大规模运用.研究人员一直在寻找可替代的催化剂.
2005年, Hinnemann等[116]开创性地利用DFT分析了MoS2的氢原子吸附能, 大约为+0.08 eV, 与Pt非常接近, 也位于“火山峰”附近.之后许多研究表明, MoSe2、WSe2等TMDCs也具有很高的HER催化活性[117~119].
许多理论和实验研究认为纳米TMDCs的催化活性位位于边缘位点上那些暴露的X (Mo, W等)原子, 而大量的表面位是催化惰性的[16]. Chorkendorff课题组[120]在研究MoS2的边缘催化位点时发现:在反应过程中, MoS2边缘位点只有四分之一结合了氢原子, 因此, 同一时刻只有四分之一的位点能释放H2, 而Pt(111) 的氢原子覆盖度大约为1 ML[121].因此, 通过调节材料边缘的电子结构来增强氢原子吸附能, 或者通过修饰纳米结构、改变电子传输结构, 提高单位面积的催化位点数量, 就能达到提升催化性能的目的.
利用碳材料做基底, 不仅可以提高HER催化剂的电子传输特性, 而且催化剂在碳基上的高度分散可以获得更多的活性位点. Li等[117]利用溶剂热法合成MoS2/rGO杂化材料用于HER研究. MoS2纳米粒在rGO层上高度分散充分暴露了MoS2的催化位点, 其过电位仅有0.1 V (Pt为0 V), Tafel斜率为41 mV·dec-1, 催化活性高于单一的MoS2催化剂, 与贵金属催化剂Pt接近(31 mV·dec-1).但不同于Pt的Volmer-Tafel机理, 该催化过程符合Volmer-Heyrovsky机理, H2的电化学解吸过程是反应的限速步骤. Xu等[122]也合成3D MoS2-rGO气凝胶, 其过电位和Tafel斜率只有105 mV和51 mV·dec-1, 交换电流密度可以达到3.28×10-5 A·cm-2, 与单一MoS2相比(120 mV和69 mV·dec-1), 催化性能有所提升.
TMDCs具有多种微观结构和形态, 例如纳米颗粒、纳米线和纳米片等, 而不同的形态将影响边缘催化位点数. Wang等[123]采用溶剂热法合成了石墨烯状的WSe2纳米片, 其独特的多孔纳米结构提供了更多的边缘催化位点, 初始过电位为150 mV, Tafel斜率78 mV·dec-1. Chen等[124]构建了MoO3-MoS2的核-壳结构纳米线, 其中MoS2壳起到催化HER和防护的作用, MoO3核提供了纵向的高比表面积模板和电荷传输通道.在电流密度为10 mA·cm-2时, 过电位为250 mV, Tafel斜率为50~60 mV·dec-1. Yang等[125]利用MoSe2纳米片分别构建了MoSe2和硫掺杂的MoSe2-x纳米多层结构, 高催化位点数和良好的导电性使其具有优异的HER催化性能.而后, Ding等[126]利用MoS2纳米片构建了片层取向更加有序的3D柱状分层结构, 不仅减少了片层团聚的情况, 而且使得片层充分暴露活性位点, 其Tafel斜率低至39 mV·dec-1.
TMDCs的多种晶体结构之间也存在非常大的差异, 例如2H-MoS2和2H-WS2相表现出半导体性质, 而将其转化成金属性1T-MoS2和1T-WS2相可以提升HER催化活性. Lukowski等[127]先在石墨基底上合成了纳米花状的2H-MoS2, 而后通过锂离子插层的方法实现了2H-MoS2向1T-MoS2的转变.在电流密度为10 mA· cm-2时, 2H-MoS2需要施加320 mV的过电位; 而在电流密度为200 mA·cm-2时, 1T-MoS2只需要施加400 mV的过电位, 表现出更高的HER催化活性. Voiry等[128]对MoS2的1T和2H相的催化效能差异进行研究, 结果发现1T和2H相的催化机理没有本质上的不同, 限制2H相催化性能的主要原因是高电阻妨碍了电子传递.在2H-MoS2中掺入单壁碳纳米管以降低其过电位, 可以提升2H-MoS2的催化性能.随后, Wang等[129]制备了一种1T/2H多相催化剂, 1T相为HER提供更多的活性位点和更优良的导电性能, 而2H相的存在有利于稳定1T亚稳定相, 当电流密度为10 mA·cm-2时, 其Tafel斜率为46 mV·dec-1, 过电位为234 mV. Li等[42]将非晶态镍钴配合物和1T相MoS2复合, 并将其用于析氢和析氧反应中, 在10 mA·cm-2处对应的过电位分别为70和235 mV, Tafel斜率为38.1和45.7 mV·dec-1.
除了催化HER反应, TMDCs还能用于催化加氢脱硫反应[130, 131]、减压渣油的加氢裂化[132]和CO甲烷化反应[133], 甚至作为制备Co3O4的衬底参与水裂解的研究也有报道[134].
5 总结与展望
过渡金属硫族化合物纳米材料具有独特的二维结构和优良的电学、光学以及催化特性, 在诸多领域表现出巨大的应用潜力.但目前的研究主要集中在MoS2, 对其他TMDCs的结构与性能探索明显关注不够.即使对MoS2的研究已取得了一定成果, 但仍有一些需要解决的问题.
首先是材料的制备方法和工艺方面.制备方法的难易程度、产量以及对材料性质的影响都是评价制备方法的依据.目前采用较多的制备TMDCs纳米材料的方法主要有机械剥离法、水(溶剂)热合成法和化学气相沉积法.其中机械剥离虽然不会改变材料本身的特性, 但产量过低, 且无法控制产物的尺寸以及厚度; 水(溶剂)热法不仅可以用于合成单组分物质, 也可以用于复合材料的制备, 但是该过程的影响因素多, 产物尺寸和厚度难以控制, 尤其是难以合成高结晶度的产品; 化学气相沉积法可以实现高质量TMDCs的批量制备, 但相对于前两种方法, 其适用性相对较窄, 还有待于进一步推广.由此可见, 制备技术仍是影响和制约TMDCs发展的重要因素, 寻求更为有效的手段是今后研究的主要方向.
其次是材料的应用性能方面. TMDCs在场效应晶体管、逻辑电路、传感器、微波吸收和催化等领域都有重要应用. TMDCs具有合适的带隙宽度, 可以实现电子器件的逻辑开关功能, 但是电子迁移率低是其固有的属性.与石墨烯等材料复合是对TMDCs的重要改性方法, 但复合材料的性能还有待进一步开发.带隙可调是TMDCs最大的优点之一, 通过调整合适带隙宽度来构建的光电探测器可以与不同波段的光耦合, 提高响应度, 同时TMDCs还可以作为太阳能电池材料, 用于构建超薄PV器件, 但是其PCE效率尚无法与传统材料相比.利用TMDCs构建传感器可以用于气体、湿度、生物分子的高灵敏度检测, 甚至可以间接测量金属离子, 但是对于一些物质的检测, 尤其是气体分子, 检测分子的特异性识别仍然存在许多问题, 制约了其实际应用.将TMDCs用于微波吸收材料是近几年出现的研究方向, 与传统材料相比, 单组分的MoS2就能表现出优异的吸波性能, 但是该领域的研究才刚起步, 吸波机理及成分、形貌对性能的影响规律尚不十分明确.当然, TMDCs在储能器件和催化方面的研究也待深入.
总而言之, 高性能的TMDCs材料及应用研究任重而道远.但机遇与挑战并存, 相信随着理论研究和实践的深入, 阻碍TMDCs发展的障碍将被慢慢移除, 新的性能将被发掘, TMDCs将以其独特的性质而得到更广泛的应用.
-
-
[1]
Novoselov, K. S.; Geim, A. K.; Morozov, S. V.; Jiang, D.; Zhang, Y.; Dubonos, S. V.; Grigorieva, I. V.; Firsov, A. A. Science 2004, 306, 666. doi: 10.1126/science.1102896
-
[2]
Nag, A.; Raidongia, K.; Hembram, K. P. S. S.; Datta, R.; Waghmare, U. V.; Rao, C. N. R. ACS Nano 2010, 4, 1539. doi: 10.1021/nn9018762
-
[3]
Behzad, S. Solid State Commun. 2016, 248, 27. doi: 10.1016/j.ssc.2016.09.007
-
[4]
Wang, Z. G.; Su, Q. L.; Yin, G. Q.; Shi, J. J.; Deng, H. Q.; Guan, J.; Wu, M. P.; Zhou, Y. L.; Lou, H. L.; Fu, Y. Q. Mater. Chem. Phys. 2014, 147, 1068. doi: 10.1016/j.matchemphys.2014.06.060
-
[5]
Tan, C. L.; Zhang, H. Chem. Soc. Rev. 2015, 44, 2713. doi: 10.1039/C4CS00182F
-
[6]
崔向红, 陈怀银, 杨涛, 化学学报, 2016, 74, 392. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345436.shtmlCui, X. H.; Chen, H. Y.; Yang, T. Acta Chim. Sinica 2016, 74, 392. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345436.shtml
-
[7]
Poizot, P.; Laruelle, S.; Grugeon, S.; Dupont, L.; Tarascon, J.-M. Nature 2000, 407, 496. doi: 10.1038/35035045
-
[8]
Naguib, M.; Mashtalir, O.; Carle, V.; Presser, V.; Hultman, L.; Gogotsi, Y.; Barsoum, M. W. ACS Nano 2012, 6, 1322. doi: 10.1021/nn204153h
-
[9]
Anasori, B.; Lukatskaya, M. R.; Gogotsi, Y. Nat. Rev. Mater. 2017, 2, 16098. doi: 10.1038/natrevmats.2016.98
-
[10]
Li, L. K.; Yu, Y. J.; Ye, G. J.; Ge, Q. Q.; Ou, X. D.; Wu, H.; Feng, D. L.; Chen, X. H.; Zhang, Y. D. Nat. Nanotechnol. 2014, 9, 372. doi: 10.1038/nnano.2014.35
-
[11]
Liu, H.; Du, Y. C.; Deng, Y. X.; Ye, P. D. Chem. Soc. Rev. 2015, 44, 2732. doi: 10.1039/C4CS00257A
-
[12]
Song, J.; Wang, J.; Lin, X. Y.; He, J. F.; Liu, H. L.; Lei, Y. P.; Chu, Z. Y. ChemElectroChem 2017, 4, 2373. doi: 10.1002/celc.v4.9
-
[13]
Marseglia, E. A. Int. Rev. Phys. Chem. 1983, 3, 177. doi: 10.1080/01442358309353343
-
[14]
Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Nat. Nanotechnol. 2011, 6, 147. doi: 10.1038/nnano.2010.279
-
[15]
Wang, Q. H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J. N.; Strano, M. S. Nat. Nanotechnol. 2012, 7, 699. doi: 10.1038/nnano.2012.193
-
[16]
Chhowalla, M.; Shin, H. S.; Eda, G.; Li, L. J.; Loh, K. P.; Zhang, H. Nat. Chem. 2013, 5, 263. doi: 10.1038/nchem.1589
-
[17]
Klein, A.; Tiefenbacher, S.; Eyert, V.; Pettenkofer, C.; Jaegermann, W. Phys. Rev. B 2001, 64, 205416. doi: 10.1103/PhysRevB.64.205416
-
[18]
Tongay, S.; Zhou, J.; Ataca, C.; Lo, K.; Matthews, T. S.; Li, J.; Grossman, J. C.; Wu, J. Nano Lett. 2012, 12, 5576. doi: 10.1021/nl302584w
-
[19]
Lee, C.; Yan, H. G.; Brus, L. E.; Heinz, T. F.; Hone, J.; Ryu, S. ACS Nano 2010, 4, 2695. doi: 10.1021/nn1003937
-
[20]
Novoselov, K. S.; Jiang, D.; Schedin, F.; Booth, T. J.; Khotkevich, V. V.; Morozov, S. V.; Geim, A. K. J. Am. Chem. Soc. 2005, 102, 10451. http://www.tandfonline.com/servlet/linkout?suffix=CIT0001&dbid=8&doi=10.1080%2F21663831.2013.824516&key=16027370
-
[21]
Li, H.; Yin, Z. Y.; He, Q.; Li, H.; Huang, X.; Lu, G.; Fam, D. W. H.; Tok, A. I. Y.; Zhang, Q.; Zhang, H. Small 2012, 8, 63. doi: 10.1002/smll.201101016
-
[22]
Hernandez, Y.; Nicolosi, V.; Lotya, M.; Blighe, F. M.; Sun, Z. Y.; De, S.; McGovern, I. T.; Holland, B.; Byrne, M.; Gun'Ko, Y. K.; Boland, J. J.; Niraj, P.; Duesberg, G.; Krishnamurthy, S.; Goodhue, R.; Hutchison, J.; Scardaci, V.; Ferrari, A. C.; Coleman, J. N. Nat. Nanotechnol. 2008, 3, 563. doi: 10.1038/nnano.2008.215
-
[23]
Coleman, J. N.; Lotya, M.; O'Neill, A.; Bergin, S. D.; King, P. J.; Khan, U.; Young, K.; Gaucher, A.; De, S.; Smith, R. J.; Shvets, I. V.; Arora, S. K.; Stanton, G.; Kim, H. Y.; Lee, K.; Kim, G. T.; Duesberg, G. S.; Hallam, T.; Boland, J. J.; Wang, J. J.; Donegan, J. F.; Grunlan, J. C.; Moriarty, G.; Shmeliov, A.; Nicholls, R. J.; Perkins, J. M.; Grieveson, E. M.; Theuwissen, K.; McComb, D. W.; Nellist, P. D.; Nicolosi, V. Science 2011, 331, 568. doi: 10.1126/science.1194975
-
[24]
Nicolosi, V.; Chhowalla, M.; Kanatzidis, M. G.; Strano, M. S.; Coleman, J. N. Science 2013, 340, 1226419. doi: 10.1126/science.1226419
-
[25]
Wang, K. P.; Wang, J.; Fan, J. T.; Lotya, M.; O'Neill, A.; Fox, D.; Feng, Y. Y.; Zhang, X. Y.; Jiang, B. X.; Zhao, Q. Z.; Zhang, H. Z.; Coleman, J. N.; Zhang, L.; Blau, W. J. ACS Nano 2013, 7, 9260. doi: 10.1021/nn403886t
-
[26]
Smith, R. J.; King, P. J.; Lotya, M.; Wirtz, C.; Khan, U.; De, S.; O'Neill, A.; Duesberg, G. S.; Grunlan, J. C.; Moriarty, G.; Chen, J.; Wang, J.; Minett, A. I.; Nicolosi, V.; Coleman, J. N. Adv. Mater. 2011, 23, 3944. doi: 10.1002/adma.v23.34
-
[27]
Joensen, P. F. R. F.; Morrison, S. R. Mater. Res. Bull. 1986, 21, 457. doi: 10.1016/0025-5408(86)90011-5
-
[28]
Matte, H. S. S. R.; Gomathi, A.; Manna, A. K.; Late, D. J.; Datta, R.; Pati, S. K.; Rao, C. N. R. Angew. Chem. 2010, 122, 4153. doi: 10.1002/ange.201000009
-
[29]
Eda, G.; Yamaguchi, H.; Voiry, D.; Fujita, T.; Chen, M.; Chhowalla, M. Nano Lett. 2011, 11, 5111. doi: 10.1021/nl201874w
-
[30]
Zeng, Z. Y.; Yin, Z. Y.; Huang, X.; Li, H.; He, Q. Y.; Lu, G.; Boey, F.; Zhang, H. Angew. Chem., Int. Ed. 2011, 50, 11093. doi: 10.1002/anie.v50.47
-
[31]
Zeng, Z. Y.; Sun, T.; Zhu, J. X.; Huang, X.; Yin, Z. Y.; Lu, G.; Fan, Z. X.; Yan, Q. Y.; Hng, H. H.; Zhang, H. Angew. Chem. 2012, 51, 9052. doi: 10.1002/anie.201204208
-
[32]
Peng, Y. Y.; Meng, Z. Y.; Zhong, C.; Lu, J.; Yu, W. C.; Jia, Y. B.; Qian, Y. T. Chem. Lett. 2001, 8, 772. http://ci.nii.ac.jp/naid/10006870510
-
[33]
Peng, Y. Y.; Meng, Z. Y.; Zhong, C.; Lu, J.; Yu, W. C.; Yang, Z. P.; Qian, Y. T. J. Solid State Chem. 2001, 159, 170. doi: 10.1006/jssc.2001.9146
-
[34]
He, H. Y. Res. Chem. Intermed. 2010, 36, 155. doi: 10.1007/s11164-010-0125-6
-
[35]
Zhang, X. H.; Yang, X. H.; Yang, F.; Xue, M. Q.; Luo, G. S. Micro Nano Lett. 2015, 10, 339. doi: 10.1049/mnl.2015.0014
-
[36]
Chakravarty, D.; Late, D. J. RSC Adv. 2015, 5, 21700. doi: 10.1039/C4RA12599A
-
[37]
Wu, J. F.; Fu, X. Mater. Lett. 2007, 61, 4332. doi: 10.1016/j.matlet.2007.01.099
-
[38]
Cao, S. X.; Liu, T. M.; Zeng, W.; Hussain, S.; Peng, X. H.; Pan, F. S. J. Mater. Sci.:Mater. Electron. 2014, 25, 4300. doi: 10.1007/s10854-014-2164-z
-
[39]
Cao, S. X.; Liu, T. M.; Hussain, S.; Zeng, W.; Peng, X. H.; Pan, F. S. Mater. Lett. 2014, 129, 205. doi: 10.1016/j.matlet.2014.05.013
-
[40]
Huang, G. C.; Chen, T.; Chen, W. X.; Wang, Z.; Chang, K.; Ma, L.; Huang, F. H.; Chen, D. Y.; Lee, J. Y. Small 2013, 9, 3693. doi: 10.1002/smll.201300415
-
[41]
Shelke, N. T.; Karche, B. R. J. Alloys Compd. 2015, 653, 298. doi: 10.1016/j.jallcom.2015.08.255
-
[42]
Li, H. Y.; Chen, S. M.; Jia, X. F.; Xu, B.; Lin, H. F.; Yang, H. Z.; Song, L.; Wang, X. Nat. Commun. 2017, 8, 15377. doi: 10.1038/ncomms15377
-
[43]
Lee, Y. H.; Zhang, X. Q.; Zhang, W. J.; Chang, M. T.; Lin, C. T.; Chang, K. D.; Yu, Y. C.; Wang, J. T. W.; Chang, C. S.; Li, L. J.; Lin, T. W. Adv. Mater. 2012, 24, 2320. doi: 10.1002/adma.201104798
-
[44]
Elías, A. L.; Perea-López, N.; Castro-Beltrán, A.; Berkdemir, A.; Lv, R. T.; Feng, S. M.; Long, A. D.; Hayashi, T.; Kim, Y. A.; Endo, M.; Gutiérrez, H. R.; Pradhan, N. R.; Balicas, L.; Mallouk, T. E.; López-Urías, F.; Terrones, H.; Terrones, M. ACS Nano 2013, 7, 5235. doi: 10.1021/nn400971k
-
[45]
Lin, Y. C.; Zhang, W. J.; Huang, J. K.; Liu, K. K.; Lee, Y. H.; Liang, C. T.; Chu, C. W.; Li, L. J. Nanoscale 2012, 4, 6637. doi: 10.1039/c2nr31833d
-
[46]
Zhan, Y. J.; Liu, Z.; Najmaei, S.; Ajayan, P. M.; Lou, J. Small 2012, 8, 966. doi: 10.1002/smll.201102654
-
[47]
Laskar, M. R.; Ma, L.; Kannappan, S.; Park, P. S.; Krishnamoorthy, S.; Nath, D. N.; Lu, W.; Wu, Y. Y.; Rajan, S. Appl. Phys. Lett. 2013, 102, 252108. doi: 10.1063/1.4811410
-
[48]
Najmaei, S.; Liu, Z.; Zhou, W.; Zou, X. L.; Shi, G.; Lei, S. D.; Yakobson, B. I.; Idrobo, J. C.; Ajayan, P. M.; Lou, J. Nat. Mater. 2013, 12, 754. doi: 10.1038/nmat3673
-
[49]
Lee, Y. H.; Yu, L.; Wang, H.; Fang, W.; Ling, X.; Shi, Y.; Lin, C. T.; Huang, J. K.; Chang, M. T.; Chang, C. S.; Dresselhaus, M.; Palacios, T.; Li, L. J.; Kong, J. Nano Lett. 2013, 13, 1852. doi: 10.1021/nl400687n
-
[50]
史建平, 马东林, 张艳锋, 刘忠范, 化学学报, 2015, 73, 877. doi: 10.3866/PKU.WHXB201503161Shi, J. P.; Ma, D. L.; Zhang, Y. F.; Liu, Z. F. Acta Chim. Sinica 2015, 73, 877. doi: 10.3866/PKU.WHXB201503161
-
[51]
Wang, B. B.; Zheng, K.; Zhong, X. X.; Gao, D.; Gao, B. J. Alloys Compd. 2017, 695, 27. doi: 10.1016/j.jallcom.2016.10.154
-
[52]
许冠辰, 卢至行, 张琪, 邱海龙, 焦丽颖, 化学学报, 2015, 73, 895. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345054.shtmlXu, G. C.; Lu, Z. X.; Zhang, Q.; Qiu, H. L.; Jiao, L. Y. Acta Chim. Sinica 2015, 73, 895. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345054.shtml
-
[53]
Mahler, B.; Hoepfner, V.; Liao, K.; Ozin, G. J. Am. Chem. Soc. 2014, 136, 14121. doi: 10.1021/ja506261t
-
[54]
Jung, W.; Lee, S.; Yoo, D.; Jeong, S.; Miro, P.; Kuc, A.; Heine, T.; Cheon, J. J. Am. Chem. Soc. 2015, 137, 7266. doi: 10.1021/jacs.5b02772
-
[55]
Guo, W. B.; Chen, Y. Z.; Wang, L. S.; Xu, J.; Zeng, D. Q.; Peng, D. L. Electrochim. Acta 2017, 231, 69. doi: 10.1016/j.electacta.2017.02.048
-
[56]
Huang, X.; Yin, Z. Y.; Wu, S. X.; Qi, X. Y.; He, Q. Y.; Zhang, Q. C.; Yan, Q. Y.; Boey, F.; Zhang, H. Small 2011, 7, 1876. doi: 10.1002/smll.201002009
-
[57]
Han, M. Y.; Ozyilmaz, B.; Zhang, Y.; Kim, P. Phys. Rev. Lett. 2007, 98, 206805. doi: 10.1103/PhysRevLett.98.206805
-
[58]
Zhang, Y.; Tang, T. T.; Girit, C.; Hao, Z.; Martin, M. C.; Zettl, A.; Crommie, M. F.; Shen, Y. R.; Wang, F. Nature 2009, 459, 820. doi: 10.1038/nature08105
-
[59]
Chen, F.; Xia, J. L.; Ferry, D. K.; Tao, N. J. Nano Lett. 2009, 9, 2571. doi: 10.1021/nl900725u
-
[60]
Konar, A.; Fang, T.; Jena, D. Phys. Rev. B 2010, 82, 115452. doi: 10.1103/PhysRevB.82.115452
-
[61]
Li, X. L.; Wang, X. R.; Zhang, L.; Lee, S.; Dai, H. J. Science 2008, 319, 1229. doi: 10.1126/science.1150878
-
[62]
Gomez, L.; Aberg, I.; Hoyt, J. L. IEEE Electron Dev. Lett. 2007, 28, 285. doi: 10.1109/LED.2007.891795
-
[63]
Kim, S. Y.; Park, S.; Choi, W. Appl. Phys. Lett. 2016, 109, 152101. doi: 10.1063/1.4964606
-
[64]
Guo, J.; Jiang, J.; Zheng, Z.; Yang, B. Org. Electron. 2017, 40, 75. doi: 10.1016/j.orgel.2016.10.043
-
[65]
Yoon, Y.; Ganapathi, K.; Salahuddin, S. Nano Lett. 2011, 11, 3768. doi: 10.1021/nl2018178
-
[66]
Ovchinnikov, D.; Allain, A.; Huang, Y. S.; Dumcenco, D.; Kis, A. ACS Nano 2014, 8, 8174. doi: 10.1021/nn502362b
-
[67]
Kwak, J. Y.; Hwang, J.; Calderon, B.; Alsalman, H.; Munoz, N.; Schutter, B.; Spencer, M. G. Nano Lett. 2014, 14, 4511. doi: 10.1021/nl5015316
-
[68]
Srivastava, A.; Fahad, M. S. Solid-State Electron. 2016, 126, 96. doi: 10.1016/j.sse.2016.09.008
-
[69]
Radisavljevic, B.; Whitwick, M. B.; Kis, A. ACS Nano 2011, 5, 9934. doi: 10.1021/nn203715c
-
[70]
Zou, X.; Huang, C. W.; Wang, L.; Yin, L. J.; Li, W.; Wang, J.; Wu, B.; Liu, Y.; Yao, Q.; Jiang, C.; Wu, W. W.; He, L.; Chen, S.; Ho, J. C.; Liao, L. Adv. Mater. 2016, 28, 2062. doi: 10.1002/adma.201505205
-
[71]
Lee, H. S.; Min, S. W.; Chang, Y. G.; Park, M. K.; Nam, T.; Kim, H.; Kim, J. H.; Ryu, S.; Im, S. Nano Lett. 2012, 12, 3695. doi: 10.1021/nl301485q
-
[72]
Wang, X. D.; Wang, P.; Wang, J. L.; Hu, W. D.; Zhou, X. H.; Guo, N.; Huang, H.; Sun, S.; Shen, H.; Lin, T.; Tang, M. H.; Liao, L.; Jiang, A. Q.; Sun, J. L.; Meng, X. J.; Chen, X. S.; Lu, W.; Chu, J. H. Adv. Mater. 2015, 27, 6575. doi: 10.1002/adma.201503340
-
[73]
Xie, Y.; Zhang, B.; Wang, S. X.; Wang, D.; Wang, A. Z.; Wang, Z. Y.; Yu, H. H.; Zhang, H. J.; Chen, Y. X.; Zhao, M. W.; Huang, B. B.; Mei, L. M.; Wang, J. Y. Adv. Mater. 2017, 29, 1605972. doi: 10.1002/adma.v29.17
-
[74]
Chang, Y. H.; Zhang, W. J.; Zhu, Y. H.; Han, Y.; Pu, J.; Chang, J. K.; Hsu, W. T.; Huang, J. K.; Hsu, C. L.; Chi, M. H.; Takenobu, T.; Li, H. N.; Wu, C. I.; Chang, W. H.; Wee, A. T. S.; Li, L. J. ACS Nano 2014, 8, 8582. doi: 10.1021/nn503287m
-
[75]
Bernardi, M.; Palummo, M.; Grossman, J. C. Nano Lett. 2013, 13, 3664. doi: 10.1021/nl401544y
-
[76]
马春燕, 傅伟飞, 黄国伟, 陈红征, 徐明生, 化学学报, 2015, 73, 949. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345057.shtmlMa, C. Y.; Fu, W. F.; Huang, G. W.; Chen, H. Z.; Xu, M. S. Acta Chim. Sinica 2015, 73, 949. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345057.shtml
-
[77]
Tsai, M. L.; Su, S. H.; Chang, J. K.; Tsai, D. S.; Chen, C. H.; Wu, C. I.; Li, L. J.; Chen, L. J.; He, J. H. ACS Nano 2014, 8, 8317. doi: 10.1021/nn502776h
-
[78]
Deng, Q. R.; Li, Y. Q.; Shen, Y. L.; Chen, L.; Wang, G. M.; Wang, S. G. Mod. Phys. Lett. B 2017, 31, 1750079. doi: 10.1142/S0217984917500798
-
[79]
Reynolds, K. J.; Barker, J. A.; Greenham, N. C.; Friend, R. H.; Frey, G. L. J. Appl. Phys. 2002, 92, 7556. doi: 10.1063/1.1522812
-
[80]
Liu, J.; Zeng, Z.; Cao, X.; Lu, G.; Wang, L. H.; Fan, Q. L.; Huang, W.; Zhang, H. Small 2012, 8, 3517. doi: 10.1002/smll.v8.22
-
[81]
Afzal, A.; Cioffi, N.; Sabbatini, L.; Torsi, L. Sens. Actuators B 2012, 171-172, 25. http://www.sciencedirect.com/science/article/pii/S0925400512004753
-
[82]
Late, D. J.; Doneux, T.; Bougouma, M. Appl. Phys. Lett. 2014, 105, 233103. doi: 10.1063/1.4903358
-
[83]
Li, X. G.; Li, X. X.; Li, Z.; Wang, J.; Zhang, J. W. Sens. Actuators B 2017, 240, 273. doi: 10.1016/j.snb.2016.08.163
-
[84]
Donarelli, M.; Prezioso, S.; Perrozzi, F.; Bisti, F.; Nardone, M.; Giancaterini, L.; Cantalini, C.; Ottaviano, L. Sens. Actuators B 2015, 207, 602. doi: 10.1016/j.snb.2014.10.099
-
[85]
Luo, Y. H.; Chen, C. Y.; Xia, K.; Peng, S. H.; Guan, H. Y.; Tang, J. Y.; Lu, H. U.; Yu, J. H.; Zhang, J.; Xiao, Y.; Chen, Z. Opt. Express 2016, 24, 8956. doi: 10.1364/OE.24.008956
-
[86]
He, S. J.; Song, B.; Li, D.; Zhu, C. F.; Qi, W. P.; Wen, Y. Q.; Wang, L. H.; Song, S. P.; Fang, H. P.; Fan, C. H. Adv. Func. Mater. 2010, 20, 453. doi: 10.1002/adfm.v20:3
-
[87]
Baby, T. T.; Aravind, S. S. J.; Arockiadoss, T.; Rakhi, R. B.; Ramaprabhu, S. Sens. Actuators B 2010, 145, 71. doi: 10.1016/j.snb.2009.11.022
-
[88]
Liu, Y. X.; Dong, X. C.; Chen, P. Chem. Soc. Rev. 2012, 41, 2283. doi: 10.1039/C1CS15270J
-
[89]
Zhu, C. F.; Zeng, Z. Y.; Li, H.; Li, F.; Fan, C. H.; Zhang, H. J. Am. Chem. Soc. 2013, 135, 5998. doi: 10.1021/ja4019572
-
[90]
Jin, K.; Xie, L. M.; Tian, Y.; Liu, D. M. J. Phys. Chem. C 2016, 120, 11204. doi: 10.1021/acs.jpcc.6b01193
-
[91]
Xiang, X.; Shi, J. B.; Huang, F. H.; Zheng, M. M.; Deng, Q. C.; Xu, J. Q. Biosens. Bioelectron. 2015, 74, 227. doi: 10.1016/j.bios.2015.06.045
-
[92]
Xi, Q.; Zhou, D. M.; Kan, Y. Y.; Ge, J.; Wu, Z. K.; Yu, R. Q.; Jiang, J. H. Anal. Chem. 2014, 86, 1361. doi: 10.1021/ac403944c
-
[93]
Wang, X. X.; Nan, F. X.; Zhao, J. L.; Yang, T.; Ge, T.; Jiao, K. Biosens. Bioelectron. 2015, 64, 386. doi: 10.1016/j.bios.2014.09.030
-
[94]
Yang, Y. Y.; Zhang, H.; Huang, C. S.; Yang, D. P.; Jia, N. Q. Biosens. Bioelectron. 2017, 89, 461. doi: 10.1016/j.bios.2016.04.019
-
[95]
Ning, M. Q.; Lu, M. M.; Li, J. B.; Chen, Z.; Dou, Y. K.; Wang, C. Z.; Rehman, F.; Cao, M. S.; Jin, H. B. Nanoscale 2015, 7, 15734. doi: 10.1039/C5NR04670J
-
[96]
Liang, X. H.; Zhang, X. M.; Liu, W.; Tang, D. M.; Zhang, B. S.; Ji, G. B. J. Mater. Chem. C 2016, 4, 6816. doi: 10.1039/C6TC02006B
-
[97]
Zhang, X. J.; Li, S.; Wang, S. W.; Yin, Z. J.; Zhu, J. Q.; Guo, A. P.; Wang, G. S.; Yin, P. G.; Guo, L. J. Phys. Chem. C 2016, 120, 22019. doi: 10.1021/acs.jpcc.6b06661
-
[98]
Ding, X.; Huang, Y.; Li, S. P.; Zhang, N.; Wang, J. G. Composites Part A 2016, 90, 424. doi: 10.1016/j.compositesa.2016.08.006
-
[99]
Zhang, X. J.; Wang, S. W.; Wang, G. S.; Li, Z.; Guo, A. P.; Zhu, J. Q.; Liu, D. P.; Yin, P. G. RSC Adv. 2017, 7, 22454. doi: 10.1039/C7RA03260A
-
[100]
Palacin, M. R. Chem. Soc. Rev. 2009, 38, 2565. doi: 10.1039/b820555h
-
[101]
Guo, G. H.; Hong, J. H.; Cong, C. J.; Zhou, X. W. J. Mater. Sci. 2005, 40, 2557. doi: 10.1007/s10853-005-2073-x
-
[102]
Du, G. D.; Guo, Z. P.; Wang, S. Q.; Zeng, R.; Chen, Z. X.; Liu, H. K. Chem. Commun. 2010, 46, 1106. doi: 10.1039/B920277C
-
[103]
Wang, P. P.; Sun, H.; Ji, Y.; Li, W.; Wang, X. Adv. Mater. 2014, 26, 964. doi: 10.1002/adma.v26.6
-
[104]
Zhou, L. Y.; Yan, S. C.; Pan, L. J.; Wang, X. R.; Wang, Y. Q.; Shi, Y. Nano Res. 2016, 9, 857. doi: 10.1007/s12274-015-0966-9
-
[105]
Seng, K. H.; Du, G. D.; Li, L.; Chen, Z. X.; Liu, H. K.; Guo, Z. P. J. Mater. Chem. 2012, 22, 16072. doi: 10.1039/c2jm32822d
-
[106]
Cao, Y.; Lin, X. G.; Zhang, C. L.; Yang, C.; Zhang, Q.; Hu, W. Q.; Zheng, M. S.; Dong, Q. F. RSC Adv. 2014, 4, 30150. doi: 10.1039/C4RA02838D
-
[107]
Chang, K.; Chen, W. X. ACS Nano 2011, 5, 4720. doi: 10.1021/nn200659w
-
[108]
Luo, Z. G.; Zhou, J.; Wang, L. R.; Fang, G. Z.; Pan, A. Q.; Liang, S. Q. J. Mater. Chem. A 2016, 4, 15302. doi: 10.1039/C6TA04390A
-
[109]
Wang, J. Y.; Zhao, X. M.; Fu, Y. S.; Wang, X. Appl. Surf. Sci. 2017, 399, 237. doi: 10.1016/j.apsusc.2016.12.029
-
[110]
Soon, J. M.; Loh, K. P. Electrochem. Solid-State Lett. 2007, 10, 250. doi: 10.1149/1.2778851
-
[111]
Zhou, J.; Fang, G. Z.; Pan, A. Q.; Liang, S. Q. ACS Appl. Mater. Interfaces 2016, 8, 33681. doi: 10.1021/acsami.6b11811
-
[112]
Zheng, N. F.; Bu, X. H.; Feng, P. Y. Nature 2003, 426, 428. doi: 10.1038/nature02159
-
[113]
Xiao, J.; Choi, D.; Cosimbescu, L.; Koech, P.; Liu, J.; Lemmon, J. P. Chem. Mater. 2010, 22.
-
[114]
Wang, J.; Wu, Z. C.; Hu, K. H.; Chen, X. Y.; Yin, H. B. J. Alloys Compd. 2015, 619, 38. doi: 10.1016/j.jallcom.2014.09.008
-
[115]
Nørskov, J. K.; Bligaard, T.; Rossmeisl, J.; Christensen, C. H. Nat. Chem. 2009, 1, 37. doi: 10.1038/nchem.121
-
[116]
Hinnemann, B.; Moses, P. G.; Bonde, J.; Jorgensen, K. P.; Nielsen, J. H.; Horch, S.; Chorkendorff, I.; Nørskov, J. K. J. Am. Chem. Soc. 2005, 127, 5308. doi: 10.1021/ja0504690
-
[117]
Li, Y. G.; Wang, H. L.; Xie, L. M.; Liang, Y. Y.; Hong, G. S.; Dai, H. J. J. Am. Chem. Soc. 2011, 133, 7296. doi: 10.1021/ja201269b
-
[118]
Li, T. S.; Galli, G. J. Phys. Chem. C 2007, 111, 16192. doi: 10.1021/jp075424v
-
[119]
Tsai, C.; Chan, K.; Abild-Pedersen, F.; Nørskov, J. K. Phys. Chem. Chem. Phys. 2014, 16, 13156. doi: 10.1039/C4CP01237B
-
[120]
Jaramillo, T. F.; Jorgensen, K. P.; Bonde, J.; Nielsen, J. H.; Horch, S.; Chorkendorff, I. Science 2007, 317, 100. doi: 10.1126/science.1141483
-
[121]
Skulason, E.; Karlberg, G. S.; Rossmeisl, J.; Bligaard, T.; Greeley, J.; Jonsson, H.; Norskov, J. K. Phys. Chem. Chem. Phys. 2007, 9, 3241. doi: 10.1039/B700099E
-
[122]
Xu, X. B.; Sun, Y.; Qiao, W.; Zhang, X.; Chen, X.; Song, X. Y.; Wu, L. Q.; Zhong, W.; Du, Y. W. Appl. Surf. Sci. 2017, 396, 1520. doi: 10.1016/j.apsusc.2016.11.201
-
[123]
Wang, X. Q.; Chen, Y. F.; Zheng, B. J.; Qi, F.; He, J. R.; Li, Q.; Li, P. J.; Zhang, W. L. J. Alloys Compd. 2017, 691, 698. doi: 10.1016/j.jallcom.2016.08.305
-
[124]
Chen, Z.; Cummins, D.; Reinecke, B. N.; Clark, E.; Sunkara, M. K.; Jaramillo, T. F. Nano Lett. 2011, 11, 4168. doi: 10.1021/nl2020476
-
[125]
Yang, Y.; Wang, S. T.; Zhang, J. C.; Li, H. Y.; Tang, Z. L.; Wang, X. Inorg. Chem. Front. 2015, 2, 931. doi: 10.1039/C5QI00126A
-
[126]
Ding, J. B.; Zhou, Y.; Li, Y. G.; Guo, S. J.; Huang, X. Q. Chem. Mater. 2016, 28, 2074. doi: 10.1021/acs.chemmater.5b04815
-
[127]
Lukowski, M. A.; Daniel, A. S.; Meng, F.; Forticaux, A.; Li, L.; Jin, S. J. Am. Chem. Soc. 2013, 135, 10274. doi: 10.1021/ja404523s
-
[128]
Voiry, D.; Salehi, M.; Silva, R.; Fujita, T.; Chen, M.; Asefa, T.; Shenoy, V. B.; Eda, G.; Chhowalla, M. Nano Lett. 2013, 13, 6222. doi: 10.1021/nl403661s
-
[129]
Wang, D. Z.; Zhang, X. Y.; Bao, S. Y.; Zhang, Z. T.; Fei, H.; Wu, Z. Z. J. Mater. Chem. A 2017, 5, 2681. doi: 10.1039/C6TA09409K
-
[130]
Lauritsen, J. V.; Nyberg, M.; Nørskov, J. K.; Clausen, B. S.; Topsøe, H.; Lægsgaard, E.; Besenbacher, F. J. Catal. 2004, 224, 94. doi: 10.1016/j.jcat.2004.02.009
-
[131]
Tsverin, Y.; Popovitz-Biro, R.; Feldman, Y.; Tenne, R.; Komarneni, M. R.; Yu, Z. Q.; Chakradhar, A.; Sand, A.; Burghaus, U. Mater. Res. Bull. 2012, 47, 1653. doi: 10.1016/j.materresbull.2012.03.053
-
[132]
Hur, Y. G.; Kim, M. S.; Lee, D. W.; Kim, S.; Eom, H. J.; Jeong, G.; No, M. H.; Nho, N. S.; Lee, K. Y. Fuel 2014, 137, 237. doi: 10.1016/j.fuel.2014.07.094
-
[133]
Zhang, C. Y.; Liu, B. N.; Wang, Y. X.; Zhao, L.; Zhang, J.; Zong, Q. Y.; Gao, J. S.; Xu, C. M. RSC Adv. 2017, 7, 11862. doi: 10.1039/C6RA27422F
-
[134]
Wang, X. D.; Zheng, Y. Y.; Yuan, J. H.; Shen, J. F.; Wang, A. J.; Niu, L.; Huang, S. T. Electrochim. Acta 2016, 212, 890. doi: 10.1016/j.electacta.2016.07.078
-
[1]
-




-



 下载:
下载:








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 518
- 文章访问数: 27230
- HTML全文浏览量: 7120
 下载:
下载:
