 图 1
不同溶解氧条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 1.
Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) with nZⅥ under different DO conditions
图 1
不同溶解氧条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 1.
Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) with nZⅥ under different DO conditions
引用本文:
夏雪芬, 滑熠龙, 黄潇月, 凌岚, 张伟贤. 纳米零价铁对水中砷和硒去除的比较研究[J]. 化学学报,
2017, 75(6): 594-601.
doi:
10.6023/A17030099

Citation: Xia Xuefen, Hua Yilong, Huang Xiaoyue, Ling Lan, Zhang Weixian. Removal of Arsenic and Selenium with Nanoscale Zero-Valent Iron (nZⅥ)[J]. Acta Chimica Sinica, 2017, 75(6): 594-601. doi: 10.6023/A17030099
Citation: Xia Xuefen, Hua Yilong, Huang Xiaoyue, Ling Lan, Zhang Weixian. Removal of Arsenic and Selenium with Nanoscale Zero-Valent Iron (nZⅥ)[J]. Acta Chimica Sinica, 2017, 75(6): 594-601. doi: 10.6023/A17030099
纳米零价铁对水中砷和硒去除的比较研究
摘要:
采用纳米零价铁去除水中的砷和硒,考察了不同溶解氧、纳米零价铁投加量、接触时间以及溶液初始pH值等条件下纳米零价铁去除水中砷和硒的效果,并比较了反应前后固体的形貌、组成、溶液pH值及反应机制.结果表明,纳米零价铁可广泛用于去除水中的砷/硒污染,去除效果顺序依次为Se(Ⅳ)> As(Ⅲ)> Se(Ⅵ)> As(Ⅴ);水中溶解氧(DO)对As(Ⅲ)和Se(Ⅳ)的去除没有显著的影响,而高浓度的DO对As(Ⅴ)和Se(Ⅵ)的去除产生了一定的抑制作用;增加纳米零价铁投加量可促进砷和硒的去除;溶液初始pH值对纳米零价铁去除As(Ⅲ),As(Ⅴ)和Se(Ⅵ)的影响较大,而对Se(Ⅳ)的去除几乎没有影响;反应后固体的形貌、组成及溶液pH值存在差异.纳米零价铁与砷、硒反应机理的不同造成了去除效果及反应后固液两相的差异.
English
Removal of Arsenic and Selenium with Nanoscale Zero-Valent Iron (nZⅥ)
Abstract:
Arsenic (As(Ⅲ/Ⅴ)) and selenium (Se(Ⅳ/Ⅵ)) are toxic inorganic contaminants in groundwater and industrial wastewater. The pollution caused by As and Se has become an environmental concern throughout the world. A variety of treatment technologies have been applied for As and Se removal from aqueous solutions. Among them, nanoscale zero-valent iron (nZⅥ) has been found to have a remarkable capability to remove As and Se from waters. Although lots of studies on the process of As and Se removal with nZⅥ are published, a systematic comparative study is still limited. In this study, the removal capacities of As(Ⅲ), As(Ⅴ), Se(Ⅳ) and Se(Ⅵ) with nZⅥ in a single-specie system were compared. The performances of nZⅥ for As(Ⅲ), As(Ⅴ), Se(Ⅳ) and Se(Ⅵ) were investigated on different conditions (including dissolved oxygen, nZⅥ dosage, contact time, and initial solution pH). The morphology and structure of fresh and spent nZⅥ were also examined by spherical aberration corrected scanning transmission electron microscopy (Cs-STEM) intergrated with energy-dispersive X-ray spectrometry (XEDS), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD). The batch experiments were conducted at room temperature in the 50-mL glass vials sealed with screwcaps. According to the speciation diagram, H3AsO30 and H2AsO4- are the respective predominant dissolved As(Ⅲ) and As(Ⅴ) species respectively at pH 5.0; while HSeO3- and SeO42- are the predominant dissolved Se(Ⅳ) and Se(Ⅵ) species respectively at pH 5.0. The results showed that the removal capacities of As and Se investigated generally followed the order of Se(Ⅳ)> As(Ⅲ)> Se(Ⅵ)> As(Ⅴ). Dissolved oxygen (DO) was found no apparent effects on the removal of As(Ⅲ) and Se(Ⅳ), while the removal performance of As(Ⅴ) and Se(Ⅵ) was inhibited at high dissolved oxygen level (>14 mg/L). The removal of As and Se were enhanced with increasing nZⅥ dosage. Initial solution pH had no significant effect on Se(Ⅳ) removal, whereas the removal of As(Ⅲ), As(Ⅴ), and Se(Ⅵ) appeared to be strongly dependent on the initial solution pH. The spent nZⅥ were different due to the different mechanisms of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) reactions with nZⅥ. The results will be useful for the application of nZⅥ to the treatment of As/Se-containing wastewater.
-
Key words:
- nanoscale zero-valent iron (nZⅥ)
- / arsenic
- / selenium
- / removal
-
1 引言
砷(As)和硒(Se)是地下水和工业废水中较常见且较难处理的两种类金属污染物, 两者均具有毒性, 时常会同时存在于工业废水中, 尤其在冶炼废水中含量较高[1].大量的砷和硒被排放到环境中, 给动植物及人类的健康和发展带来极大威胁.因此, 含砷/硒废水造成的污染成为世界关注的焦点[2].
硒是氧化还原敏感元素, 其形态、价态及毒性很大程度受环境的pH值和氧化还原条件(Eh)的影响[3].环境中的硒主要有-2, 0, +4和+6四种价态.水中的硒主要以硒酸根[Se(Ⅵ)]和亚硒酸根[Se(Ⅳ)]含氧阴离子的形式存在, 其中Se(Ⅳ)的毒性大于Se(Ⅵ)[4], Se(Ⅵ)的化学稳定性和生物可利用性强于Se(Ⅳ)[5].砷也是氧化还原敏感元素, 在环境中主要有-3, 0, +3和+5四种价态.水中的砷主要以砷酸根[As(Ⅴ)]和亚砷酸根[As(Ⅲ)]的形式存在, 其中As(Ⅲ)的毒性大于As(Ⅴ)的毒性[6].据报道, 环境中小剂量(10~100 μg•L-1)砷和硒的慢性暴露即可造成水生生物基因变异甚至罹患各种癌症[7].为此, 世界各国均制定了严格的砷和硒排放标准.例如, 中国环保部规定工业排水砷和硒含量均不得高于0.5 mg• L-1, 城市污水处理厂出水砷和硒含量均不得高于0.1 mg•L-1; 美国环保局(USEPA)为保护水生生物将火力发电厂废水中硒的日排放最高浓度限值从10 μg•L-1降低至5 μg•L-1, 此外还将饮用水中砷的最高允许浓度从50 μg•L-1降低至10 μg•L-1.这些严格标准的出台无疑给含砷/硒废水处理提出了新的挑战.
含砷/硒废水的传统处理方法主要有吸附、离子交换、化学沉淀/絮凝、膜分离以及电化学处理法等[8], 但这些方法存在处理污染物单一、速度慢、不能回收资源、投资费用昂贵、产生大量有毒化学污泥等问题[9].例如, 化学沉淀/絮凝法, 往废水加入钙盐、铝盐或铁盐形成沉淀可将砷和硒从水中去除, 但处理过程中会产生大量的有毒化学污泥给后续处理带来较大的困难[10].相比于传统材料, 纳米零价铁具有粒径小、比表面积大、表面活性高、环境友好等优点, 因而纳米零价铁处理含砷/硒废水具有速度快、效能高、绿色环保等优势[11~24].例如, Kanel等[13, 16]研究发现纳米零价铁去除水中As(Ⅲ)和As(Ⅴ)的速率比微米铁粉快许多倍; 而纳米零价铁去除水中Se(Ⅵ)的速率比传统铁粉快4倍[11], 去除水中Se(Ⅳ)的速率则是微米铁粉、铁氧化物、二氧化钛等传统材料的3.3~11倍, 且Se(Ⅳ)被还原生成元素硒Se(0) 纳米颗粒[14, 17, 18], 有利于硒资源的回收再利用.
我们课题组[14, 15, 17~22, 24]前期研究发现纳米零价铁与水中As(Ⅲ), As(Ⅴ), Se(Ⅳ)的反应是发生在纳米零价铁表面的反应, Olegario等[11]的研究也表明纳米零价铁与水中Se(Ⅵ)的反应是表面反应.在表面反应过程中, 诸多因素如水中溶解氧(DO)、溶液pH值、纳米零价铁投加量等均会影响纳米零价铁对污染物质的去除效果[25].因此, 本工作较系统地研究并比较了不同溶解氧、纳米零价铁投加量、接触时间以及溶液初始pH值(pHini)等条件下纳米零价铁去除水中砷和硒的效果以及反应后固体形貌、组成、溶液pH值及反应机制.研究结果可为纳米零价铁处理实际含砷/硒废水提供依据.
2 结果与讨论
2.1 不同溶解氧条件下的去除
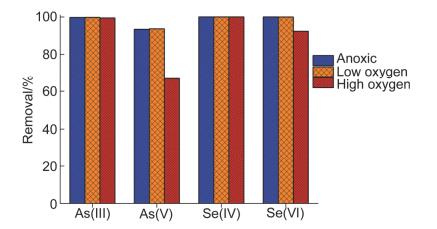
图 1为不同溶解氧条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除.
图 1
不同溶解氧条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 1.
Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) with nZⅥ under different DO conditions
从图 1可看出, 三种溶解氧条件下纳米零价铁对水中As(Ⅲ)和Se(Ⅳ)的去除没有显著变化.反应24 h, 水中As(Ⅲ)和Se(Ⅳ)在三种溶解氧条件下都能被完全去除.溶解氧对水中Se(Ⅳ)的去除速率也没有显著影响, 三种溶解氧条件下纳米零价铁都能将水中Se(Ⅳ)快速去除(图S1).缺氧(DO<0.5 mg•L-1)和低溶解氧(DO~7 mg•L-1)条件对As(Ⅴ)和Se(Ⅵ)的去除也没有显著影响; 但高溶解氧(DO>14 mg•L-1)条件下, As(Ⅴ)的去除率从93.5%下降至66.8%, 说明高溶解氧对纳米零价铁去除As(Ⅴ)有极大的影响; 而高溶解氧条件下Se(Ⅵ)的去除率则从100%略下降至92.4%, 即高溶解氧对Se(Ⅵ)的去除有轻微影响, 这与Klas和Kirk[26]的研究结果一致. Klas和Kirk研究了不同溶解氧和pH条件下零价铁对Se(Ⅵ)的去除, 结果表明, 酸性且厌氧条件下零价铁能将Se(Ⅵ)直接还原, 适量增加水中溶解氧可促进Se(Ⅵ)的去除, 但溶解氧过量会导致去除速率下降.
在实际操作中, 水中溶解氧的含量通常在低溶解氧(DO≈7 mg•L-1)范围, 而低溶解氧条件对纳米零价铁去除水中的As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)都没有显著的影响.因此, 纳米零价铁能高效去除水中As和Se污染.
2.2 不同纳米零价铁投加量下的去除
图 2为不同纳米零价铁投加量条件下As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除.由图 2可见, 当纳米零价铁投加量为0.2 g•L-1时, As(Ⅲ), As(Ⅴ), Se(Ⅳ)和Se(Ⅵ)的去除率分别为87.9%, 72.6%, 42.2%和30.4%, 即去除效率的顺序为: As(Ⅲ)>As(Ⅴ)>Se(Ⅳ)>Se(Ⅵ).这表明, 在纳米零价铁表面活性位点不足的情况下, 纳米零价铁对As的去除能力要大于Se的去除, 这可能是由于纳米零价铁去除As和Se的机理不同造成的.
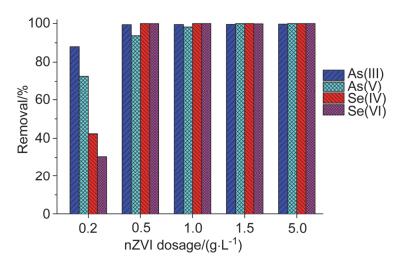 图 2
不同纳米零价铁投加量条件下As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 2.
Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) with different nZⅥ dosages
图 2
不同纳米零价铁投加量条件下As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 2.
Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) with different nZⅥ dosages
当纳米零价铁投加量增加至0.5 g•L-1时, As(Ⅲ), As(Ⅴ), Se(Ⅳ)和Se(Ⅵ)的去除率分别上升至99.6%, 93.7%, 100%和100%;此时As(Ⅲ), Se(Ⅳ)和Se(Ⅵ)的去除率基本达到稳定, 此后再增加投加量, 去除效率也没有随之显著增加, 证明0.5 g•L-1的投加量已经足够.而对于As(Ⅴ), 纳米零价铁投加量增加至1.5 g•L-1时, 去除率也可达到100%, 表明增加纳米零价铁投加量, 可大幅增加可供反应的表面活性位点[27], 从而提高污染物质的去除效率.
许多学者采用零价铁进行了去除水中Se(Ⅵ)的研究[28~30], 结果表明零价铁可将Se(Ⅵ)逐步还原去除, 但零价铁投加量较大(≈50 g•L-1).而本研究中0.5 g•L-1的纳米零价铁即可将水中1.33 mmol•L-1 (≈105 mg• L-1)的Se(Ⅵ)完全去除, 表明纳米零价铁去除Se(Ⅵ)具有经济、高效的特性.
2.3 不同接触时间下的去除
图 3为不同接触时间条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除.
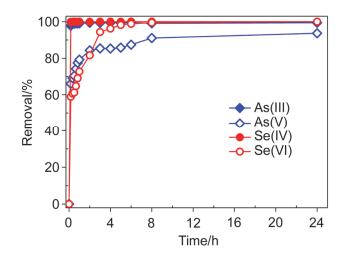 图 3
不同接触时间条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 3.
Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) as functions of time
图 3
不同接触时间条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 3.
Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) as functions of time
由图 3可看出, 纳米零价铁对水中As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除能力总体趋势为Se(Ⅳ)>As(Ⅲ)>Se(Ⅵ)>As(Ⅴ). 0.5 g•L-1的纳米零价铁可在10 min内将水中Se(Ⅳ)完全去除; 反应10 min, 纳米零价铁对As(Ⅲ)的去除即可达97.8%以上; 而对于As(Ⅴ)的去除, 反应速率在前1 h内相对较快, 而后速率减慢, 反应8 h后达到平衡.纳米零价铁对Se(Ⅵ)的去除在前4 h上升较快, 去除率可达96.2%以上, 而后去除率缓慢上升, 反应8 h时去除率可达到100%. Yoon等[31]采用1 g•L-1的零价铁去除水中10 mg•L-1的Se(Ⅵ), 反应10 h后Se(Ⅵ)的去除率仅为72%.由此可见, 纳米零价铁去除水中Se(Ⅵ)的效能明显好于零价铁.
2.4 不同初始pH值下的去除
图 4为不同初始pH值条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除.
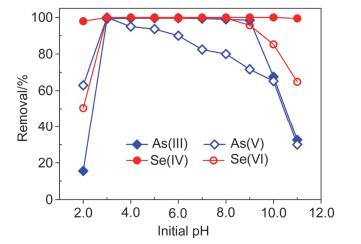 图 4
不同溶液初始pH值条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 4.
Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) with nZⅥ at different initial solution pH
图 4
不同溶液初始pH值条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 4.
Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) with nZⅥ at different initial solution pH
由图 4可见, 在不同初始pH值条件下, 纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除能力有所不同.溶液初始pH值对纳米零价铁去除水中的Se(Ⅳ)几乎没有影响, 在pHini=2.0~11.0的范围内, Se(Ⅳ)的去除率都很高, 基本可达到完全去除(≈100%).而Se(Ⅵ)的去除受溶液初始pH的影响较大: pHini=2.0时, Se(Ⅵ)的去除率仅为50.5%; pHini=3.0~8.0时, Se(Ⅵ)的去除率最高, 接近100%;而pHini>8.0时, Se(Ⅵ)的去除率则随pH值的上升而下降, pHini=11.0时去除率已降到64.9%.尽管如此, 纳米零价铁去除Se(Ⅵ)的有效pH范围依然明显大于文献中零价铁的工作范围(pH=4.0~7.2)[32].
纳米零价铁对As(Ⅲ)和As(Ⅴ)的去除受溶液初始pH值的影响也较大.对于As(Ⅲ)的去除: pHini=2.0时, As(Ⅲ)的去除率较低, 仅为15.5%;而当pHini上升为3.0时, 去除率则急剧升高到接近100%; pHini=3.0~9.0时As(Ⅲ)的去除率始终保持在99%以上; 而当pHini>9.0时As(Ⅲ)的去除率则随pHini值的上升而下降.对于As(Ⅴ)的去除:当溶液初始pH值从2.0上升至3.0时, As(Ⅴ)的去除率从62.9%升高到100%, 而后As(Ⅴ)的去除随着pHini值的上升而逐渐下降, 当pHini>10.0时则出现急剧下降.
溶液初始pH对纳米零价铁去除As和Se的影响的原因可作如下解释.当pHini=2.0时, 纳米零价铁去除As和Se的效率都较低, 这是因为此时溶液呈强酸性造成了纳米零价铁的溶解, 导致纳米零价铁投加量相对不足, 进而造成了表面反应活性位点的短缺.而高pHini值时去除效率的下降则是因为高pHini值造成了纳米零价铁表面的快速钝化, 阻碍了来自Fe(0) 核的电子的传递, 从而抑制了As和Se在纳米零价铁表面的反应导致去除率下降.而溶液初始pH值对As(Ⅴ)的影响, 主要是因为厌氧或缺氧状态下纳米零价铁表面的铁氧化物对As(Ⅴ)的吸附是随pHini值的升高而降低的, 而酸性介质(如pHini=3.0) 能促进纳米零价铁与As(Ⅴ)的还原反应, 因此造成了As(Ⅴ)在pHini=3.0时的去除率最高.
2.5 反应后固液两相变化
以上结果表明, 在相同实验条件下纳米零价铁去除水中As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的效果各有不同.而反应后固体的形貌(图 5)、组成(图 6)以及溶液pH值(图 7)也有较大的差异(图S2~S7).
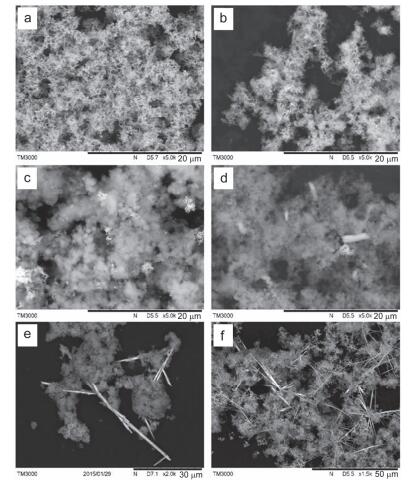 图 5
新鲜nZⅥ, (b) DI water, (c) As(Ⅲ), (d) As(Ⅴ), (e) Se(Ⅳ)和(f) Se(Ⅵ)反应前后固体SEM图
Figure 5.
SEM images of fresh nZⅥ (a) and spent nZⅥ ((b) DI water, (c) As(Ⅲ), (d) As(Ⅴ), (e) Se(Ⅳ) and (f) Se(Ⅵ))
图 5
新鲜nZⅥ, (b) DI water, (c) As(Ⅲ), (d) As(Ⅴ), (e) Se(Ⅳ)和(f) Se(Ⅵ)反应前后固体SEM图
Figure 5.
SEM images of fresh nZⅥ (a) and spent nZⅥ ((b) DI water, (c) As(Ⅲ), (d) As(Ⅴ), (e) Se(Ⅳ) and (f) Se(Ⅵ))
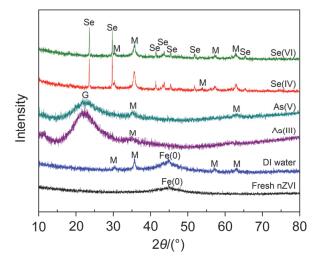 图 6
反应前后固体XRD图
Figure 6.
XRD pattern of fresh and spent nZⅥ
图 6
反应前后固体XRD图
Figure 6.
XRD pattern of fresh and spent nZⅥ
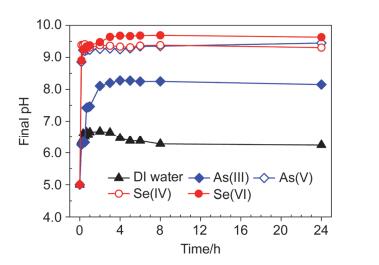 图 7
纳米零价铁与As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)反应后溶液pH值
Figure 7.
Final solution pH after reactions between nZⅥ and As(Ⅲ/Ⅴ)/ Se(Ⅳ/Ⅵ)
图 7
纳米零价铁与As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)反应后溶液pH值
Figure 7.
Final solution pH after reactions between nZⅥ and As(Ⅲ/Ⅴ)/ Se(Ⅳ/Ⅵ)
由图 5a可见, 新鲜纳米零价铁因表面张力较大而团聚成典型的链状结构; 与去离子水反应24 h后纳米零价铁形貌略有变化, 链状结构依然存在, 但新出现一些絮体和颗粒状物质(图 5b), 表明纳米零价铁在去离子水中发生腐蚀(老化)反应, 老化使得纳米零价铁的铁氧化物层增厚, 并经演变、转化生成了新的铁(羟基)氧化物. XRD(图 6)结果证实, 老化24 h后新生成的颗粒状物质为磁铁矿或磁赤铁矿, 而絮状物质则可能为晶体转化的前驱物(例如Fe(OH)2和FeOOH等).
纳米零价铁与水反应后溶液的pH值变化较小, 仅从5.0上升至6.5左右(图 7), 这是因为反应(Eq. 1) 引起了纳米零价铁氧化物壳层厚度的增加, 当氧化物层厚度增加至一定程度时会阻碍电子传递, 从而保护Fe(0) 核不被继续腐蚀而保持高活性, 这也是纳米零价铁即使老化后也能高效去除污染物质的原因[33].
纳米零价铁与As(Ⅲ)反应24 h后固体整体形状呈棉花团状絮体(图 5c), 溶液pH值稳定在8.2左右(图 7); XRD结果显示反应后腐蚀产物主要为磁铁矿/磁赤铁矿和针铁矿, 但衍射峰较宽, 表明矿物结晶度不好, 呈无定形态(图 6); 序批式化学提取法结果显示纳米零价铁可将水中As(Ⅲ)还原为As(0)(图S6).而纳米零价铁与As(Ⅴ)反应24 h后的溶液pH值稳定在9.4左右(图 7), 反应后固体呈絮状夹杂有片状物质, 其边界较为模糊(图 5d); XRD结果表明主要产物可能为无定形态的磁铁矿和针铁矿.纳米零价铁与As(Ⅲ)和As(Ⅴ)反应后固体形貌有差异(图S5), 表明不同氧化态As对形成铁腐蚀产物的影响有所不同[34].
纳米零价铁与Se(Ⅳ)和Se(Ⅵ)反应24 h后溶液pH值分别稳定在9.3和9.6左右(图 7); 反应后固体中均观测到固相新物质的生成(图 5e, 5f), 这些新物质具有规则结构, 其形貌明显异于磁铁矿、针铁矿等铁腐蚀产物.采用球差校正扫描透射电镜(Cs-STEM)联合X射线能量散射谱(XEDS)的表征结果显示, 新生成物质的直径在纳米级范畴, 为结构密实的单质硒Se(0) 纳米颗粒(图 8a~8e). XRD结果表明这些Se(0) 纳米颗粒主要为以P3121空间群排布的三方晶系灰硒晶体(t-Se)(图 6).反应中除生成t-Se纳米颗粒外, 还可生成无定形红硒(a-Se)纳米球(图S3和S4)[17]. XPS表征(图 8f)和序批式化学提取法(图S7) 结果也证实水中Se(Ⅳ)可被纳米零价铁还原为硒单质Se(0), 也可进一步还原为硒化物[Se(-Ⅱ)].纳米零价铁与Se(Ⅳ/Ⅵ)反应后的铁腐蚀产物主要为晶形较好的磁铁矿/磁赤铁矿(图 6).
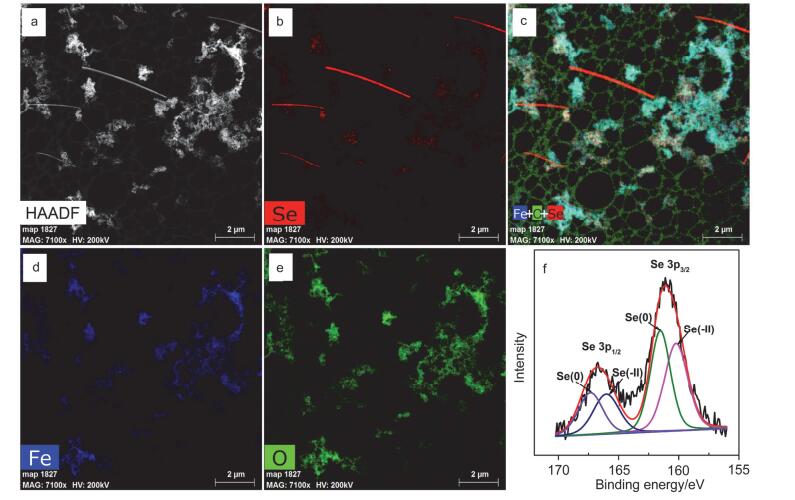 图 8
纳米零价铁与Se(Ⅳ)反应产物STEM-XEDS [(a) HAADF像、(b) Se元素面扫描图、(c) Fe+O+Se三元素面扫描叠加图、(d) Fe元素面扫描图、(e) O元素面扫描图]和(f) XPS Se 3p分析图
Figure 8.
STEM-XEDS mappings [(a) HAADF, (b) Se, (c) Fe+O+Se color overlay, (d) Fe, (e) O] and (f) XPS survey of the Se 3p regions of spent nZⅥ
图 8
纳米零价铁与Se(Ⅳ)反应产物STEM-XEDS [(a) HAADF像、(b) Se元素面扫描图、(c) Fe+O+Se三元素面扫描叠加图、(d) Fe元素面扫描图、(e) O元素面扫描图]和(f) XPS Se 3p分析图
Figure 8.
STEM-XEDS mappings [(a) HAADF, (b) Se, (c) Fe+O+Se color overlay, (d) Fe, (e) O] and (f) XPS survey of the Se 3p regions of spent nZⅥ
2.6 去除机理比较
纳米零价铁与水中As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)反应的效率、形貌组成等的不同可能是由反应机理的差异性造成的(图 9).
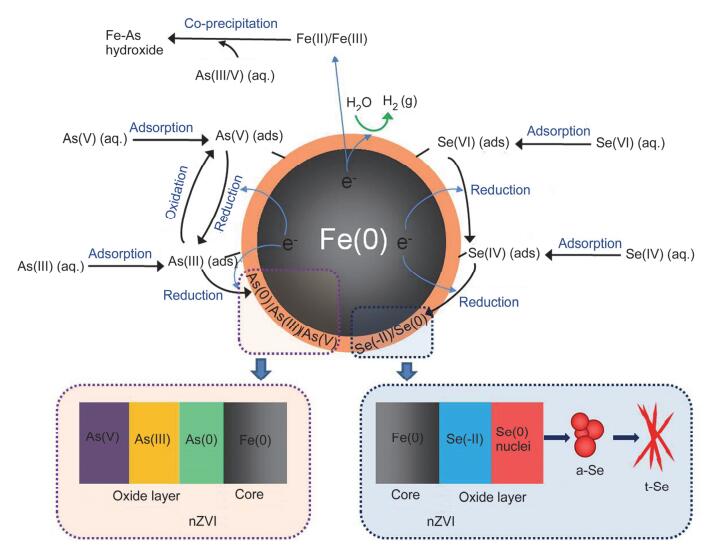 图 9
纳米零价铁与水中As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的反应机理图
Figure 9.
Mechanisms of nZⅥ reactions with aqueous As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ)
图 9
纳米零价铁与水中As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的反应机理图
Figure 9.
Mechanisms of nZⅥ reactions with aqueous As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ)
2.6.1 砷的去除机理
许多学者对零价铁和纳米零价铁与水中砷反应的机理进行了研究, 发现纳米零价铁的氧化物壳层可将水中的砷吸附到颗粒表面[13], 而后被吸附的砷会逐步向壳层内扩散, 扩散过程中逐步发生断键、还原反应[20], 高浓度的砷还会与溶解的铁发生共沉淀作用[35, 36].因此在吸附、还原和共沉淀三种机制的作用下纳米零价铁可将水中的As(Ⅲ)和As(Ⅴ)快速去除, 但去除过程略有不同.
研究发现, 水中的As(Ⅴ)特别容易吸附于铁氧化物表面, 形成内层络合物[37, 38], As(Ⅲ)也容易与纳米零价铁的氧化物壳层形成内层络合物[13, 39, 40].但随着As(Ⅴ)在铁氧化物表面吸附量的不同可能形成不同类型的表面络合物(如单齿、双齿)[37].通常在单齿络合物中Fe-As键的结合能较弱, 使得单齿络合物中的As(Ⅴ)比双齿络合物更容易解吸[38].
纳米零价铁的标准氧化还原电位为-0.440 V [Fe(Ⅱ)/Fe(0)], 而As(Ⅴ)和As(Ⅲ)的标准氧化还原电位分别为0.560 V (As(Ⅴ)/As(Ⅲ))和0.240 V (As(Ⅲ)/As(0))(表S1).因此吸附到纳米零价铁表面的As(Ⅴ)和As(Ⅲ)可被纳米零价铁还原为更低的价态. Ramos等[12]通过XPS表征分析发现, As(Ⅴ)可被纳米零价铁还原为As(Ⅲ)或As(0), 剩余的As(Ⅴ)吸附在纳米铁颗粒的外层氧化铁中, 形成的As(Ⅲ)则吸附或共沉于纳米颗粒的表面. Yan等[24]采用多线分析法对XPS数据进行了分析, 也发现不同形态的砷在纳米零价铁颗粒内呈分层分布, As(Ⅴ)主要在氧化壳层的外表面, As(Ⅲ)和As(0) 则富集在靠近Fe(0) 核的亚表层.而XAS, EXAFS的表征分析也证实了这种分层现象[20], 采用球差校正扫描透射电镜(Cs-STEM)联合XEDS, EELS的表征分析则更为直观地“看”到了反应后砷在单个纳米零价铁颗粒内的空间分布[19, 22].
在纳米零价铁与As(Ⅲ)的反应中, 除还原[As(0)]外, 吸附在铁氧化物壳层上的As(Ⅲ)还可能发生同步氧化[As(Ⅴ)]反应[15], 这可能是纳米零价铁的腐蚀产物Fe(Ⅱ)和针铁矿(α-FeOOH)共存造成的[41].而有溶解氧存在时, As(Ⅲ)的氧化则可能是类芬顿反应的结果[42].
考虑到本研究所用的As(Ⅲ/Ⅴ)浓度较高(≈1.33 mmol•L-1), 纳米零价铁还可通过共沉淀的机制将其从水中去除[35, 36].纳米零价铁在水中发生经典的腐蚀反应(Eq. 1), 释放出Fe(Ⅱ)[18]; Fe(Ⅱ)可被进一步氧化为Fe(Ⅲ) (Eqs. 2, 3), 形成磁铁矿、针铁矿等铁(羟基)氧化物[43].这些铁(羟基)氧化物在自身形成的过程中可通过共沉淀的方式将水中的As(Ⅲ/Ⅴ)固定在其结构内部, 形成Fe-As(羟基)氧化物, 达到从水中去除As(Ⅲ/Ⅴ)的目的[35].这也可能是在纳米零价铁表面活性位点不足的情况下, 纳米零价铁对As的去除能力要大于Se的主要原因.
溶液中砷的去除行为主要取决于砷的氧化态[As(Ⅲ), As(Ⅴ)]以及纳米零价铁表面壳层中铁(羟基)氧化物的种类及性质[44].研究表明[34], As(Ⅲ)与As(Ⅴ)对铁(羟基)氧化物的形成具有类似的抑制作用, 均可强烈抑制磁铁矿和针铁矿晶体的形成(图 6).但As(Ⅴ)比As(Ⅲ)对铁(羟基)氧化物的种类、结构和形貌的影响更大: As(Ⅴ)会导致所形成的铁(羟基)氧化物的结构、种类的多样性更丰富; As(Ⅴ)存在的体系中会形成空心结构的铁(羟基)氧化物、绿锈以及不同形态的砷酸铁[34].因此, 在同等实验条件下As(Ⅴ)的去除行为与As(Ⅲ)以及Se(Ⅳ/Ⅵ)等存在较大的差别.
2.6.2 硒的去除机理
纳米零价铁去除水中Se(Ⅳ)和Se(Ⅵ)的机制主要为吸附和还原[11, 14].课题组前期对纳米零价铁去除水中Se(Ⅳ)的微观机理研究结果表明, 纳米零价铁去除水中Se(Ⅳ)包含硒在纳米零价铁颗粒内的扩散与还原和零价硒纳米颗粒的形成两个过程[14, 17, 18], 即水中的Se(Ⅳ)首先被吸附到纳米零价铁表面形成双齿双核内层络合物[45~47], 在Fe(0) 核释放的电子的驱使下络合分子通过壳层上存在的瑕疵位点向颗粒内扩散, 并在扩散的过程中逐步发生断键、还原反应, 水中的Se(Ⅳ)最终被还原为Se晶核; Se晶核不断累积并长大形成球形纳米颗粒即无定形红硒(a-Se), 不稳定的a-Se会随时间逐渐转化为更稳定的三方晶系灰硒(t-Se)纳米颗粒.在电子供应充足的情况下, 部分Se核子会被进一步还原为Se(-Ⅱ), 形成Fe-Se化合物沉积在纳米零价铁Fe(0) 核与氧化物壳层之间.
纳米零价铁与水中Se(Ⅵ)的反应亦是逐步还原的过程[11], 即水中的Se(Ⅵ)首先被吸附至纳米零价铁表面, 而后被还原为Se(Ⅳ), 在电子充足的情况下Se(Ⅳ)可被进一步还原为Se(0) 和Se(-Ⅱ).由于Se(Ⅳ)与材料(如铁氧化物等)表面的亲和力优于Se(Ⅵ), 因此纳米零价铁去除Se(Ⅳ)的效果略好于Se(Ⅵ).
3 结论
纳米零价铁可广泛用于去除水中的砷/硒污染.一般而言, 在单元素体系中纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除效果顺序依次为: Se(Ⅳ)>As(Ⅲ)>Se(Ⅵ)>As(Ⅴ).水中溶解氧(DO)对As(Ⅲ)和Se(Ⅳ)的去除没有显著的影响, 而高浓度的DO对As(Ⅴ)和Se(Ⅵ)的去除产生了一定的抑制作用.增加纳米零价铁投加量可促进As和Se的去除.溶液初始pH值对纳米零价铁去除As(Ⅲ), As(Ⅴ)和Se(Ⅵ)的影响稍有不同.去除机理的不同造成了纳米零价铁与As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)反应后的固体形貌、组成及溶液pH值存在差异.在As/Se共存体系中, 纳米零价铁对As/Se的去除较复杂(图S8), 具体反应机制还需进一步研究并可采用色谱-原子光谱联用技术(LC-HG-AFS)分析反应后液相中As/Se形态结合固相表征确定[48].
4 实验部分
4.1 纳米零价铁的制备
研究所用纳米零价铁均采用NaBH4液相还原FeCl3法合成制备.具体合成步骤可在支持信息及文献[14, 49]中找到.反应完成后采用真空抽滤收集纳米零价铁, 并用大量去离子水和无水乙醇反复冲洗, 而后将纳米零价铁保存在无水乙醇中置于4 ℃冰箱备用.液相还原法合成的纳米零价铁的平均粒径约为60 nm, BET比表面积约为25~35 m2•g-1[17].
4.2 水中砷和硒的去除
纳米零价铁去除水中砷和硒的实验是在50 mL血清瓶中进行的.除投加量实验和初始浓度实验外, 所有实验中砷和硒的浓度均为1.33 mmol•L-1, 纳米零价铁的投加量均为0.5 g•L-1.为减少溶液中砷和硒的形态进而简化整个反应系统, 反应溶液的初始pH值(pHini)在实验开始前用0.1 mol•L-1 HCl或NaOH调整到5.0.当pHini=5.0时, As(Ⅲ)的主要形态为H3AsO30, As(Ⅴ)的主要形态为H2AsO4-, Se(Ⅳ)的主要形态为HSeO3-, Se(Ⅵ)的主要形态为SeO42-.实验过程中没有添加缓冲剂来保持pH值的恒定.具体实验步骤为:将35 mL的砷或硒溶液加入到相应的血清瓶中, 而后加入一定量的纳米零价铁, 加盖密封后置于25 ℃恒温摇床中震荡反应, 相应取样时间时将血清瓶从摇床取下, 样品经高速离心并经0.22 μm滤膜过滤后测定滤液中残留总砷、总硒的浓度.考察高溶解氧的影响时, 在加入纳米零价铁之前往溶液中通入30 min高纯氧气(纯度>99.9%); 模拟缺氧状态时, 则在加入纳米零价铁之前往溶液中通入30 min高纯氮气(纯度>99.9%)以排除溶液中的溶解氧; 其他实验则忽略溶解氧的影响.
4.3 分析方法
采用电感耦合等离子体发射光谱(ICP-720ES, 安捷伦公司, 美国)测定水中总砷和总硒的浓度.溶液pH值由pHS-4S型pH计测定, 溶解氧(DO)由BENNET便携式溶氧仪测定.采用球差校正扫描透射电镜(Cs-STEM, FEI公司, 美国)联合X射线能量散射谱(XEDS)对反应后纳米零价铁固体的形貌、结构和元素分布进行表征.采用X射线光电子能谱(XPS)探测固体表面的化学组成及元素化学态, 本研究采用XPSPEAK 41软件对Se 3p进行分峰拟合, 拟合过程中峰形遵循高斯(80%)-洛伦兹(20%)规则.采用X射线衍射光谱(XRD, 布鲁克公司, 德国)和TM3000扫描电镜(SEM, 东芝公司, 日本)对反应前后纳米零价铁的晶相结构和形貌进行表征.
-
-
[1]
Mirza, A.; Ramachandran, V. Removal of Arsenic and Selenium from Wastewaters——A Review, Minerals, Metals & Materials Society, Warrendale, PA, United States, 1996.
-
[2]
崔光, 付云飞, 罗军, 刘培生, 丁爱中, 化学学报, 2012, 70, 1059. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract341279.shtmlCui, G.; Fu, Y.; Luo, J.; Liu, P.; Ding, A. Acta Chim. Sinica 2012, 70, 1059. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract341279.shtml
-
[3]
Torres, J.; Pintos, V.; Gonzatto, L.; Domínguez, S.; Kremer, C.; Kremer, E. Chem. Geol. 2011, 288, 32. http://www.sciencedirect.com/science/article/pii/S0009254111002610
-
[4]
Missana, T.; Alonso, U.; Scheinost, A.; Granizo, N.; García-Gutiérrez, M. Geochim. Cosmochim. Acta 2009, 73, 6205. doi: 10.1016/j.gca.2009.07.005
-
[5]
Winkel, L. H. E.; Johnson, C. A.; Lenz, M.; Grundl, T.; Leupin, O. X.; Amini, M.; Charlet, L. Environ. Sci. Technol. 2012, 46, 571. doi: 10.1021/es203434d
-
[6]
Korte, N. E.; Fernando, Q. Crit. Rev. Environ. Control 1991, 21, 1. doi: 10.1080/10643389109388408
-
[7]
Hamilton, S. J. Sci. Total Environ. 2004, 326, 1. doi: 10.1016/j.scitotenv.2004.01.019
-
[8]
崔光, 郭宜娇, 刘培生, 化学学报, 2012, 70, 2525. doi: 10.3866/PKU.WHXB201208222Cui, G.; Guo, Y.; Liu, P. Acta Chim. Sinica 2012, 70, 2525. doi: 10.3866/PKU.WHXB201208222
-
[9]
Yamani, J. S.; Lounsbury, A. W.; Zimmerman, J. B. Water Res. 2016, 88, 889. doi: 10.1016/j.watres.2015.11.017
-
[10]
Mohan, D.; Pittman, C. U. J. Hazard. Mater. 2007, 142, 1. doi: 10.1016/j.jhazmat.2007.01.006
-
[11]
Olegario, J. T.; Yee, N.; Miller, M.; Sczepaniak, J.; Manning, B. J. Nanopart. Res. 2010, 12, 2057. doi: 10.1007/s11051-009-9764-1
-
[12]
Ramos, M. A.; Yan, W.; Li, X. Q.; Koel, B. E.; Zhang, W. X. J. Phys. Chem. C 2009, 113, 14591.
-
[13]
Kanel, S. R.; Manning, B.; Charlet, L.; Choi, H. Environ. Sci. Technol. 2005, 39, 1291. doi: 10.1021/es048991u
-
[14]
Ling, L.; Pan, B.; Zhang, W. X. Water Res. 2015, 71, 274. doi: 10.1016/j.watres.2015.01.002
-
[15]
Yan, W.; Ramos, M. A. V.; Koel, B. E.; Zhang, W. X. J. Phys. Chem. C 2012, 116, 5303. doi: 10.1021/jp208600n
-
[16]
Kanel, S. R.; Greneche, J. M.; Choi, H. Environ. Sci. Technol. 2006, 40, 2045. doi: 10.1021/es0520924
-
[17]
Xia, X.; Ling, L.; Zhang, W. X. Environ. Sci.:Nano 2017, 4, 52. doi: 10.1039/C6EN00231E
-
[18]
Xia, X.; Ling, L.; Zhang, W. X. Chemosphere 2017, 168, 1597. doi: 10.1016/j.chemosphere.2016.11.150
-
[19]
Ling, L.; Zhang, W. X. Environ. Sci. Technol. Lett. 2014, 1, 305. doi: 10.1021/ez5001512
-
[20]
Yan, W.; Vasic, R.; Frenkel, A. I.; Koel, B. E. Environ. Sci. Technol. 2012, 46, 7018. doi: 10.1021/es2039695
-
[21]
Yan, W.; Lien, H.-L.; Koel, B. E.; Zhang, W. X. Environ. Sci.:Proc. Impacts 2013, 15, 63. doi: 10.1039/C2EM30691C
-
[22]
Ling, L.; Zhang, W. X. Environ. Sci. Technol. 2017, 51, 2288. doi: 10.1021/acs.est.6b04315
-
[23]
Li, S.; Wang, W.; Liang, F.; Zhang, W. X. J. Hazard. Mater. 2017, 322, Part A, 163. http://europepmc.org/abstract/MED/26861641
-
[24]
Yan, W.; Ramos, M. A.; Koel, B. E.; Zhang, W. X. Chem. Commun. 2010, 46, 6995. doi: 10.1039/c0cc02311f
-
[25]
O'Carroll, D.; Sleep, B.; Krol, M.; Boparai, H.; Kocur, C. Adv. Water Res. 2013, 51, 104. doi: 10.1016/j.advwatres.2012.02.005
-
[26]
Klas, S.; Kirk, D. W. Sep. Purif. Technol. 2013, 116, 222. doi: 10.1016/j.seppur.2013.05.044
-
[27]
Kanel, S. R.; Greneche, J. M.; Choi, H. Environ. Sci. Technol. 2006, 40, 2045. doi: 10.1021/es0520924
-
[28]
Zhang, Y. Q.; Wang, J. F.; Amrhein, C.; Frankenberger, W. T. J. Environ. Qual. 2005, 34, 487. doi: 10.2134/jeq2005.0487
-
[29]
Tang, C.; Huang, Y. H.; Zeng, H.; Zhang, Z. Water Res. 2014, 67, 166. doi: 10.1016/j.watres.2014.09.016
-
[30]
Tang, C.; Huang, Y. H.; Zeng, H.; Zhang, Z. Chem. Eng. J. 2014, 244, 97. doi: 10.1016/j.cej.2014.01.059
-
[31]
Yoon, I.-H.; Kim, K.-W.; Bang, S.; Kim, M. G. Appl. Catal. B:Environ. 2011, 104, 185. doi: 10.1016/j.apcatb.2011.02.014
-
[32]
Liang, L.; Guan, X.; Huang, Y.; Ma, J.; Sun, X.; Qiao, J.; Zhou, G. Sep. Purif. Technol. 2015, 156, 1064. doi: 10.1016/j.seppur.2015.09.062
-
[33]
Sarathy, V.; Tratnyek, P. G.; Nurmi, J. T.; Baer, D. R.; Amonette, J. E.; Chun, C. L.; Penn, R. L.; Reardon, E. J. J. Phys. Chem. C 2008, 112, 2286. doi: 10.1021/jp0777418
-
[34]
Song, J.; Jia, S.-Y.; Yu, B.; Wu, S.-H.; Han, X. J. Hazard. Mater. 2015, 294, 70. doi: 10.1016/j.jhazmat.2015.03.048
-
[35]
Lien, H.-L.; Wilkin, R. T. Chemosphere 2005, 59, 377. doi: 10.1016/j.chemosphere.2004.10.055
-
[36]
Morin, G.; Wang, Y.; Ona-Nguema, G.; Juillot, F.; Calas, G.; Menguy, N.; Aubry, E.; Bargar, J. R.; Brown Jr, G. E. Langmuir 2009, 25, 9119. doi: 10.1021/la900655v
-
[37]
Fendorf, S.; Eick, M. J.; Grossl, P.; Sparks, D. L. Environ. Sci. Technol. 1997, 31, 315. doi: 10.1021/es950653t
-
[38]
Grossl, P. R.; Eick, M.; Sparks, D. L.; Goldberg, S.; Ainsworth, C. C. Environ. Sci. Technol. 1997, 31, 321. doi: 10.1021/es950654l
-
[39]
Kanel, S. R.; Nepal, D.; Manning, B.; Choi, H. J. Nanopart. Res. 2007, 9, 725. doi: 10.1007/s11051-007-9225-7
-
[40]
Jegadeesan, G.; Mondal, K.; Lalvani, S. B. Environ. Prog. 2005, 24, 289. doi: 10.1002/(ISSN)1547-5921
-
[41]
Amstaetter, K.; Borch, T.; Larese-Casanova, P.; Kappler, A. Environ. Sci. Technol. 2009, 44, 102. doi: 10.1021/es901274s
-
[42]
Hug, S. J.; Leupin, O. Environ. Sci. Technol. 2003, 37, 2734. doi: 10.1021/es026208x
-
[43]
Tuček, J.; Prucek, R.; Kolařík, J.; Zoppellaro, G.; Petr, M.; Filip, J.; Sharma, V. K.; Zbořil, R. ACS Sustain. Chem. Eng. 2017, 5, 3027. doi: 10.1021/acssuschemeng.6b02698
-
[44]
Dixit, S.; Hering, J. G. Environ. Sci. Technol. 2003, 37, 4182. doi: 10.1021/es030309t
-
[45]
Jordan, N.; Foerstendorf, H.; Weiß, S.; Heim, K.; Schild, D.; Brendler, V. Geochim. Cosmochim. Acta 2011, 75, 1519. doi: 10.1016/j.gca.2011.01.012
-
[46]
Myneni, S.; Tokunaga, T. K.; Brown, G. Science 1997, 278, 1106. doi: 10.1126/science.278.5340.1106
-
[47]
Duc, M.; Lefevre, G.; Fedoroff, M.; Jeanjean, J.; Rouchaud, J.; Monteil-Rivera, F.; Dumonceau, J.; Milonjic, S. J. Environ. Radioact. 2003, 70, 61. doi: 10.1016/S0265-931X(03)00125-5
-
[48]
Huang, L.; Yao, L.; He, Z.; Zhou, C.; Li, G.; Yang, B.; Deng, X. Chemosphere 2014, 100, 57. doi: 10.1016/j.chemosphere.2013.12.074
-
[49]
Sun, Y. P.; Li, X. Q.; Cao, J.; Zhang, W. X.; Wang, H. P. Adv. Colloid Interface Sci. 2006, 120, 47. doi: 10.1016/j.cis.2006.03.001
-
[1]
-

图 1 不同溶解氧条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 1 Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) with nZⅥ under different DO conditions
Reaction conditions: [nZⅥ]=0.5 g•L-1, [As(Ⅲ/Ⅴ)]0=[Se(Ⅳ/Ⅵ)]0=1.33 mmol•L-1, pHini=5.0, time=24 h, anoxic (DO < 0.5 mg•L-1), low oxygen (DO≈7 mg•L-1), high oxygen (DO > 14 mg•L-1).

图 2 不同纳米零价铁投加量条件下As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 2 Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) with different nZⅥ dosages
Reaction conditions: [nZⅥ]=0.2~5.0 g•L-1, [As(Ⅲ/Ⅴ)]0=[Se(Ⅳ/Ⅵ)]0=1.33 mmol•L-1, pHini=5.0, time=24 h.

图 3 不同接触时间条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 3 Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) as functions of time
Reaction conditions: [nZⅥ]=0.5 g•L-1, [As(Ⅲ/Ⅴ)]0=[Se(Ⅳ/Ⅵ)]0=1.33 mmol•L-1, pHini=5.0.

图 4 不同溶液初始pH值条件下纳米零价铁对As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)的去除
Figure 4 Removal of As(Ⅲ/Ⅴ) and Se(Ⅳ/Ⅵ) with nZⅥ at different initial solution pH
Reaction conditions: [nZⅥ]=0.5 g•L-1, [As(Ⅲ/Ⅴ)]0=[Se(Ⅳ/Ⅵ)]0=1.33 mmol•L-1, time=24 h.

图 5 新鲜nZⅥ, (b) DI water, (c) As(Ⅲ), (d) As(Ⅴ), (e) Se(Ⅳ)和(f) Se(Ⅵ)反应前后固体SEM图
Figure 5 SEM images of fresh nZⅥ (a) and spent nZⅥ ((b) DI water, (c) As(Ⅲ), (d) As(Ⅴ), (e) Se(Ⅳ) and (f) Se(Ⅵ))
Reaction conditions: [nZⅥ]=0.5 g•L-1, [As(Ⅲ/Ⅴ)]0=[Se(Ⅳ/Ⅵ)]0=1.33 mmol•L-1, pHini=5.0, time=24 h.

图 6 反应前后固体XRD图
Figure 6 XRD pattern of fresh and spent nZⅥ
Reaction conditions: [nZⅥ]=0.5 g•L-1, [As(Ⅲ/Ⅴ)]0=[Se(Ⅳ/Ⅵ)]0=1.33 mmol•L-1, pHini=5.0, time=24 h. Peaks are referred to magnetite/maghemite (M), geothite (G), element selenium Se(0) and Fe(0).

图 7 纳米零价铁与As(Ⅲ/Ⅴ)和Se(Ⅳ/Ⅵ)反应后溶液pH值
Figure 7 Final solution pH after reactions between nZⅥ and As(Ⅲ/Ⅴ)/ Se(Ⅳ/Ⅵ)
Reaction conditions: [nZⅥ]=0.5 g•L-1, [As(Ⅲ/Ⅴ)]0=[Se(Ⅳ/Ⅵ)]0=1.33 mmol•L-1, pHini=5.0

图 8 纳米零价铁与Se(Ⅳ)反应产物STEM-XEDS [(a) HAADF像、(b) Se元素面扫描图、(c) Fe+O+Se三元素面扫描叠加图、(d) Fe元素面扫描图、(e) O元素面扫描图]和(f) XPS Se 3p分析图
Figure 8 STEM-XEDS mappings [(a) HAADF, (b) Se, (c) Fe+O+Se color overlay, (d) Fe, (e) O] and (f) XPS survey of the Se 3p regions of spent nZⅥ
-


 下载:
下载:








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 15
- 文章访问数: 3047
- HTML全文浏览量: 526
 下载:
下载:
