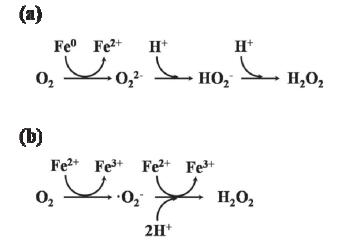
 图 1
(a)双电子分子氧活化过程; (b)连续单电子分子氧活化过程[7]
Figure 1.
(a) Two-electron molecular oxygen activation with Fe0; (b) A sequential single-electron molecular oxygen activation with Fe2+[7]
图 1
(a)双电子分子氧活化过程; (b)连续单电子分子氧活化过程[7]
Figure 1.
(a) Two-electron molecular oxygen activation with Fe0; (b) A sequential single-electron molecular oxygen activation with Fe2+[7]
引用本文:
穆毅, 贾法龙, 艾智慧, 张礼知. 纳米零价铁活化分子氧原理及降解有机污染物性能增强策略[J]. 化学学报,
2017, 75(6): 538-543.
doi:
10.6023/A17020047

Citation: Mu Yi, Jia Falong, Ai Zhihui, Zhang Lizhi. Molecular Oxygen Activation with Nano Zero-valent Iron for Aerobic Degradation of Organic Contaminants and the Performance Enhancement[J]. Acta Chimica Sinica, 2017, 75(6): 538-543. doi: 10.6023/A17020047
Citation: Mu Yi, Jia Falong, Ai Zhihui, Zhang Lizhi. Molecular Oxygen Activation with Nano Zero-valent Iron for Aerobic Degradation of Organic Contaminants and the Performance Enhancement[J]. Acta Chimica Sinica, 2017, 75(6): 538-543. doi: 10.6023/A17020047
纳米零价铁活化分子氧原理及降解有机污染物性能增强策略
摘要:
纳米零价铁直接还原降解有机污染物运行长效性差,且不能矿化有机污染物.利用纳米零价铁还原活化分子氧生成活性氧物种可以氧化甚至矿化有机污染物.在最近的研究中,作者提出了纳米零价铁活化分子氧的双途径机理,即铁核电子转移到氧化铁壳表面的双电子还原活化分子氧途径和氧化铁表面结合态亚铁离子的单电子还原活化分子氧途径,阐释了纳米零价铁核壳结构依赖的分子氧活化降解有机污染物性能机制及性能增强策略.证实在纳米零价铁活化分子氧体系添加少量亚铁离子能在零价铁表面形成更多的结合态亚铁,显著增强纳米铁表界面活性氧物种生成量;同时,在纳米零价铁活化分子氧体系中引入少量有机或无机配体亦可提高活性氧物种产生效率,从而增强有机污染物降解性能.最后讨论了典型环境因素如pH值、共存离子、天然有机物等影响纳米零价铁活化分子氧降解有机污染物性能的规律.
English
Molecular Oxygen Activation with Nano Zero-valent Iron for Aerobic Degradation of Organic Contaminants and the Performance Enhancement
Abstract:
Nano zero-valent iron (nZVI) is a special kind of iron with large specific surface area, strong reduction activity, and the environmental friendliness. nZVI was usually used to reductively degrade organic pollutants, but its long-term performance was poor and the organic pollutants could not be mineralized. Nano zero-valent iron can reductively activate molecular oxygen to generate reactive oxygen species for oxidation or even mineralization of organic pollutants. Recently, we found the core-shell structure dependent aerobic degradation of organic pollutants by nZVI and proposed a new physical insight into the molecular oxygen activation mechanism of the aerobic nZVI process, where the outward electrons transfer from the iron core initiate the two-electron molecular oxygen activation and surface bound ferrous ions on iron oxide shell favor the single-electron molecular oxygen activation. Several strategies have also been proposed to enhance the production of reactive oxidants by nZVI-induced oxygen activation. We confirmed that addition of extra ferrous ions into the nZVI/O2 system could generate more surface bound ferrous ions for significantly enhancing the generation of reactive oxygen species. Meanwhile, the introduction of some inorganic or organic ligands in the aerobic nZVI system could also improve the active oxygen species generation efficiency. Finally main typical environmental factors including of the pH value, coexisting ions, natural organic matter on the organic pollutants degradation with the aerobic nZVI were discussed. By the way, we also investigated the anoxic Cr(Ⅵ) removal with nZVI. It was found the Cr(Ⅵ) removal rate constant was mainly attributed to the reduction of Cr(Ⅵ) by the surface bound Fe(Ⅱ) besides the reduction of Cr(Ⅵ) adsorbed on the iron oxide shell via the electrons transferred from the iron core. We also demonstrated that the presence of oxygen molecule can inhibit Cr(Ⅵ) removal with nZVI, which was attributed to that the oxygen molecular activation could compete with Cr(Ⅵ) for the consumption of surface bond Fe(Ⅱ) and donor electrons transferred from Fe0 core.
-
1 引言
纳米零价铁(Nano zero-valent iron, nZVI)具有还原能力强, 比表面积大, 环境友好等优点.自20世纪末以来, 纳米零价铁、负载型零价铁、双金属零价铁等被广泛用于还原修复卤代烷烃/烯烃、卤代苯/联苯、DDT、硝基芳香化合物、重金属等污染地下水或土壤[1~3].但利用nZVI的还原特性修复地下水或土壤有两点备受争议:其一, nZVI的腐蚀和损耗导致其运行长效性差; 其二, 仅利用nZVI的还原性不能够矿化有机物, 修复过程中甚至产生毒性更大的副产物[4].相较于nZVI还原降解有机污染物, nZVI活化分子氧产生活性氧物种氧化矿化有机污染物更受青睐.
氧气是一种最为绿色经济的氧化剂, 在空气中含量高达21%.但是氧气分子的电子排布特性使它和有机分子在基态反应时受到自旋限制, 难以在常温常压下氧化降解有机污染物.零价铁具备较低的还原电势[E0(Fe2+/Fe0)=-0.44 VNHE]. O2得到一个电子还原成•O2-的还原电位是E0=-0.33 VNHE, 而O2得到两个电子还原成H2O2的还原电位是E0=-0.695 VNHE.因此零价铁能够活化分子氧产生超氧自由基(•O2-), 双氧水(H2O2) (式1~3). H2O2与Fe2+发生Fenton反应进一步生成活性氧物种(ROS)羟基自由基(•OH)和Fe(Ⅳ) (式4, 5)[5, 6], ROS能够氧化甚至矿化有机污染物至无毒的小分子酸或二氧化碳(式6).但纳米零价铁的电子利用率低, 零价铁活化分子氧过程中会伴随着大量不利于活性氧物种生成的副反应, 如典型的产氢反应(式7), 实际•OH生成效率仅为理论值的10%左右[7], 如何增强纳米零价铁活化分子氧体系ROS生成效率, 促进其氧化甚至矿化有机污染物性能是目前亟待解决的问题.
作者系统地总结了近年来围绕纳米零价铁活化分子氧氧化降解有机污染物研究成果, 揭示了纳米零价铁活化分子氧原理以及通过在反应体系中加入亚铁离子、络合剂, 或改变纳米零价铁结构、引入双金属型(纳米零价铁)等策略增强活性氧物种生成效率, 促进有机污染物降解方面的工作, 同时评论了自然环境因素如pH值、共存离子、天然有机物等对纳米零价铁活化分子氧的影响规律.借此加深理解纳米零价铁活化分子氧原理, 并为该技术的工程应用提供理论支持.
2 纳米零价铁活化分子氧
2.1 纳米零价铁活化分子氧原理
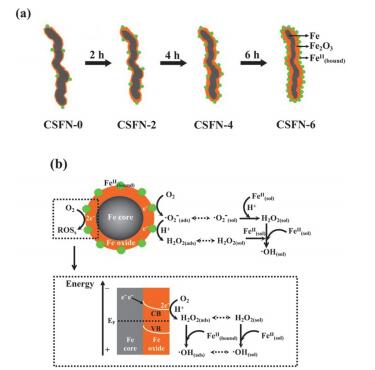
近年来, 加州伯克利大学Sedlak教授等[7]和澳洲新南威尔士大学Waite教授等[6]提出利用纳米零价铁与分子氧反应生成活性氧物种氧化降解有机污染物, 发现分子氧的存在不仅能加快零价铁降解有机污染物, 而且能实现有机污染物的深度氧化.纳米零价铁nZVI活化分子氧过程中产生的ROS及其后续反应是有机污染物氧化降解的源动力.一般来说, nZVI活化分子氧(nZVI/Air)产生ROS可以通过两条电子转移途径完成[7].第一条途径称为“双电子分子氧活化途径”(图 1a), 即分子氧从nZVI直接得到两个电子还原生成双氧水(式1)[8, 9].第二条途径称为“连续单电子分子氧活化途径”, 即nZVI在与O2反应过程中产生Fe(Ⅱ), Fe(Ⅱ)单电子活化分子氧生成•O2-(式2), •O2-在水相中继续和Fe(Ⅱ)反应生成H2O2(式3) (图 1b)[10].而生成的双氧水随后会发生芬顿(Fenton)反应生成•OH和Fe(Ⅳ) (式4, 5). •OH和Fe(Ⅳ)具有强氧化性,能够氧化甚至矿化有机污染物(式6).与此同时, 铁是一种活泼金属, 会与溶剂水(水合质子)发生反应生成气态H2释放(式7).由于H2的生成会消耗零价铁的电子, 因此被认为是nZVI/Air体系活化分子氧的副反应, 会抑制ROS的生成及后续有机污染物的氧化降解(式6).
图 1
(a)双电子分子氧活化过程; (b)连续单电子分子氧活化过程[7]
Figure 1.
(a) Two-electron molecular oxygen activation with Fe0; (b) A sequential single-electron molecular oxygen activation with Fe2+[7]
目前研究还尚未确认到底是哪条电子转移途径是nZVI活化分子氧产生双氧水的主要途径.从热力学的角度来说, 分子氧两电子还原成H2O2的电位(E0=0.695 VSHE)比单电子还原成•O2-的电位(E0=-0.33 VSHE)正得多, 因此分子氧还原成H2O2在热力学上更容易发生, 但由于生成•O2-仅需要一个电子, 因此•O2-的生成具有动力学上的优势.活化分子氧生成•O2-是连续单电子转移途径的决速步骤.普遍认为纳米零价铁活化分子氧主要是通过双电子还原分子氧生成双氧水这一途径实现.为了阐明零价铁活化分子氧机理, 我们设计合成出表面氧化铁层厚度可控且在空气中稳定的高活性纳米零价铁——Fe@Fe2O3核壳结构纳米线[11].在利用核壳结构纳米Fe@Fe2O3活化分子氧深度氧化4-氯酚时, X射线光电子能谱表征实验发现, Fe@Fe2O3表面Fe2+离子的浓度会随着陈放时间的增加而增加.电子顺磁共振表征实验证明了表面吸附的Fe2+能够促进单电子还原分子氧生成超氧负离子.而且, 电子还能够从纳米铁核经氧化铁导带传递到表面双电子还原分子氧生成双氧水, 因此我们认为单电子活化途径和双电子活化途径能够同时发生.并发现双途径活化分子氧的协同效应能加速4-氯酚的深度氧化(图 2)[12].我们认为纳米零价铁双途径分子氧活化机理具有两个重要的环境意义.其一, 该机理能提供增强分子氧活化新途径; 其二, 该机理将有利于我们调控活性氧物种实现难降解有机污染物的深度氧化.
2.2 活性氧物种的生成效率
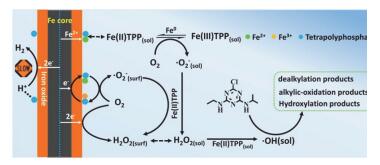
纳米零价铁活化分子氧生成活性氧物种效率十分低下, 实际•OH生成效率仅为理论值的10%左右[7].这是因为nZVI在活化分子氧的过程中会伴随着大量副反应, 如nZVI与氧气和水的副反应分别生成水和氢气(式7, 8).而且, 分子氧在nZVI表面发生反应, 生成的一些铁氧化物也会导致nZVI钝化(图 3) (式9~13).同时, 由于nZVI表面活性过高, 新生成的H2O2会马上在表面还原生成水(式14).使得H2O2与Fe2+反应几率减少, 从而导致活性氧物种的产量大大减少[13, 14].
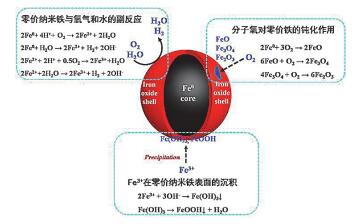 图 3
零件纳米铁活化分子氧过程中一些不利于活性氧物种生成的副反应
Figure 3.
The side reaction occurred during the molecular oxygen activation with nZVI
图 3
零件纳米铁活化分子氧过程中一些不利于活性氧物种生成的副反应
Figure 3.
The side reaction occurred during the molecular oxygen activation with nZVI
3 增强纳米零价铁活化分子氧降解有机污染物策略
3.1 核壳结构纳米零价铁
材料的结构, 尤其是表面结构, 在许多情况下甚至比其尺寸和形态的影响更为重要.如果在纳米材料表面包裹一层其它材料, 制备成为核壳结构的纳米复合材料, 有可能改善其表面化学性质、热力学性质、光电性能、催化性能以及磁性质等. Fe@Fe2O3核壳结构纳米线是一种表面氧化铁层厚度可控, 且高活性的纳米零价铁, Fe@Fe2O3比单一的Fe或者Fe2O3在中性条件下超声-Fenton或者电-Fenton降解染料废水具有更高的效率[15, 16].在纳米零价铁双途径分子氧活化机理指导下, 我们诠释了Fe@Fe2O3纳米线核壳结构依赖的分子氧活化深度氧化4-氯酚特性(图 2).发现随着nZVI表面铁氧化层厚度增加, 核内Fe0向外传输电子的过程受阻, 不利于双电子分子氧活化生成H2O2, 导致4-氯酚的降解效率随nZVI的氧化层厚度增加呈现先增加后降低的趋势; 同时, 随着核壳结构纳米线氧化层增厚, 氧化层表面吸附的Fe(Ⅱ)增加, 有利于单电子活化分子氧生成•O2-(式15)[12].
3.2 添加亚铁离子
纳米零价铁暴露在空气或者水溶液中表面会形成氧化铁层, 导致纳米铁表面本身是一个复杂的“多价态铁”体系, 特定条件下溶出的Fe(Ⅲ)和Fe(Ⅱ)使纳米铁具备潜在的氧化还原能力.同时, 纳米铁的铁氧化物外层表面存在着台阶、空位和扭折位点能够吸附络合溶液中的Fe(Ⅱ)并与其发生复杂的交互反应, 形成Fe(Ⅱ)/铁氧化物表面结合铁物种.一般认为, 铁氧化物表面的吸附位点≡FeⅢOH与游离态FeⅡ(OH)+吸附生成表面≡FeⅢOFeⅡOH物种(式16), 再通过电子转移生成被认为是具有增强还原性能的活性混合价态表面结合铁物种≡FeⅡOFeⅢOH(式17), 此时电子转移停留在固相表面, 与有机物接触性最强; 继而在铁氧化物表面形成无定形氧化铁沉淀≡FeⅡOFeⅢ (AFO) (式18), 最后表面生成Fe3O4或其他形态混合价态铁氧化物(式19).对于Fe(Ⅱ)与纳米零价铁界面相互作用还伴随着两个逆方向的电子转移过程, 即表层电子不断向内层迁移和内层铁源源不断地向外层提供电子; 同时, 纳米零价铁电子传输和迁移与表层氧化铁的半导体本质密切相关.
研究已证实铁氧化物表面结合Fe(Ⅱ)具有比游离的Fe(Ⅱ)更强的还原能力[18].游离态Fe(Ⅱ)通过离子交换形式进入矿物表面形成吸附态Fe(Ⅱ), 吸附态Fe(Ⅱ)在铁氧化物表面彼此接近, 增大了电子云密度, 从而增强了Fe(Ⅱ)/铁氧化物的还原能力[19].利用nZVI表面氧化层吸附的Fe(Ⅱ)比游离态Fe(Ⅱ)具备更高的还原活性的特性, 能够调控nZVI表界面Fe(Ⅱ)量, 达到调控ROS的目的.我们研究发现在nZVI活化分子氧体系添加少量亚铁离子能在nZVI表面形成更多的结合态亚铁, 显著增强nZVI界面ROS特别是•OH的生成量, 将nZVI活化分子氧降解西玛津的速率提高了5倍; 以陈放时间为2 h的Fe@Fe2O3纳米线为例, 吸附在Fe@Fe2O3纳米线氧化层表面Fe(Ⅱ)的连续单电子分子氧活化途径对H2O2生成的贡献比率可以高达60%, 高于Fe0的两电子分子氧活化途径对H2O2生成的贡献比率(图 4)[17].
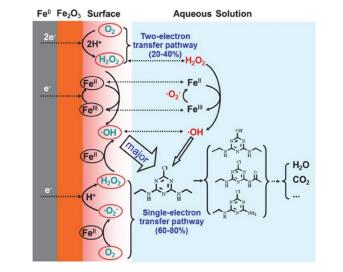 图 4
根据双氧水检测结果, 连续单电子分子氧活化路径对双氧水产量的贡献率高达60%, 高于双电子分子氧活化路径对双氧水生成的贡献率[17]
Figure 4.
According to the H2O2 detection results, the contribution of the sequential single-electron transfer pathway on the H2O2 generation was more than 60%, higher than that of the two-electron molecular oxygen activation pathway[17]
图 4
根据双氧水检测结果, 连续单电子分子氧活化路径对双氧水产量的贡献率高达60%, 高于双电子分子氧活化路径对双氧水生成的贡献率[17]
Figure 4.
According to the H2O2 detection results, the contribution of the sequential single-electron transfer pathway on the H2O2 generation was more than 60%, higher than that of the two-electron molecular oxygen activation pathway[17]
3.3 加入配体
体系中加入铁螯合剂配体是一种比较常见的提高nZVI活化分子氧产生活性氧物种效率的方法. Cheng等[20]报道了乙二胺四乙酸(EDTA)辅助的nZVI空气体系在温和条件下可以完全矿化4-氯酚和五氯酚; Sedlak等[13]发现在nZVI活化分子氧反应体系中加入草酸, EDTA, 次氮基三乙酸(NTA), 活性氧物种的生成量分别提高了2到7倍.这是因为配体能够与Fe(Ⅱ)或Fe(Ⅲ)发生螯合作用, 阻碍铁沉积物在nZVI表面的形成, 从而在一定程度上缓解了nZVI表面钝化, 有利于分子氧活化的持续进行, 同时, 有机配体与Fe(Ⅱ)、Fe(Ⅲ)配位后能够加速Fenton反应, 也促进了ROS的生成.但在体系中加入以EDTA为代表的有机配体会发生配体自降解, 消耗ROS, 同时也可能引入新的污染源.近期我们研究了环境友好的多氨羧络合剂二乙基三胺五乙酸(DTPA)促进核壳结构Fe@Fe2O3纳米线活化分子氧降解4-CP性能, 发现DTPA的存在促进核壳结构Fe@Fe2O3纳米线活化分子氧产生更多的•OH, 从而促进DTPA和4-CP的快速氧化矿化[21].
以四聚磷酸盐(TPP), 多金属氧酸盐(POMs)为代表的无机配体因其在反应过程中能够稳定存在受到研究者的关注.如以PW12O403-和SiW12O404-为代表的多金属氧酸盐作为典型电子穿梭体[22, 23], 能促进nZVI的电子传递, 有利于氧气得电子生成H2O2.同时, POMs还能抑制H2O2在零价纳米铁表面进一步被还原成H2O, 增加H2O2参与Fenton反应的几率.最近, 我们研究发现TPP能够促进Fe@Fe2O3纳米铁活化分子氧产生ROS, 阿特拉津氧化降解的效率提高了955倍, 其增强效果是文献中常用配体EDTA效果的10倍[24]; 我们认为TPP主要起到了三方面的作用(图 5):第一, TPP能够抑制Fe@Fe2O3纳米铁的产氢反应, 更多的电子可以参与活化分子氧, 从而提高了电子利用率; 第二, TPP能够与Fe(Ⅱ)形成可溶性的Fe(Ⅱ)-TPP配合物, 不仅防止铁沉积, 同时还能够促进Fenton反应产生更多的•OH; 第三, Fe(Ⅱ)-TPP还能够促进连续单电子分子氧活化过程[24, 25].
3.4 双金属纳米零价铁体系
在纳米零价铁中引入某些金属可以有效减缓nZVI的表面钝化, 提高其活化分子氧降解有机污染物活性.这些金属包括钯(Pd), 铂(Pt), 银(Ag), 铜(Cu), 镍(Ni)[26].如研究者发现Fe-Ni合金纳米颗粒([Ni]/[Fe]摩尔浓度比为0.28) 或表面掺Ni的nZVI颗粒([Ni]/[Fe]摩尔浓度比为0.035) 氧化甲醇的能力相较于nZVI都能分别提高了40%和85%[27].鉴于表面Ni掺杂nZVI颗粒含Ni量远远少于Fe-Ni合金纳米颗粒, 表面Ni掺杂nZVI颗粒更加环境友好且更具有实用价值. Ni的引入促进nZVI活化分子氧因于两个方面的原因:第一, Ni表面掺杂后nZVI的表面活性降低, 不利于H2O2进一步还原成H2O (式14), 这也促使了H2O2与Fe(Ⅱ)发生反应生成活性氧物种(式5).第二, 在中性条件下, Ni-Fe表面还能够促进Fe(Ⅱ)连续单电子活化分子氧.事实上, 我们已发现在nZVI/Air体系中镍离子的加入能同时促进超氧负离子和羟基自由基的生成, 将nZVI活化分子氧降解阿特拉津(20 mg•L-1)的速率提高6倍[28].
4 自然因素的影响
典型环境因素, 如pH值、金属离子、无机阴离子以及天然有机物等会影响纳米零价铁表界面性质, 从而影响nZVI的电子传输和活化分子氧降解有机污染物性能.电对O2/•O2-的半反应式中有H+或OH-离子参加, 因而氧化还原电位与pH值有关, 在强碱性溶液(pH=14) 中, Eθ=0.401 V (式20);而在中性(pH=7) 和强酸性(pH=0) 溶液中, Eθ分别为0.815 V和l.229 V (式21). Fe(Ⅱ)与O2反应生成•O2-是连续单电子分子氧活化过程中的控速步, 反应速率会随着体系pH增大而变快.一般情况下, 反应体系的pH最好要高于6.6才能保证反应式2快速进行[29].研究发现, 当pH=7, 体系中氧气的浓度为0.25 mmol/L时, Fe(Ⅱ)的半衰期大约为10 min.而且, pH的变化还能影响nZVI表面的电荷以及Fe(Ⅱ)的吸附[29].譬如, 在pH值为6.0时, nZVI对Fe(Ⅱ)吸附量较少, 而在pH超过7.5时, Fe(Ⅱ)会以Fe(OH)2的形式存在, Fe(OH)2可能在nZVI表面形成新的活性位点.
水中金属阳离子和无机阴离子可能会吸附到nZVI表面的氧化层, 影响nZVI活化分子氧的性能. Liu及其同事[30, 31]发现, 一些金属阳离子如Mg2+, Mn2+, Co2+, Ni2+, Zn2+, Cu2+和Pb2+吸附到nZVI表面后, 会破坏nZVI表面氧化层, 从而有利于三氯乙烯(TCE)的降解. TCE的降解效果与这些金属阳离子和nZVI表面氧化层的亲和力(log K)呈正相关(图 6).但是, 水体中常见的无机阴离子如NO3-, HCO3-, SO42-和PO43-会占据还原分子氧的活性位点, 同时形成了一层钝化膜阻碍nZVI的电子向外传递, 降低nZVI去除有机污染物的活性[32].同时, 腐植酸、富里酸、草酸等天然有机物本身就是电子的穿梭体, 金属离子的配体, 并且具有一定的光敏性[33].有些天然有机物能够直接还原含铁矿物生成溶解性的Fe(Ⅱ)从而影响着铁的氧化还原性能.天然有机物可以提高Fe(Ⅱ)活化分子氧生成氧活性物种降解污染物的效率[34].
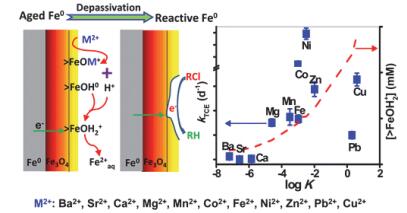 图 6
左图:金属阳离子诱导老化零价纳米铁去钝化过程的原理图.右图:零价纳米铁降解三氯乙烯的准一级动力学常数与金属阳离子和nZVI表面氧化层的亲和力(log K)的线性相关度[30]
Figure 6.
Left: Schematic illustrations of the cation-induced depassivation process of aged nZVI. Right: Correlation of the pseudo-first order rate constants (k) of TCE degradation with log K values of different cations on hydrous ferric oxide (HFO, 1 g•L-1, 600 m2•g-1)[30]
图 6
左图:金属阳离子诱导老化零价纳米铁去钝化过程的原理图.右图:零价纳米铁降解三氯乙烯的准一级动力学常数与金属阳离子和nZVI表面氧化层的亲和力(log K)的线性相关度[30]
Figure 6.
Left: Schematic illustrations of the cation-induced depassivation process of aged nZVI. Right: Correlation of the pseudo-first order rate constants (k) of TCE degradation with log K values of different cations on hydrous ferric oxide (HFO, 1 g•L-1, 600 m2•g-1)[30]
5 结论与展望
纳米零价铁活化分子氧产生活性氧物种为一种新型的高级氧化技术其应用潜力有目共睹.在环境友好且价格低廉的nZVI和自然界中最常见的绿色氧化剂氧气的共同作用下, 能够将难降解的有机污染物氧化甚至矿化至小分子酸和CO2, 也可望解决目前低价铁还原修复技术存在的问题.本文系统总结了有关nZVI活化分子氧产生活性氧物种的研究成果, 这些成果丰富了nZVI活化分子氧的理论基础, 也为基于nZVI/O2的绿色高效有机污染物控制技术提供了理论支持.但是, nZVI活化分子氧在应用上的短板也十分明显, 表现在价格较高, 反应过程中nZVI容易钝化, 活性氧物种的生成效率低, 缺乏实际工程数据等.因此, 今后的研究还应该通过挖掘一些绿色、经济、简便的方法来发展廉价的nZVI, 增强纳米零价铁和空气(氧气)体系活性氧物种的生成效率, 并且获取其实际工程示范数据等.
-
-
[1]
Phillips, D. H.; Nooten, T. V.; Bastiaens, L.; Russell, M. I.; Dickson, K.; Plant, S.; Ahad, J. M. E.; Newton, T.; Elliot, T.; Kalin, R. M. Environ. Sci. Technol. 2010, 44, 3861. doi: 10.1021/es902737t
-
[2]
Yin, W.; Wu, J.; Li, P.; Wang, X.; Zhu, N.; Wu, P.; Yang, B. Chem. Eng. J. 2012, 184, 198. doi: 10.1016/j.cej.2012.01.030
-
[3]
Mu, Y.; Ai, Z.; Zhang, L.; Song, F. ACS Appl. Mater. Inter. 2015, 7, 1997. doi: 10.1021/am507815t
-
[4]
Li, X. Q.; Elliott, D. W.; Zhang, W. X. Crit. Rev. Solid State Mater. Sci. 2006, 31, 111. doi: 10.1080/10408430601057611
-
[5]
Ona-Nguema, G.; Morin, G.; Wang, Y.; Foster, A. L.; Juillot, F.; Calas, G.; Brown, G. E. Environ. Sci. Technol. 2010, 44, 5416. doi: 10.1021/es1000616
-
[6]
Joo, S. H.; Feitz, A. J.; Waite, T. D. Environ. Sci. Technol. 2004, 38, 2242. doi: 10.1021/es035157g
-
[7]
Keenan, C. R.; Sedlak, D. L. Environ. Sci. Technol. 2008, 42, 1262. doi: 10.1021/es7025664
-
[8]
Zečević, S.; Dražić, D. M.; Gojković, S. J. Electroanal. Chem. 1989, 265, 179. doi: 10.1016/0022-0728(89)80188-3
-
[9]
Fu, F.; Dionysiou, D. D.; Liu, H. J. Hazard. Mater. 2014, 267, 194. doi: 10.1016/j.jhazmat.2013.12.062
-
[10]
Stumm, W.; Lee, G. F. Ind. Eng. Chem. 1961, 53, 143. doi: 10.1021/ie50614a030
-
[11]
Lu, L.; Ai, Z.; Li, J.; Zheng, Z.; Li, Q.; Zhang, L. Cryst. Growth Des. 2007, 7, 459. doi: 10.1021/cg060633a
-
[12]
Ai, Z.; Gao, Z.; Zhang, L.; He, W.; Yin, J. J. Environ. Sci. Technol. 2013, 47, 5344. doi: 10.1021/es4005202
-
[13]
Keenan, C. R.; Sedlak, D. L. Environ. Sci. Technol. 2008, 42, 6936. doi: 10.1021/es801438f
-
[14]
Lee, H.; Lee, H. J.; Kim, H. E.; Kweon, J.; Lee, B. D.; Lee, C. J. Hazard. Mater. 2014, 265, 201. doi: 10.1016/j.jhazmat.2013.11.066
-
[15]
Ai, Z.; Lu, L.; Li, J.; Zhang, L.; Qiu, J.; Wu, M. J. Phys. Chem. C 2007, 111, 7430. doi: 10.1021/jp070412v
-
[16]
Ai, Z.; Mei, T.; Liu, J.; Li, J.; Jia, F.; Zhang, L.; Qiu, J. J. Phys. Chem. C 2007, 111, 14799. doi: 10.1021/jp073617c
-
[17]
Liu, W.; Ai, Z.; Cao, M.; Zhang, L. Appl. Catal., B 2014, 150-151, 1. doi: 10.1016/j.apcatb.2013.11.034
-
[18]
Pecher, K.; Haderlein, S. B.; Schwarzenbach, R. P. Environ. Sci. Technol. 2002, 36, 1734. doi: 10.1021/es011191o
-
[19]
Amonette, J. E.; Workman, D. J.; Kennedy, D. W.; Fruchter, J. S.; Gorby, Y. A. Environ. Sci. Technol. 2000, 34, 4606. doi: 10.1021/es9913582
-
[20]
Noradoun, C.; Engelmann, M. D.; McLaughlin, M.; Hutcheson, R.; Breen, K.; Paszczynski, A.; Cheng, I. F. Ind. Eng. Chem. Res. 2003, 42, 5024. doi: 10.1021/ie030076e
-
[21]
Huang, Q.; Cao, M.; Ai, Z.; Zhang, L. Appl. Catal., B 2015, 162, 326. http://www.sciencedirect.com/science/article/pii/s0926337314003853
-
[22]
Lee, C.; Keenan, C. R.; Sedlak, D. L. Environ. Sci. Technol. 2008, 42, 4921. doi: 10.1021/es800317j
-
[23]
Lee, J.; Kim, J.; Choi, W. Environ. Sci. Technol. 2007, 41, 3335. doi: 10.1021/es062430g
-
[24]
Wang, L.; Cao, M.; Ai, Z.; Zhang, L. Environ. Sci. Technol. 2014, 48, 3354. doi: 10.1021/es404741x
-
[25]
Kim, H. H.; Lee, H.; Kim, H. E.; Seo, J.; Hong, S. W.; Lee, J. Y.; Lee, C. Water Res. 2015, 86, 66. doi: 10.1016/j.watres.2015.06.016
-
[26]
Cwiertny, D. M.; Bransfield, S. J.; Roberts, A. L. Environ. Sci. Technol. 2007, 41, 3734. doi: 10.1021/es062993s
-
[27]
Lee, C.; Sedlak, D. L. Environ. Sci. Technol. 2008, 42, 8528. doi: 10.1021/es801947h
-
[28]
Ai, Z.; Jia, F.; Zhang, L. Environ. Chem. 2016, 35, 1977.
-
[29]
King, D. W.; Lounsbury, H. A.; Millero, F. J. Environ. Sci. Technol. 1995, 29, 818. doi: 10.1021/es00003a033
-
[30]
Liu, T.; Li, X.; Waite, T. D. Environ. Sci. Technol. 2014, 48, 14564. doi: 10.1021/es503777a
-
[31]
Liu, T.; Li, X.; Waite, T. D. Environ. Sci. Technol. 2013, 47, 7350. doi: 10.1021/es400362w
-
[32]
Liu, Y.; Phenrat, T.; Lowry, G. V. Environ. Sci. Technol. 2007, 41, 7881. doi: 10.1021/es0711967
-
[33]
Wittbrodt, P. R.; Palmer, C. D. Environ. Sci. Technol. 1995, 29, 255. doi: 10.1021/es00001a033
-
[34]
Wang, Z.; Zhang, L.; Zhao, J.; Xing, B. Environ. Sci.-Nano 2016, 3, 240. doi: 10.1039/C5EN00230C
-
[1]
-



图 3 零件纳米铁活化分子氧过程中一些不利于活性氧物种生成的副反应
Figure 3 The side reaction occurred during the molecular oxygen activation with nZVI

图 4 根据双氧水检测结果, 连续单电子分子氧活化路径对双氧水产量的贡献率高达60%, 高于双电子分子氧活化路径对双氧水生成的贡献率[17]
Figure 4 According to the H2O2 detection results, the contribution of the sequential single-electron transfer pathway on the H2O2 generation was more than 60%, higher than that of the two-electron molecular oxygen activation pathway[17]


图 6 左图:金属阳离子诱导老化零价纳米铁去钝化过程的原理图.右图:零价纳米铁降解三氯乙烯的准一级动力学常数与金属阳离子和nZVI表面氧化层的亲和力(log K)的线性相关度[30]
Figure 6 Left: Schematic illustrations of the cation-induced depassivation process of aged nZVI. Right: Correlation of the pseudo-first order rate constants (k) of TCE degradation with log K values of different cations on hydrous ferric oxide (HFO, 1 g•L-1, 600 m2•g-1)[30]
-


 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 132
- 文章访问数: 10303
- HTML全文浏览量: 2056
 下载:
下载:
