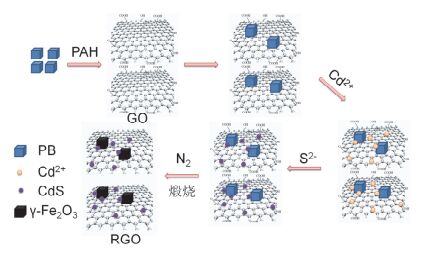
 图1
CdS/RGO/γ-Fe2O3复合物的合成示意图
Figure1.
Illustration of the synthesis procedure of CdS/RGO/γ-Fe2O3 composites
图1
CdS/RGO/γ-Fe2O3复合物的合成示意图
Figure1.
Illustration of the synthesis procedure of CdS/RGO/γ-Fe2O3 composites
引用本文:
吴佳佳, 季振源, 沈小平, 缪绪立, 徐克强. γ-Fe2O3纳米立方块修饰的Graphene/CdS复合光催化材料的合成及性能研究[J]. 化学学报,
2017, 75(12): 1207-1214.
doi:
10.6023/A17050220

Citation: Wu Jiajia, Ji Zhenyuan, Shen Xiaoping, Miao Xuli, Xu Keqiang. Synthesis of γ-Fe2O3 Nanocubes Decorated Graphene/CdS Nanocomposites with Enhanced Photocatalytic Performance[J]. Acta Chimica Sinica, 2017, 75(12): 1207-1214. doi: 10.6023/A17050220
Citation: Wu Jiajia, Ji Zhenyuan, Shen Xiaoping, Miao Xuli, Xu Keqiang. Synthesis of γ-Fe2O3 Nanocubes Decorated Graphene/CdS Nanocomposites with Enhanced Photocatalytic Performance[J]. Acta Chimica Sinica, 2017, 75(12): 1207-1214. doi: 10.6023/A17050220
γ-Fe2O3纳米立方块修饰的Graphene/CdS复合光催化材料的合成及性能研究
摘要:
利用普鲁士蓝(PB)作为γ-Fe2O3前驱体,先依次在氧化石墨烯(GO)片上负载PB和CdS纳米粒子,再将其置于惰性气体(N2)氛围下进行煅烧,成功制备出CdS/RGO/γ-Fe2O3三元复合光催化剂.通过改变PB负载量可以得到不同γ-Fe2O3含量的三元光催化剂,且PB的方块状形貌在煅烧后得以保持.利用XRD、EDS、TEM、FT-IR、UV-vis等手段对所制备的样品进行组成、结构、形貌、光吸收等的分析表征,并以罗丹明B(RhB)为模拟污染物研究上述催化剂对有机污染物的光催化降解性能.与二元复合物CdS/RGO相比,三元CdS/RGO/γ-Fe2O3光催化剂表现出更强的可见光催化活性,这说明γ-Fe2O3在光催化过程中起了重要作用.而且,由于γ-Fe2O3具有铁磁性,在外加磁场的作用下可以将光催化剂从反应体系中快速分离回收.同时,还研究了CdS/RGO/γ-Fe2O3光催化剂的降解动力学过程,并通过光催化剂的荧光表征和活性基团捕获实验,提出了光催化降解的机理.
English
Synthesis of γ-Fe2O3 Nanocubes Decorated Graphene/CdS Nanocomposites with Enhanced Photocatalytic Performance
Abstract:
With Prussian blue (PB) as the precursor for γ-Fe2O3, the tri-component CdS/RGO/γ-Fe2O3 photocatalyst was prepared through loading PB nanocubes and CdS nanoparticles on graphene oxide (GO) nanosheets, followed by a calcination process in inert atmosphere (N2). The content of γ-Fe2O3 in the CdS/RGO/γ-Fe2O3 photocatalyst can be adjusted by changing the loading amount of PB, and the cubic morphology of PB was maintained after the calcination. The composition, structure, morphology and light absorption of the as-prepared products were investigated by X-ray diffraction (XRD), X-ray energy dispersive spectroscopy (EDS), field emission scanning electron microscopy (SEM), transmission electron microscopy (TEM), infrared spectroscopy (FT-IR), Raman spectroscopy and ultraviolet-visible (UV-vis) spectroscopy. The photocatalytic activity of the ternary photocatalysts was evaluated by the degradation of the organic pollutant of Rhodamine B (RhB) under visible-light irradiation. It was found that the degradation process of RhB follows pseudo-first-order kinetics. Compared to the bi-component CdS/RGO photocatalyst, the tri-component CdS/RGO/γ-Fe2O3 exhibited greatly enhanced photocatalytic activity, demonstrating that the γ-Fe2O3 played an important role in the photocatalytic process. The CdS/RGO/γ-Fe2O3 composite with PB loading amount of 12 mg exhibits the highest photocatalytic degradation efficiency of about 99.8% and the highest apparent reaction rate constant (k) value of about 0.03289 min-1, which is almost 2.9 times and 1.8 times higher than that of CdS and CdS/RGO, respectively. This result indicates that a suitable loading amount of γ-Fe2O3 is important to optimize the photocatalytic performance of the CdS/RGO/γ-Fe2O3 composites. Moreover, owing to the ferromagnetism of γ-Fe2O3, the CdS/RGO/γ-Fe2O3 photocatalyst could be easily separated from the reaction solution for recycling by a magnet. A possible photocatalytic mechanism was also proposed based on the photoluminescence (PL) characterization and the active species capture experiment. It was demonstrated that the enhanced photocatalytic degradation properties of CdS/RGO/γ-Fe2O3 composites can be ascribed to the excellent conductivity of RGO and the construction of Z-scheme heterostructure between CdS and γ-Fe2O3, which facilitate the transport and separation of photogenerated carriers.
-
Key words:
- γ-Fe2O3
- / graphene
- / CdS
- / photocatalysis
- / mechanism
-
1 引言
目前, 半导体光催化技术在解决环境污染和能源危机方面已经取得了长足的发展[1].半导体光催化过程是光催化剂吸收光子产生光生电子-空穴对(e--h+pairs), 其中电子可以引发还原反应, 而空穴可以引发氧化反应[1~7]. 1976年, Carey等[8]报道TiO2在紫外灯照射下, 成功催化降解有机污染物氯联苯, 由此开启了光催化降解有机污染物的研究.在随后的研究中, 研究人员已开发出了各种各样的光催化剂用于有机污染物的降解.常见的是半导体氧化物(TiO2[9~16]、WO3[17, 18]、ZnO[19, 20])和硫化物(CdS[21~23]、ZnS[24~26])光催化剂.然而, 光催化技术的实际应用还需克服多方面的障碍.首先, 催化剂光响应范围窄.光催化技术的最终目的是对可再生能源太阳光的利用, 太阳光包含紫外光、可见光、近红外光等.然而, 大多数光催化过程仅能发生在紫外光条件下, 对太阳能的利用率低.其次, 光催化的效率低.研究人员发现半导体材料在光催化过程中常面临光生电荷快速复合这一难题, 造成仅有少部分光生电荷可以参与氧化还原反应, 减少了催化活性组分的生成, 致使催化效率降低.另外, 半导体材料在完成降解过程后的回收效率低, 不利于重复利用.因此, 为了实现光催化技术的实际应用, 人们正在开发新型可见光催化剂以拓宽催化剂的光响应范围和研发多组分催化剂, 发挥各个组分的协同作用, 降低光生电荷复合率及提高催化剂回收利用效率.
硫化镉(CdS)是一种直接带隙半导体, 其禁带宽度约为2.4 eV, 可作为可见光响应的催化剂[27~30]. CdS的价带位置不仅能满足光催化分解水、CO2光还原、降解有机污染物的要求, 而且CdS的导带电位比传统半导体如TiO2、ZnO更负, 在光催化过程中CdS导带电子具有更强的还原能力.然而, 单纯的CdS光催化剂有诸多不足, 如对反应物的吸附能力弱, 光稳定性差, 粒子团聚严重, 不利于增大催化剂的表面积, 光生电荷的分离效率低, 造成光催化活性降低.为了改善单组分CdS的光催化性能, 一般采用将CdS与其他材料复合以发挥协同优势. Zhou等[31]利用水热和高温煅烧相结合的方法, 制备了g-C3N4包裹的CdS纳米棒复合光催化剂, 并研究了其光电化学分解水产氢性能.发现CdS与g-C3N4之间形成Ⅱ型异质结构, 提高了光生电荷的分离效率, CdS@g-C3N4复合催化剂的产氢量是单组分CdS的2.5倍, 稳定性也得到提高. Liu等[32]利用微波辅助法制备了CdS/RGO复合光催化剂, 研究了该催化剂光照还原Cr (Ⅵ)的性能.发现在可见光照射下CdS/RGO复合物的光催化还原效率达到92%, 远高于单组分CdS (效率为79%).其原因为RGO的引入增强了催化剂对可见光的吸收, 促进了光生电子的转移和分离[33~37].
为了便于回收半导体催化剂, 研究人员通常将光催化剂与具有磁性的材料如Fe3O4[38], γ-Fe2O3[39], NiFe2O4[40]等进行复合.磁性组分的引入不仅使催化剂可以进行方便的磁分离回收, 还可与催化活性组分形成异质结, 协同提高光催化活性.目前, 许多具有磁性的复合光催化材料已经被报道, 如Fe3O4/TiO2[41]、Fe3O4/CdS[42]等.楼雄文课题组[43]利用微波加热法成功制备了铁磁性壳核结构γ-Fe2O3/ZnO纳米复合光催化剂.通过研究降解有机污染物亚甲基蓝(MB)、罗丹明B (RhB)和甲基橙(MO)的活性, 发现γ-Fe2O3/ZnO复合光催化剂的性能要远高于单组分的ZnO, 且具有更好的循环稳定性.
在本论文中, 我们合成了一种新型可磁分离的CdS/RGO/γ-Fe2O3三元复合可见光催化剂, 并对催化剂进行了系统表征和光催化降解RhB的性能研究.结果表明, 与CdS和CdS/RGO光催化剂相比, CdS/RGO/ γ-Fe2O3三元光催化剂表现出更高的光催化降解活性和稳定性, 且在外磁场作用下催化剂能够实现有效的磁分离, 系统考察了该三元光催化剂的催化反应动力学及光催化机理.
2 结果与讨论
2.1 样品的合成和表征
在本文中, 我们采用自组装、原位生长与煅烧相结合的方法成功制备了CdS/RGO/γ-Fe2O3三元复合光催化剂, 合成路线如图 1所示.首先采用水热法合成了立方块状普鲁士蓝(PB)纳米颗粒, 并用聚丙烯胺盐酸盐(PAH)修饰PB颗粒使其带上正电荷, 利用静电作用将之与带负电荷的氧化石墨烯(GO)纳米片进行自组装, 得到GO/PB复合物.然后再将CdS纳米粒子原位生长在GO纳米片上, 得到CdS/GO/PB复合物.最后将样品置于N2氛围中煅烧, 使PB分解形成γ-Fe2O3, 同时GO被热还原为RGO, 从而得到CdS/RGO/γ-Fe2O3复合物.将PB的加入量为9、12、15 mg所合成的最终样品分别标记为IOC-1, IOC-2, IOC-3 (见实验部分).
图1
CdS/RGO/γ-Fe2O3复合物的合成示意图
Figure1.
Illustration of the synthesis procedure of CdS/RGO/γ-Fe2O3 composites
采用XRD对合成的PB, CdS/RGO-3%, γ-Fe2O3/ RGO及IOC-2样品进行相结构表征.如图 2a所示, 样品CdS/RGO-3%(见支持信息)所有衍射峰与立方相CdS (JCPDS No. 10-0454)相吻合. PB的所有衍射峰(图S1)与JCPDS No. 73-0687相吻合[44], 对应于立方相的Fe4[Fe(CN)6]3.在γ-Fe2O3/RGO的XRD图中, 2θ值为30.24°, 35.63°, 43.28°, 53.73°, 57.27°和62.925°的衍射峰分别对应于立方相γ-Fe2O3 (JCPDS No. 39-1346)的(220), (311), (400), (422), (511)和(440)晶面, 表明γ-Fe2O3被成功合成.而在IOC-2的XRD图中, 除了γ-Fe2O3的衍射峰, 其余峰均可归属于立方相CdS (JCPDS No. 10-0454).但在CdS/RGO-3%、γ-Fe2O3/RGO和CdS/RGO/γ-Fe2O3样品的XRD图中, 均没有发现RGO的衍射峰, 这可归因于RGO低的含量和相对弱的衍射强度.样品IOC-1和IOC-3的XRD谱图与IOC-2一致, 均能观察到γ-Fe2O3和CdS的衍射峰(图S1). 图 2b为样品IOC-2的EDS能谱图.图中可观察到Fe、O、C、Cd和S元素, 与CdS/RGO/γ-Fe2O3三元复合物的组成相一致.其中C元素来源于RGO, 而O元素来源于γ-Fe2O3及RGO表面残留的含氧官能团.
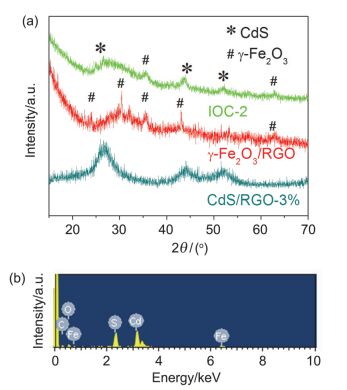 图2
(a) CdS/RGO-3%、γ-Fe2O3/RGO、IOC-2的XRD图, (b) IOC-2的EDS图
Figure2.
(a) XRD patterns of the as-prepared samples, (b) EDS spectrum of IOC-2
图2
(a) CdS/RGO-3%、γ-Fe2O3/RGO、IOC-2的XRD图, (b) IOC-2的EDS图
Figure2.
(a) XRD patterns of the as-prepared samples, (b) EDS spectrum of IOC-2
我们采用SEM与TEM来系统表征制备样品的形貌、尺寸和显微结构. 图 3a为PB的SEM图.从图中可以看出PB为大小均一的立方块, 粒径约为200 nm.由PB/GO复合物的TEM图(图 3b)可知, PB立方块均匀地负载在GO片上, 表明PB与GO能够很好地复合在一起; 图 3c, 3d为煅烧后样品γ-Fe2O3/RGO的TEM图和HRTEM图.从TEM图可以清楚地看出, 经过煅烧后, γ-Fe2O3很好地维持了原有的立方块形貌, 但粒径略有缩小. HRTEM图中晶面间距d=0.252 nm, 对应于γ-Fe2O3的(311)晶面.图 3e, 3f为样品IOC-2的TEM和HRTEM图.从TEM图中可以看出RGO片上均匀分布着γ-Fe2O3立方块和CdS纳米粒子. HRTEM图(图 3f)中晶面间距d=0.252 nm和d=0.336 nm分别对应于γ-Fe2O3的(311)晶面和CdS的(111)晶面.同样的, 在CdS/RGO-3%样品的SEM图和TEM图中(图S2), 可以清楚地看到负载于RGO片上的CdS纳米粒子.
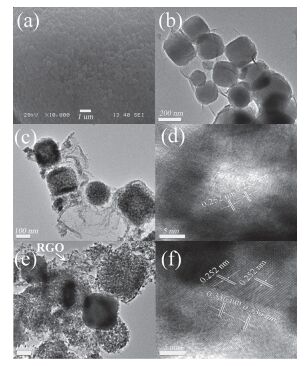 图3
(a) PB的SEM图, (b) PB/GO的TEM图, (c, d) γ-Fe2O3/RGO的TEM和HRTEM图, (e, f) IOC-2的TEM和HRTEM图
Figure3.
(a) SEM image of PB, (b) TEM image of PB/GO, (c, d) TEM and HRTEM images of γ-Fe2O3/RGO, (e, f) TEM and HRTEM images of IOC-2
图3
(a) PB的SEM图, (b) PB/GO的TEM图, (c, d) γ-Fe2O3/RGO的TEM和HRTEM图, (e, f) IOC-2的TEM和HRTEM图
Figure3.
(a) SEM image of PB, (b) TEM image of PB/GO, (c, d) TEM and HRTEM images of γ-Fe2O3/RGO, (e, f) TEM and HRTEM images of IOC-2
图 4a为氧化石墨、γ-Fe2O3/RGO和IOC-2复合物的FT-IR光谱图.氧化石墨在1386和3420 cm-1处的特征吸收峰分别对应O—H的变形与伸缩振动.在1107、1230、1726 cm-1处的吸收峰分别对应COOH基团的C—O、C—OH、C=O伸缩振动.石墨的碳骨架振动和氧化石墨表面吸附的水分子的特征吸收峰约在1626 cm-1处[45].而在γ-Fe2O3/RGO和IOC-2复合物的FT-IR光谱图中, 氧化石墨中含氧基团的特征峰强度明显减弱甚至消失, 这表明GO被较好的还原.在约为670 cm-1处的吸收峰对应于γ-Fe2O3的Fe—O伸缩振动, 表明γ-Fe2O3的存在[46].由于Cd—S的伸缩振动峰(约在644 cm-1)与Fe—O伸缩振动峰位置接近, 故在IOC-2复合物中仅能观察到一个峰[47].
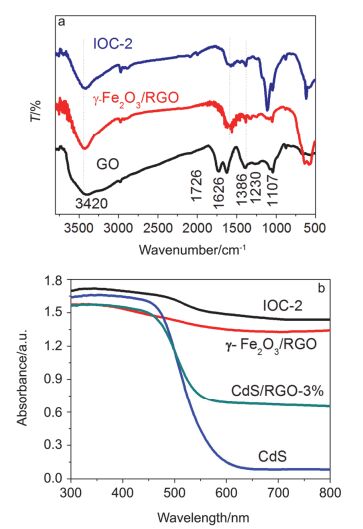 图4
(a) 所合成样品的FT-IR图, (b)所合成样品的UV-vis漫反射光谱
Figure4.
(a) FT-IR spectra of as-prepared samples, (b) UV-vis diffuse reflectance spectra of as-prepared samples
图4
(a) 所合成样品的FT-IR图, (b)所合成样品的UV-vis漫反射光谱
Figure4.
(a) FT-IR spectra of as-prepared samples, (b) UV-vis diffuse reflectance spectra of as-prepared samples
图 4b为样品的紫外-可见漫反射光谱图. CdS在紫外-可见光区均有吸收, 吸收边约位于570 nm, 由计算可得CdS的能带隙Eg=2.2 eV (图S3).与CdS相比, CdS/RGO-3%复合材料在可见光区域的吸收增强, 这可归因于RGO的引入.对比γ-Fe2O3/RGO与IOC-2的UV-vis图, 这两个复合物都没有明显的吸收边, 在紫外和可见光区内吸收强度相差不大, 表明两者均表现出增强的可见光吸收.比较IOC-1、IOC-2和IOC-3样品的UV-vis漫反射光谱(图S4), 可看出样品在可见光区均有较强的吸收, 这说明加入γ-Fe2O3有利于催化剂对可见光的吸收.且IOC-2样品在可见光范围内具有最高的吸收强度, 因此能够更好地利用可见光, 有利于提高光催化活性.
Raman光谱是表征碳材料缺陷与无序度的有效且广泛应用的手段. 图 5为GO、γ-Fe2O3/RGO、CdS/RGO-3%和IOC-2复合材料的Raman谱图.所有的光谱在1000~2000 cm-1范围内均有两个主要的峰, 对应于碳的D带和G带.其中D带约在1359 cm-1处, 与碳材料的结构缺陷和无序度有关, 而G带, 约在1590 cm-1处, 为sp2 C原子的E2g振动模式[48]. γ-Fe2O3/RGO、CdS/RGO-3%和IOC-2中D带和G带的出现证明了RGO的存在, 与TEM的结果相一致. D带与G带的强度比(ID/IG)与石墨结构的无序度和缺陷相关. sp2杂化范围越小, 比值越大. γ-Fe2O3/RGO、CdS/RGO-3%, IOC-2复合材料的ID/IG值比氧化石墨的大, 这主要是因为在煅烧过程中共轭石墨烯网状结构的重建及纳米粒子的附着导致缺陷增加.因此GO在煅烧过程中可以被有效的去氧还原.另外, γ-Fe2O3/RGO样品的Raman图, 在低频区域存在几个强度较小的峰, 它们可归因于γ-Fe2O3[46, 49].相似地, CdS/RGO-3%样品的Raman光谱图, 在约300和600 cm-1处有两个强峰, 在850 cm-1处有一个较弱的峰, 它们均为CdS纳米粒子的特征峰[50].对于IOC-2样品的Raman图, 除了RGO的特征峰外, 由于γ-Fe2O3峰的位置与CdS的接近, 且峰强度较低, 故在三元材料中仅可以观察到CdS的特征峰.
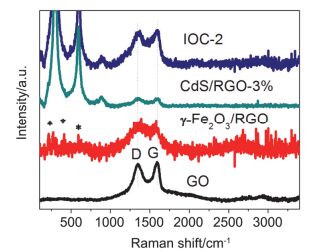 图5
所合成样品的Raman图
Figure5.
Raman spectra of as-prepared samples
图5
所合成样品的Raman图
Figure5.
Raman spectra of as-prepared samples
2.2 光催化性能的研究
在室温下, 我们通过在可见光下RhB的降解来评价样品的光催化活性, 结果如图 6和图S5所示.以CdS作为光催化剂时, 在可见光光照80 min后约有67.2%的RhB被降解.二元催化剂CdS/RGO的光催化降解性能较单组分CdS有所提高, 表明CdS可以通过结合RGO来提高光催化性能(图S5).随着RGO含量的增加, CdS/RGO的光催化降解活性表现为先上升后下降, 当RGO含量约为3 wt%时, 光催化活性最高, 在80 min内约有83.4%的RhB被降解.该结果表明, 适当的RGO含量对优化光催化性能十分重要. RGO的存在可减少CdS纳米粒子的团聚, 并且加快CdS光生电子的转移与利用, 从而可提高CdS/RGO光催化活性.但当RGO含量过高时, 催化活性组分CdS的含量将相应的减少, 且过多的碳材料为光生电荷复合提供了位点, 两者都不利于提高光催化活性. CdS/RGO复合物的光催化反应速率遵循准一级反应动力学方程: -ln(C/C0)=kt, 其中C和C0分别为照射时间为t(min)和t=0时RhB的浓度; k(min-1)为反应速率常数.如图S5(b)所示, CdS/RGO-3%具有最高的k值, 约为0.01855 min-1, 比单组分CdS高约1.67倍.由此可见, 与RGO复合可提高CdS的光催化活性.
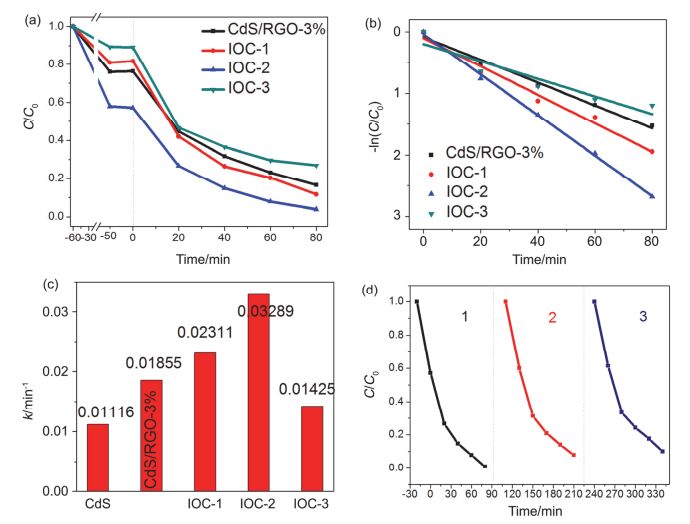 图6
(a) CdS/RGO/γ-Fe2O3系列催化剂的光催化性能, (b) CdS/RGO/γ-Fe2O3系列催化剂降解RhB的一级拟合动力学; (c)动力学k值, (d) IOC-2循环三次的性能图
Figure6.
(a) The photocatalytic activity of the as-prepared CdS/RGO/γ-Fe2O3, (b) Kinetic curves of the photocatalytic degradation of RhB with CdS/RGO/γ-Fe2O3; (c) The rate constants (k) of the photodegradation with different photocatalysts, (d) Cycle experiments of IOC-2 for photocatalytic degradation of RhB under visible-light irradiation
图6
(a) CdS/RGO/γ-Fe2O3系列催化剂的光催化性能, (b) CdS/RGO/γ-Fe2O3系列催化剂降解RhB的一级拟合动力学; (c)动力学k值, (d) IOC-2循环三次的性能图
Figure6.
(a) The photocatalytic activity of the as-prepared CdS/RGO/γ-Fe2O3, (b) Kinetic curves of the photocatalytic degradation of RhB with CdS/RGO/γ-Fe2O3; (c) The rate constants (k) of the photodegradation with different photocatalysts, (d) Cycle experiments of IOC-2 for photocatalytic degradation of RhB under visible-light irradiation
图 6a为CdS/RGO/γ-Fe2O3光催化剂的催化降解性能图.随着γ-Fe2O3含量的增加, 三元复合材料的光催化性能表现为先提高后下降.当PB加入量为12 mg时, CdS/RGO/γ-Fe2O3表现出最好的光催化性能.在可见光照射80 min后, 约有99.2%的RhB被降解.当PB用量为9 mg时, CdS/RGO/γ-Fe2O3光催化活性较IOC-2要低, 但仍高于CdS/RGO-3%.而当PB加入量为15 mg时, CdS/RGO/γ-Fe2O3的光催化性能明显下降, 甚至低于CdS/RGO-3%.这是由于适当的γ-Fe2O3含量有利于光生电子-空穴分离, 而γ-Fe2O3含量过高会覆盖催化活性表面, 反而降低光催化活性.如图 6b所示. CdS/RGO/ γ-Fe2O3的光催化反应速率也遵循准一级动力学方程. 图 6c为样品的k值比较图.从图中可知, IOC-2具有最高的k值, 为0.03289 min-1, 比CdS/RGO-3%高约1.8倍.由此可见, CdS/RGO的光催化活性可以通过与γ-Fe2O3的复合得到进一步的提高.
在光催化过程中, 光催化剂的循环稳定性对实际应用至关重要.在相同条件下, 我们利用IOC-2光催化剂对RhB进行三次降解来测试其稳定性.在每次降解实验后, 我们用磁铁将催化剂吸出, 并超声清洗后供下一轮催化实验使用.如图 6d所示, 在三次循环实验之后, IOC-2复合材料的光催化活性仅有轻微的降低, 表明CdS/RGO/γ-Fe2O3复合材料具有良好的光催化稳定性.
图 7给出了CdS/RGO/γ-Fe2O3的磁滞回线, 说明该材料具有铁磁性质, 是一种软磁体.如图 7中的插图所示, 当磁铁靠近反应溶液时, IOC-2催化剂可以快速的吸到磁铁一边, 同时溶液变得澄清.这一结果表明, CdS/RGO/γ-Fe2O3催化剂可以很容易通过磁铁从反应体系中分离出来.
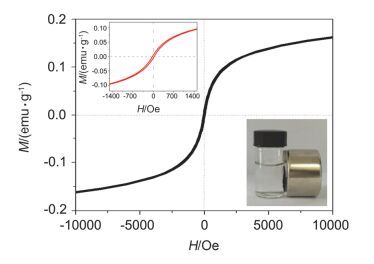 图7
IOC-2催化剂的室温磁滞回线, 插图为分离效果的数码照片
Figure7.
Magnetic hysteresis loop of IOC-2 photocatalyst at 300 K. The inset shows the magnetic separation of the photocatalyst
图7
IOC-2催化剂的室温磁滞回线, 插图为分离效果的数码照片
Figure7.
Magnetic hysteresis loop of IOC-2 photocatalyst at 300 K. The inset shows the magnetic separation of the photocatalyst
2.3 光电化学性能的研究
图 8为样品CdS、CdS/RGO-3%以及IOC-2在偏压为-0.9 V的光电流-时间曲线.通常, 较大的光电流对应于较高的光生电荷的分离效率和较好的光催化活性.显然, 相对于CdS、CdS/RGO-3%来说, IOC-2的光电流强度最高, 表明IOC-2的光生电子-空穴分离效果最好.
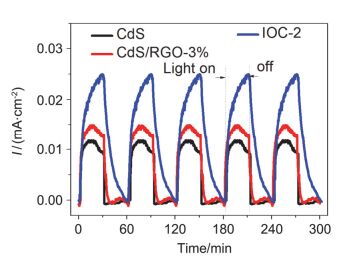 图8
CdS, CdS/RGO-3%和IOC-2样品的光电流响应图
Figure8.
Photocurrent response curves of the as-prepared CdS, CdS/RGO-3% and IOC-2
图8
CdS, CdS/RGO-3%和IOC-2样品的光电流响应图
Figure8.
Photocurrent response curves of the as-prepared CdS, CdS/RGO-3% and IOC-2
2.4 CdS/RGO/γ-Fe2O3的催化机理研究
一般来说, 光生电荷的有效分离在光催化、光电化学反应过程中起着重要的作用.我们利用荧光光谱来研究所合成样品的电子-空穴情况.通常较高的荧光强度意味着较高的电子-空穴复合率, 会导致光催化活性的降低. 图 9a为CdS、CdS/RGO-3%及IOC-2样品的PL光谱图.从图中可以看出, 单组分CdS的峰强度最高, 表明电子-空穴复合率最高.当与RGO进行复合形成CdS/RGO之后, 峰强度有所下降, 表明RGO的存在抑制了光生电荷的复合.而IOC-2复合材料的峰强度最低, 说明三元复合材料具有最高的光生电荷分离效率, 使得光催化性能最好.
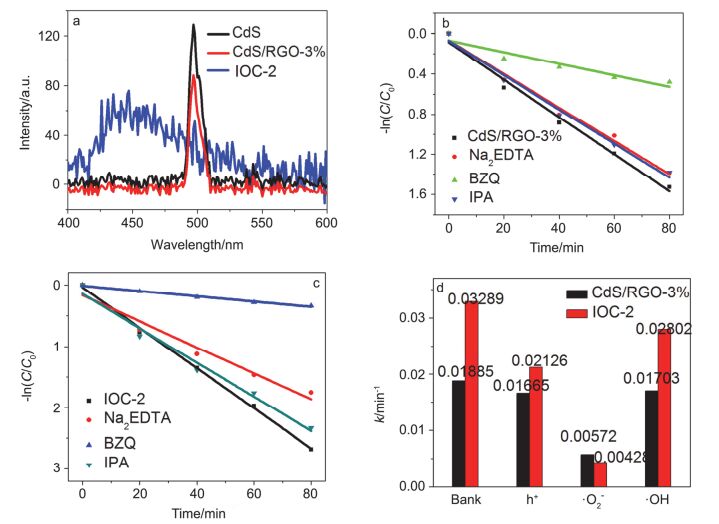 图9
(a) 样品的PL光谱图; (b) CdS/RGO-3%和(c) IOC-2加入活性物种清除剂后光催化降解RhB的拟一级动力学图; (d)动力学k值比较图
Figure9.
(a) PL spectra of the as-prepared samples, Kinetic curves described by pseudo-first-order kinetics after the adding of various scavengers for (b) CdS/RGO-3% and (c) IOC-2; (d) The rate constants k of the photodegradation after the adding of various scavengers
图9
(a) 样品的PL光谱图; (b) CdS/RGO-3%和(c) IOC-2加入活性物种清除剂后光催化降解RhB的拟一级动力学图; (d)动力学k值比较图
Figure9.
(a) PL spectra of the as-prepared samples, Kinetic curves described by pseudo-first-order kinetics after the adding of various scavengers for (b) CdS/RGO-3% and (c) IOC-2; (d) The rate constants k of the photodegradation after the adding of various scavengers
众所周知, 在光催化降解反应过程中会产生•O2、h+和•OH活性基团.为了研究CdS/RGO/γ-Fe2O3催化剂的光催化机理.我们在光催化过程中, 分别加入了不同的活性基团清除剂.不同活性基团在光催化过程中的作用可以根据光催化效率的变化来确定.在该研究中, 对苯醌(BZQ)、异丙醇(IPA)和乙二胺四乙酸钠(Na2EDTA)分别被用来作为•O2、•OH和h+的清除剂. 图 9b为CdS/RGO-3%光催化体系中加入各种清除剂后RhB的光催化降解效率.从图中可以看出, 反应速率常数(k)在加入Na2EDTA或IPA后只稍微下降, 而当加入BZQ后, k值明显降低, 从0.01885 min-1变为0.00572 min-1, 表明•O2在CdS/RGO-3%光催化过程中起主要作用.当清除剂BZQ、Na2EDTA和IPA分别加入到IOC-2光催化体系中, 反应速率常数k从0.03289 min-1分别降至0.00428 min-1、0.02126 min-1和0.02802 min-1.该结果表明, 在IOC-2的光催化降解过程中, •O2-仍然为主要的活性物种, 且作用更为显著; 同时h+的作用也有明显提高.
基于上述结果, 我们提出了光催化降解的机理.如图 10所示, 其中图 10a和10b分别为CdS/RGO和CdS/RGO/γ-Fe2O3的光催化降解的机理图.从图 10a可知, 当CdS/RGO吸收光子后, 产生光生电子-空穴对, 而电子由于RGO优异的导电性能可以从CdS导带上快速转移至RGO上, 参与氧化还原反应, 生成了活性基团•O2, 从而进行光催化降解过程[51]. 图 10b为IOC-2样品光催化降解的机理图.根据γ-Fe2O3[52]和CdS[53]的导带价带位置和活性基团实验结果, 我们推测在三元复合物CdS/RGO/γ-Fe2O3的光催化过程中, 半导体γ-Fe2O3与CdS通过RGO形成了类似Z型异质结结构, 其光生电子-空穴的分离过程为γ-Fe2O3上的电子经由RGO转移至CdS的价带, 与空穴复合; 从而留下CdS导带的光生电子与γ-Fe2O3价带上的空穴参与氧化还原反应.由于CdS导带的光生电子具有足够强的还原能力, 能还原O2形成活性基团•O2; 而γ-Fe2O3价带上的空穴具有强的氧化能力, 可直接氧化RhB.与Ⅱ型异质结相比, Z型异质结不仅能促进光生电子与空穴的有效分离, 而且其光生电子与空穴具有更强的氧化还原能力, 从而更有利于RhB的降解.
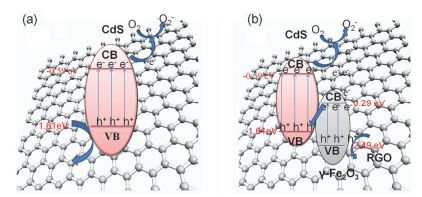 图10
(a) CdS/RGO-3%在光照条件下电荷转移情况, (b) IOC-2在光照条件下电荷转移情况
Figure10.
(a) The charge transfer behaviors of CdS/RGO-3% upon visible-light illumination. (b) The charge transfer behaviors of IOC-2 upon visible-light illumination
图10
(a) CdS/RGO-3%在光照条件下电荷转移情况, (b) IOC-2在光照条件下电荷转移情况
Figure10.
(a) The charge transfer behaviors of CdS/RGO-3% upon visible-light illumination. (b) The charge transfer behaviors of IOC-2 upon visible-light illumination
3 结论
我们成功制备了CdS/RGO/γ-Fe2O3三元复合光催化剂, 其中CdS和γ-Fe2O3纳米颗粒在RGO纳米片上分散均匀, 且γ-Fe2O3呈规则的立方块形貌.制备的光催化剂具有增强的光催化活性, 这是由于RGO和γ-Fe2O3的存在不仅增强了催化剂对可见光的吸收, 而且提高了对光生电荷的分离效率.由于γ-Fe2O3的铁磁性, 该催化剂能够利用磁铁方便地从反应体系中分离出来, 有利于催化剂的回收再利用.
4 实验部分
本实验采用的试剂均为分析纯试剂, 购自上海国药集团.
4.1 样品的制备
氧化石墨的制备:参见支持信息.
普鲁士蓝(PB)纳米立方块的制备:称取0.044 g铁氰化钾[K4Fe(CN)6]和1.52 g PVP加入到20 mL 0.1 mol•L-1 HCl溶液中, 搅拌30 min后, 将溶液加入至25 mL的反应釜中, 加热至80 ℃, 并在此温度下保持10 h.自然冷却后, 用去离子水洗涤, 干燥待用.
CdS/RGO/γ-Fe2O3的制备:将一定量(9 mg、12 mg、15 mg)的PB超声分散于20 mL去离子水中, 在搅拌下依次加入1 mL的聚丙烯胺盐酸盐(PAH)(1 g•L-1)溶液(搅拌3 h)、10 mL的氧化石墨烯(GO)(2.4 mg)溶液(搅拌12 h)、最后加入101.7 mg CdCl2 (搅拌3 h)和133 mg Na2S•9H2O (搅拌3 h).离心收集沉淀产物, 并用去离子水洗涤, 在45 ℃下真空干燥, 得到CdS/GO/PB复合物.将该产物置于管式炉中, 在N2氛围下以2 ℃•min-1升温速率至350 ℃, 并保持2 h, 自然冷却至室温, 得到最终产物CdS/RGO/γ-Fe2O3.
对比实验参见支持信息.
4.2 仪器表征
样品相结构通过X-射线衍射仪(XRD, D8 ADVANCE)进行表征, 扫描速率为4 (°)•min-1.样品成分用X-射线能量分散光谱(EDS)测定.样品的形貌和尺寸用扫描电子显微镜(SEM)和透射电子显微镜(TEM, JEM2100)进行表征.其他表征手段有红外光谱(FT-IR, Nicolet Nexus 470), 紫外-可见光谱(UV, Shimadzu UV-2450), 拉曼光谱(Raman, DXR), 荧光光谱(PL, Varian Cary Eclipse)等.
4.3 光催化实验
光催化反应实验在GHX-3型光催化反应仪中进行, 用250 W Xe灯作为光源, 并加入滤光片除去紫外光(<420 nm), 光源与反应器的距离约为10 cm.光反应系统与光源之间通循环水以除去Xe灯光照时产生的热量.光催化反应在室温下进行, 具体操作步骤如下:将30 mg催化剂分散于60 mL 5 mg/L的罗丹明B (RhB)溶液中.将溶液在暗态下搅拌1 h, 使染料在催化剂的表面达到吸附-脱附平衡.之后打开光源, 开始光催化反应.反应过程中, 每隔一定时间移取4 mL反应液, 离心分离, 取上层清液, 通过紫外-可见分光光度计测定其吸收光谱.
4.4 光电化学实验
在室温下采用CHI 760D (上海辰华仪器有限公司)电化学工作站对制备的光催化剂进行光电化学性能测试.测试体系为三电极体系, 光源为300 W Xe灯, 光源与光电极之间的距离为25 cm.铂丝和Ag/AgCl电极分别作为对电极和参比电极.电解液为0.5 mol•L-1 Na2SO4溶液.工作电极制备方法如下:取8 mg的样品溶于990 μL乙醇中, 加入10 μL萘酚混合均匀.取上述液体30 μL涂抹于1 cm×1 cm导电玻片上, 45 ℃下真空干燥24 h.
-
-
[1]
Aarthi, T.; Narahari, P.; Madras, G. J. Hazard. Mater. 2007, 149, 725. doi: 10.1016/j.jhazmat.2007.04.038
-
[2]
Gu, S. H.; Wang, L. Z.; Zhang, J. L. Chin. J. Chem. 2017, 35, 153. doi: 10.1002/cjoc.v35.2
-
[3]
Higashimoto, S.; Hikita, K.; Azuma, M. Chin. J. Chem. 2017, 35, 165. doi: 10.1002/cjoc.v35.2
-
[4]
Wang, J. T.; Xiao, C.; Wu, X. Y. Chin. J. Chem. 2017, 35, 189. doi: 10.1002/cjoc.v35.2
-
[5]
Li, X. D.; Zhang, Q. H.; Wang, H. Z. Chin. J. Chem. 2017, 35, 196. doi: 10.1002/cjoc.v35.2
-
[6]
Qin, H. X.; Bian, Y. Y.; Zhang, Y. X. Chin. J. Chem. 2017, 35, 203. doi: 10.1002/cjoc.v35.2
-
[7]
崔素珍, 杨汉培, 孙慧华, 聂坤, 吴俊明, 化学学报, 2016, 74, 995.Cui, S. Z.; Yang, H. P.; Sun, H. H. Acta Chim. Sinica 2016, 74, 995.
-
[8]
Carey, J. H.; Lawrence, J.; Tosine, H. M. B. Environ. Contam. Tox. 1976, 16, 697. doi: 10.1007/BF01685575
-
[9]
王恩君, 杨辉云, 曹亚安, 化学学报, 2009, 67, 2759. doi: 10.3321/j.issn:0567-7351.2009.24.001Wang, E. J.; Yang, H. Y.; Cao, Y. A. Acta Chim. Sinica 2009, 67, 2759(in Chinese). doi: 10.3321/j.issn:0567-7351.2009.24.001
-
[10]
Bae, E.; Choi, W. Environ. Sci. Technol 2003, 37, 147. doi: 10.1021/es025617q
-
[11]
Wang, Y. W.; Zhu, Y. H.; Yang, X. L. Chin. J. Chem. 2017, 35, 949. doi: 10.1002/cjoc.v35.6
-
[12]
Chang, J.; Zhang, W. J.; Hong, C. Y. Chin. J. Chem. 2017, 35, 1016. doi: 10.1002/cjoc.v35.6
-
[13]
Jiang, L. P.; Wang, S. J.; Shi, L. Y. Chin. J. Chem. 2017, 35, 183. doi: 10.1002/cjoc.v35.2
-
[14]
Cheng, J. S.; Wang, W. H.; Zhu, W. J. Chin. J. Chem. 2016, 34, 53. doi: 10.1002/cjoc.201500339
-
[15]
王大彬, 赵利霞, 郭良宏, 张辉, 万斌, 杨郁, 化学学报, 2015, 73, 388. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344896.shtmlWang, D. B.; Zhao, L. X.; Guo, L. H.; Zhang, H.; Wan, B.; Yang, Y. Acta Chim. Sinica 2015, 73, 388(in Chinese). http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344896.shtml
-
[16]
Bi, F.; Muhammad, F.; Liu, W. Chin. J. Chem. 2015, 33, 112. doi: 10.1002/cjoc.v33.1
-
[17]
Abe, R.; Takata, T.; Sugihara, H. Chem. Commun. 2005, 30, 3829.
-
[18]
Higashi, M.; Abe, R.; Teramura, K. Chem. Phys. Lett. 2008, 452, 120. doi: 10.1016/j.cplett.2007.12.021
-
[19]
李长全, 罗来涛, 熊光伟, 化学学报, 2010, 68, 1023. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract338884.shtmlLi, C. Q.; Luo, L. T.; Xiong, G. W. Acta Chim. Sinica 2010, 68, 1023(in Chinese). http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract338884.shtml
-
[20]
Ba-Abbad, M. M.; Kadhum, A. A. H.; Mohamad, A. B. Int. J. Therm. Environ. Eng. 2010, 1, 37. doi: 10.5383/ijtee.
-
[21]
Xie, Y. P.; Yu, Z. B.; Liu, G.; Ma, X. L.; Cheng, H. M. Energy. Environ. Sci. 2014, 7, 1895. doi: 10.1039/c3ee43750g
-
[22]
Zhang, N.; Zhang, Y.; Pan, X.; Yang, M. Q.; Xu, Y. J. J. Phys. Chem. C, 2012, 116, 18023. doi: 10.1021/jp303503c
-
[23]
Ye, X. J.; Dai, X.; Meng, S. G. Chin. J. Chem. 2017, 35, 217. doi: 10.1002/cjoc.v35.2
-
[24]
Kashiath, L.; Namratha, K.; Byrappa, K. J. Alloy. Compd. 2016, 695, 799.
-
[25]
Lee, J.; Kim, Y.; Kim, J. K.; Kim, S.; Min, D.; Jang, D. Appl. Catal. B 2017, 205, 433. doi: 10.1016/j.apcatb.2016.12.063
-
[26]
Khan, S.; Han, J. S.; Lee, S. Y. Chin. J. Chem. 2017, 35, 159. doi: 10.1002/cjoc.v35.2
-
[27]
丛日敏, 罗运军, 于怀清, 化学学报, 2012, 68, 1971. doi: 10.3866/PKU.WHXB201206111Cong, R. M.; Luo, Y. J.; Yu, H. Q. Acta Chim. Sinica 2012, 68, 1971(in Chinese). doi: 10.3866/PKU.WHXB201206111
-
[28]
Li, H. J.; Zhou, Y.; Chen, L.; Luo, W. J.; Xu, Q. F.; Wang, X. Y.; Xiao, M.; Zou, Z. G. Nanoscale 2013, 5, 11933. doi: 10.1039/c3nr03493c
-
[29]
Vaquero, F.; Navarro, R. M.; Fierro, J. L. G. Appl. Catal. B 2017, 753.
-
[30]
Li, Q.; Guo, B. D.; Yu, J. G.; Ran, J. R.; Zhang, B. H.; Yan, H. J.; Gong, J. R. J. Am. Chem. Soc. 2011, 133, 10878. doi: 10.1021/ja2025454
-
[31]
Li, Y. G.; Wei, X. L.; Li, H. J.; Wang, R. R.; Feng, J.; Yun, H.; Zhou, A. N. RSC Adv. 2015, 5, 14704. doi: 10.1039/C4RA13400A
-
[32]
Liu, X. J.; Pan, L. K.; Lv, T.; Zhu, G.; Sun, Z.; Sun, C. Q. Chem. Commun. 2011, 47, 11984. doi: 10.1039/c1cc14875c
-
[33]
Zhang, Y. H.; Tang, Z. R.; Fu, X. Z.; Xu, Y. J. ACS Nano 2010, 4, 7303. doi: 10.1021/nn1024219
-
[34]
Zhang, N.; Yang, M. Q.; Liu, S. Q.; Sun, Y. G.; Xu, Y. J. Chem. Rev. 2015, 115, 10307. doi: 10.1021/acs.chemrev.5b00267
-
[35]
Quan, Q.; Lin, X.; Zhang, N.; Xu, Y. J. Nanoscale 2017, 9, 2398. doi: 10.1039/C6NR09439B
-
[36]
Han, C.; Zhang, N.; Xu, Y. J. Nano Today 2016, 11, 351. doi: 10.1016/j.nantod.2016.05.008
-
[37]
Yuan, L.; Yang, M. Q.; Xu, Y. J. Nanoscale 2014, 6, 6335. doi: 10.1039/c4nr00116h
-
[38]
Liu, Y.; Zhou, L.; Hu, Y.; Guo, C. F.; Qian, H. S.; Zhang, F. M.; Lou, X. W. J. Mater. Chem. 2011, 21, 18359. doi: 10.1039/c1jm13789a
-
[39]
Li, N.; Zhang, J.; Tian, Y.; Zhao, J. H.; Zuo, W. Chem. Eng. J. 2017, 308, 377. doi: 10.1016/j.cej.2016.09.093
-
[40]
Chen, Y.; Liu, K. R. J. Alloy. Compd. 2017, 697, 161. doi: 10.1016/j.jallcom.2016.12.153
-
[41]
Jia, X. H.; Dai, R. R.; Lian, D. D.; Han, S.; Wu, X. Y.; Song, H. J. Appl. Surf. Sci. 2017, 392, 268. doi: 10.1016/j.apsusc.2016.09.014
-
[42]
Wang, L.; Wei, H. W.; Fan, Y. J.; Gu, X.; Zhan, J. H. J. Phys. Chem. C 2009, 113, 14119. doi: 10.1021/jp902866b
-
[43]
Liu, Y.; Yu, L.; Hu, Y.; Guo, C. F.; Zhang, F. M.; Lou, X. W. Nanoscale 2012, 4, 183. doi: 10.1039/C1NR11114K
-
[44]
Zhang, L.; Wu, H. B.; Madhavi, S., ; Hng, H. H.; Lou, X. W. J. Am. Chem. Soc. 2012, 134, 17388. doi: 10.1021/ja307475c
-
[45]
He, H.; Klinowski, J.; Forster, M. Chem. Phys. Lett. 1998, 287, 53. doi: 10.1016/S0009-2614(98)00144-4
-
[46]
Singh, A. P.; Mishra, M.; Sambyal, P.; Gupta, B. K.; Singh, A.; Dhawan, S. K. J. Mater. Chem. A 2014, 2, 3581. doi: 10.1039/C3TA14212D
-
[47]
Meng, N. N.; Zhou, Y. F.; Nie, W. Y.; Chen, P. P. J. Nanopart. Res. 2016, 18, 241. doi: 10.1007/s11051-016-3522-y
-
[48]
Kudin, K. N.; Ozbas, B.; Schniepp, H. C. Nano Lett. 2008, 8, 36. doi: 10.1021/nl071822y
-
[49]
Sirivisoot, S.; Harrison, B. S. Int. J. Nanomedicine 2015, 10, 4447.
-
[50]
Xu, J.; Wang, L.; Cao, X. J. Chem. Eng. J. 2016, 283, 816. doi: 10.1016/j.cej.2015.08.018
-
[51]
Jia, L.; Wang, D. H.; Huang, Y. X.; Xu, A. W.; Yu, H. Q. J. Phys. Chem. C 2011, 115, 11466.
-
[52]
Guo, R. Q.; Fang, L.; Dong, W.; Zheng, F. G.; Shen, M. R. J. Mater. Chem. 2011, 21, 18645. doi: 10.1039/c1jm13072b
-
[53]
Mondal, S.; Sunhu, S.; Bhattacharya, S.; Saha, S. K. J. Phys. Chem. C 2015, 119, 27749. doi: 10.1021/acs.jpcc.5b08116
-
[1]
-

图 1 CdS/RGO/γ-Fe2O3复合物的合成示意图
Figure 1 Illustration of the synthesis procedure of CdS/RGO/γ-Fe2O3 composites

图 2 (a) CdS/RGO-3%、γ-Fe2O3/RGO、IOC-2的XRD图, (b) IOC-2的EDS图
Figure 2 (a) XRD patterns of the as-prepared samples, (b) EDS spectrum of IOC-2

图 3 (a) PB的SEM图, (b) PB/GO的TEM图, (c, d) γ-Fe2O3/RGO的TEM和HRTEM图, (e, f) IOC-2的TEM和HRTEM图
Figure 3 (a) SEM image of PB, (b) TEM image of PB/GO, (c, d) TEM and HRTEM images of γ-Fe2O3/RGO, (e, f) TEM and HRTEM images of IOC-2

图 4 (a) 所合成样品的FT-IR图, (b)所合成样品的UV-vis漫反射光谱
Figure 4 (a) FT-IR spectra of as-prepared samples, (b) UV-vis diffuse reflectance spectra of as-prepared samples

图 6 (a) CdS/RGO/γ-Fe2O3系列催化剂的光催化性能, (b) CdS/RGO/γ-Fe2O3系列催化剂降解RhB的一级拟合动力学; (c)动力学k值, (d) IOC-2循环三次的性能图
Figure 6 (a) The photocatalytic activity of the as-prepared CdS/RGO/γ-Fe2O3, (b) Kinetic curves of the photocatalytic degradation of RhB with CdS/RGO/γ-Fe2O3; (c) The rate constants (k) of the photodegradation with different photocatalysts, (d) Cycle experiments of IOC-2 for photocatalytic degradation of RhB under visible-light irradiation

图 7 IOC-2催化剂的室温磁滞回线, 插图为分离效果的数码照片
Figure 7 Magnetic hysteresis loop of IOC-2 photocatalyst at 300 K. The inset shows the magnetic separation of the photocatalyst

图 8 CdS, CdS/RGO-3%和IOC-2样品的光电流响应图
Figure 8 Photocurrent response curves of the as-prepared CdS, CdS/RGO-3% and IOC-2

图 9 (a) 样品的PL光谱图; (b) CdS/RGO-3%和(c) IOC-2加入活性物种清除剂后光催化降解RhB的拟一级动力学图; (d)动力学k值比较图
Figure 9 (a) PL spectra of the as-prepared samples, Kinetic curves described by pseudo-first-order kinetics after the adding of various scavengers for (b) CdS/RGO-3% and (c) IOC-2; (d) The rate constants k of the photodegradation after the adding of various scavengers
-


 下载:
下载:









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 7
- 文章访问数: 2564
- HTML全文浏览量: 377
 下载:
下载:
