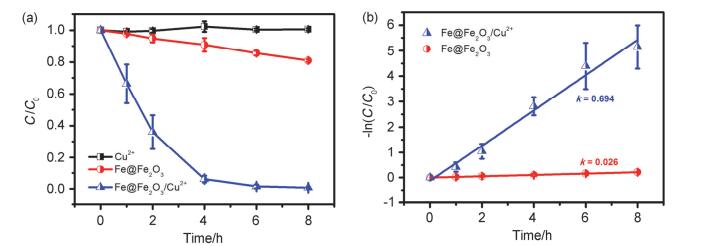
 图 1
(a)不同体系中阿特拉津降解的效率图; (b) -ln (C/C0)随时间的变化曲线
Figure 1.
(a) Time profiles of the aerobic atrazine degradation in Cu2+, Fe@Fe2O3 and Fe@Fe2O3/Cu2+ systems; (b) plots of ln(C/C0) versus time for the aerobic atrazine degradation in different systems
图 1
(a)不同体系中阿特拉津降解的效率图; (b) -ln (C/C0)随时间的变化曲线
Figure 1.
(a) Time profiles of the aerobic atrazine degradation in Cu2+, Fe@Fe2O3 and Fe@Fe2O3/Cu2+ systems; (b) plots of ln(C/C0) versus time for the aerobic atrazine degradation in different systems
引用本文:
贾法龙, 刘娟, 张礼知. 铜离子促进Fe@Fe2O3纳米线活化分子氧降解阿特拉津的研究[J]. 化学学报,
2017, 75(6): 602-607.
doi:
10.6023/A17010004

Citation: Jia Falong, Liu Juan, Zhang Lizhi. Copper Ions Promoted Aerobic Atrazine Degradation by Fe@Fe2O3 Nanowires[J]. Acta Chimica Sinica, 2017, 75(6): 602-607. doi: 10.6023/A17010004
Citation: Jia Falong, Liu Juan, Zhang Lizhi. Copper Ions Promoted Aerobic Atrazine Degradation by Fe@Fe2O3 Nanowires[J]. Acta Chimica Sinica, 2017, 75(6): 602-607. doi: 10.6023/A17010004
铜离子促进Fe@Fe2O3纳米线活化分子氧降解阿特拉津的研究
摘要:
阿特拉津是一种持久性含氯有机污染物,难以生物降解,因此有必要开发高效技术清除环境中残留的阿特拉津.近来纳米铁材料的发展为降解阿特拉津提供了一种可供选择的新方法,但降解过程中纳米铁活性逐渐减弱的问题仍需改进.本论文研究了铜离子(Cu2+)存在条件下Fe@Fe2O3纳米线活化分子氧降解阿特拉津的过程,并探讨了Cu2+的作用机理.研究结果表明,少量Cu2+的存在就可以显著促使Fe@Fe2O3生成溶解态Fe(Ⅱ),从而有助于分子氧活化并产生更多·OH等活性氧物种.在降解过程中,阿特拉津首先被氧化,进而发生脱氯上羟基反应、侧链氧化以及脱侧链反应.
-
关键词:
- 阿特拉津
- / 降解
- / Fe@Fe2O3纳米线
- / 分子氧活化
- / 铜离子
English
Copper Ions Promoted Aerobic Atrazine Degradation by Fe@Fe2O3 Nanowires
Abstract:
As a persistent chlorinated organic pollutant, Atrazine (2-chloro-4-(ethylamino)-6-isopropylamino-s-triazine) in the environment brings harm to natural environment as well as the human health. Since Atrazine is difficult to be degraded biologically, various strategies have been developed to realize efficient and environmentally-friendly removal of Atrazine. Recently, nanoscaled iron has been extensively applied for the remediation/treatment of wastewater contaminated with various organic and inorganic pollutants and exhibits superior activity than that of bulk iron. But its removal efficiency would decrease along with reaction time. In this study, we report that copper ions could efficiently promote atrazine degradation with Fe@Fe2O3 nanowires via the molecular oxygen activation processes. As indicated by the electron spin resonance analysis (ESR) and X-ray photoelectron spectroscopic analysis (XPS) results, the addition of Cu2+ ions could promote the release of dissolved Fe(Ⅱ) from Fe@Fe2O3. During the degradation process, the concentration of Fe(Ⅱ) in the solution with Cu2+ ions is maintained at a much higher level than that without Cu2+ ions. At the same time, Cu2+ ions were reduced to low valence states (Cu0), which further promoted the release of Fe2+. The generated Fe2+ would then activate the molecular oxygen via the single-electron or double-electron transfer route. As a result, more reactive oxygen species such as ·OH were generated to degrade atrazine. Under room temperature and aerobic condition, the Atrazine removal rate constant in Fe@Fe2O3/Cu2+ system was 0.694 h-1, which was almost 23 times that in Fe@Fe2O3 system. Moreover, the Fe@Fe2O3/Cu2+ catalytic system also remains superior activity in the pH range of 2~5. The intermediates of atrazine degradation were detected and the atrazine degradation in the Fe@Fe2O3/Cu2+ catalytic system was accompanied with alkylic oxidation, dealkylation and dechlorination. This study provides a new way to enhance molecular oxygen activation by core-shell Fe@Fe2O3 nanowires, and also deepens our understanding of the molecular oxygen activation processes by Fe@Fe2O3 for the aerobic pollutant degradation.
-
Key words:
- atrazine
- / degradation
- / Fe@Fe2O3 nanowires
- / molecular oxygen activation
- / copper ions
-
1 引言
阿特拉津(Atrazine)化学名称为2-氯-4-乙氨基-6-异丙氨基-1, 3, 5-三嗪, 是一种重要的含氯有机农药, 且在全球范围内广泛使用.它可通过吸附及扩散等方式在自然环境中进行迁移, 最终危害生态环境及人类健康[1].阿特拉津被认为是一种内分泌干扰物质及C类致癌物, 被学术界界定为新持久性有机污染物(POPs), 已经成为了水体质量管理的重点监控对象.
近年来的研究已证明铁[2~4]、锌[5]等金属可以有效去除多种环境污染物, 而零价铁因其廉价及环境友好等特点而得到广泛应用.例如, 零价铁能还原降解水体中含氯有机污染物(四氯化碳、三氯乙烯及多氯联苯)[6], 零价铁还被用于降解硝基芳香族化合物而生成相应的苯胺.零价铁在环境污染治理中的另一个重要应用就是Fenton氧化体系.例如, Boussahel等[7]利用零价铁的Fenton体系降解杀虫剂DDT, 发现该反应体系对有机污染物有较好的降解效果.
伴随着污染物的降解, 铁逐渐腐蚀生成的氧化物在表面附着, 从而阻碍了后续还原反应的进行.因此, 如何保持零价铁活性就成为污染物处理急需解决的难题.针对于此, 研究人员提出了一些解决办法, 其中包括增加溶液酸性、机械剥离腐蚀层、微波辅助以及施加弱磁场等[4].譬如, Kim等[8]发现在酸性体系中阿特拉津的脱氯效果较好, 他们认为降解环境呈酸性是土壤中阿特拉津脱氯必不可少的条件.同济大学关小红研究组[9]证实弱磁场可使零价铁表面产生非均匀感应磁场, 可缓解零价铁的钝化并活化已钝化的氧化层, 从而使零价铁与污染物的反应速率大幅度提高.
为了进一步提高污染物的处理效率, 研究者们开始尝试使用更小粒径的纳米零价铁代替铁粉, 并发现纳米铁具有较高的还原能力及反应速率, 为水体中有机污染物的有效治理带来了新的机遇[10~12].例如, 张伟贤等[13]开展了纳米铁处理实际水样的中试实验, 发现氯化有机物均可被快速完全降解; 李益民等[14]在有机膨润土上负载纳米零价铁用于阿特拉津的去除; 我们课题组[15, 16]近年来着力于研究零价铁活化分子氧技术, 合成了一种具有核壳结构Fe@Fe2O3纳米线, 发现该纳米线可以活化分子氧有效降解4-氯酚.然而, 水体中常见的金属离子(如Cu2+)对Fe@Fe2O3纳米线活化分子氧降解有机污染物性能的影响尚不得而知, 值得系统研究.
基于本课题组前期关于Fe@Fe2O3纳米线活化分子氧机理的工作基础, 本论文探讨了铜离子存在下Fe@Fe2O3纳米线活化分子氧降解阿特拉津的过程.我们发现低浓度Cu2+离子的引入可以促使Fe@Fe2O3生成更多溶解态Fe(Ⅱ), 从而有效活化分子氧并产生•OH等活性氧物种降解阿特拉津.
2 结果与讨论
2.1 阿特拉津的降解
图 1a结果显示在8 h之内Fe@Fe2O3体系中有近20%的阿特拉津被降解, 结合氩气条件下阿特拉津的去除效果(图S1), 可以认为氧气对于铁纳米线降解阿特拉津起着至关重要的作用.当仅有单独Cu2+离子存在时阿特拉津几乎没有降解, 但是Fe@Fe2O3体系中加入Cu2+离子后阿特拉津就可以完全降解. Fe@Fe2O3以及Fe@Fe2O3/Cu2+体系中阿特拉津的降解速率经拟合后均符合一级动力学(图 1b), 其中Fe@Fe2O3/Cu2+体系降解阿特拉津的速率常数约为Fe@Fe2O3体系的22.7倍, 这说明Cu2+极大地提高了Fe@Fe2O3降解阿特拉津的速率.
图 1
(a)不同体系中阿特拉津降解的效率图; (b) -ln (C/C0)随时间的变化曲线
Figure 1.
(a) Time profiles of the aerobic atrazine degradation in Cu2+, Fe@Fe2O3 and Fe@Fe2O3/Cu2+ systems; (b) plots of ln(C/C0) versus time for the aerobic atrazine degradation in different systems
考虑到零价铁活化分子氧降解污染物的能力与环境pH密切相关, 我们检测了不同体系中pH的变化, 结果如图 2a所示. Cu2+、Fe@Fe2O3和Fe@Fe2O3/Cu2+这三个体系pH值分别维持在4.4、5.3和4.9左右.为了确定降解速率的不同是否与体系pH有关, 我们将这三个体系的初始pH分别调节至2.4、3.8、4.8、5.2和5.4左右(pH>5.5时Cu2+会水解形成沉淀), 结果表明Fe@Fe2O3/Cu2+体系中的阿特拉津的降解速率均明显高于Cu2+和Fe@Fe2O3这两个体系(图S2和2b), 这说明Fe@Fe2O3/Cu2+体系中阿特拉津的高效降解并不是由于初始pH值的改变.
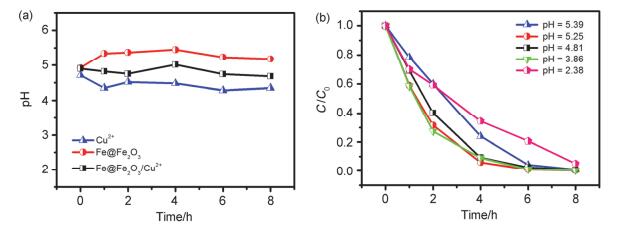 图 2
(a)不同体系pH值的变化; (b) Fe@Fe2O3/Cu2+体系调节不同pH值后降解阿特拉津的效率图
Figure 2.
(a) Time profiles of the aerobic atrazine degradation in Cu2+, Fe@Fe2O3 and Fe@Fe2O3/Cu2+systems; (b) Time profiles of atrazine degradation in Fe@Fe2O3/Cu2+ systems at different initial pH values
图 2
(a)不同体系pH值的变化; (b) Fe@Fe2O3/Cu2+体系调节不同pH值后降解阿特拉津的效率图
Figure 2.
(a) Time profiles of the aerobic atrazine degradation in Cu2+, Fe@Fe2O3 and Fe@Fe2O3/Cu2+systems; (b) Time profiles of atrazine degradation in Fe@Fe2O3/Cu2+ systems at different initial pH values
2.2 活性氧物种的捕获
为了探究Fe@Fe2O3/Cu2+体系中产生的活性氧物种种类, 我们进行了活性氧物种的捕获实验, 其中异丙醇(Isopropanol)用来捕获羟基自由基(•OH), 超氧化物歧化酶(SOD)用来捕获超氧自由基(•O2-), 过氧化氢酶(CAT)用以捕获H2O2.从图 3中可以看出, 加入异丙醇后阿特拉津的降解被明显抑制, 在8 h内仅有20%被降解, 这说明Fe@Fe2O3/Cu2+体系降解阿特拉津的主要活性物种为羟基自由基; 加入过氧化氢酶后阿特拉津的降解效率有部分减弱, 表明H2O2是导致阿特拉津的次要活性氧物种; 而加入SOD之后阿特拉津的降解效率反而有一定提高, 这说明•O2-可能并不直接作用于阿特拉津.根据课题组[16]之前的研究, SOD可与•O2-发生歧化反应生成H2O2, 而产生的H2O2进一步发生Fenton过程而产生•OH[17], 从而促进阿特拉津的降解.这一现象也从侧面进一步证实•OH是Fe@Fe2O3/Cu2+体系降解阿特拉津的主要活性物种.
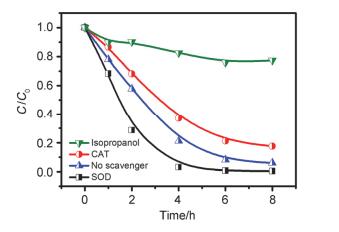 图 3
Fe@Fe2O3/Cu2+/Air体系加入不同捕获剂后阿特拉津的降解图
Figure 3.
Degradation of atrazine in the Fe@Fe2O3/Cu2+/Air system after adding different scavengers
图 3
Fe@Fe2O3/Cu2+/Air体系加入不同捕获剂后阿特拉津的降解图
Figure 3.
Degradation of atrazine in the Fe@Fe2O3/Cu2+/Air system after adding different scavengers
随后, 我们检测了不同体系中产生的•OH及H2O2含量, 结果表明单独Cu2+体系基本不能产生羟基自由基(图 4a), 而Fe@Fe2O3/Cu2+体系以及Fe@Fe2O3体系中都产生了羟基自由基, 羟基自由基含量均随着反应进行而增加.并且, Fe@Fe2O3/Cu2+体系的羟基自由基产生量远远大于Fe@Fe2O3体系, 图 4b中ESR图谱数据进一步证实了该实验结果.
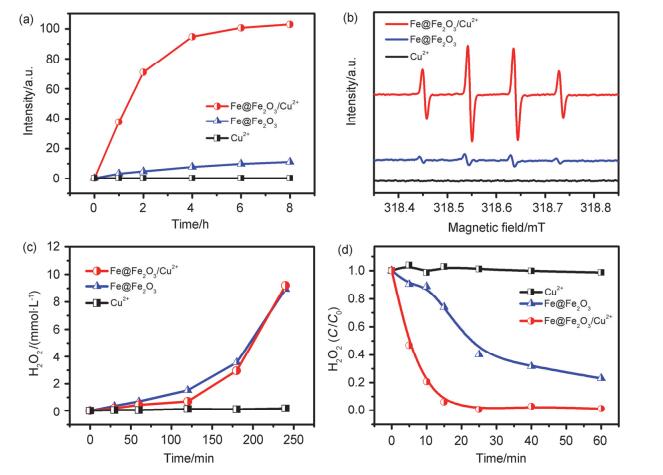 图 4
不同体系中活性氧物种的测定: (a)产生•OH量随时间变化曲线; (b) •OH的ESR图谱; (c)产生H2O2量随时间变化曲线; (d) 5 mmol•L-1 H2O2的分解速度
Figure 4.
Detection of reactive oxygen species under different systems: (a) amount of generated •OH radical versus time; (b) ESR spectra for •OH species; (c) generated H2O2 versus time; (d) Time profiles of the H2O2 decomposition
图 4
不同体系中活性氧物种的测定: (a)产生•OH量随时间变化曲线; (b) •OH的ESR图谱; (c)产生H2O2量随时间变化曲线; (d) 5 mmol•L-1 H2O2的分解速度
Figure 4.
Detection of reactive oxygen species under different systems: (a) amount of generated •OH radical versus time; (b) ESR spectra for •OH species; (c) generated H2O2 versus time; (d) Time profiles of the H2O2 decomposition
图 4c证实单独的Cu2+并不能活化分子氧至双氧水, 而Fe@Fe2O3/Cu2+体系以及Fe@Fe2O3体系中的双氧水含量差别很小.考虑到产生的双氧水可能被消耗而转化为•OH, 我们分别对三个体系进行了双氧水的分解实验.在起始双氧水浓度相同的条件下, 三个体系分解双氧水的能力差异较大(图 4d), 单独Cu2+基本不能分解双氧水, 而Fe@Fe2O3/Cu2+分解双氧水的速率明显快于Fe@Fe2O3.因此在Fe@Fe2O3/Cu2+体系中产生的H2O2有相当一部分会被快速分解, 导致积累的H2O2浓度与Fe@Fe2O3体系相差不大(图 4c).
2.3 阿特拉津降解中间产物的分析
虽然我们已经证实Cu2+可以有效促进Fe@Fe2O3降解阿特拉津, 但是尚不能确定Cu2+是否会影响阿特拉津的降解途径.已有研究表明•OH进攻阿特拉津的反应一般包括侧链烷基脱除和羟基取代氯等过程[18], 因此我们首先检测了三种脱侧链烷基中间产物(2-氯-4, 6-二乙氨基-1, 3, 5-三嗪、脱乙基阿特拉津以及脱异丙基阿特拉津).在两个体系中均能检测到上述中间产物(图 5a, 5b和5c), 在Fe@Fe2O3体系中产物的浓度呈现逐渐增加的趋势.而在Fe@Fe2O3/Cu2+体系中这三种中间产物的浓度却先增加后降低, 这是由于脱侧链烷基中间产物会进一步转化(实验中检测到了对应的上羟基脱氯产物), 从而会出现上述的浓度变化趋势.
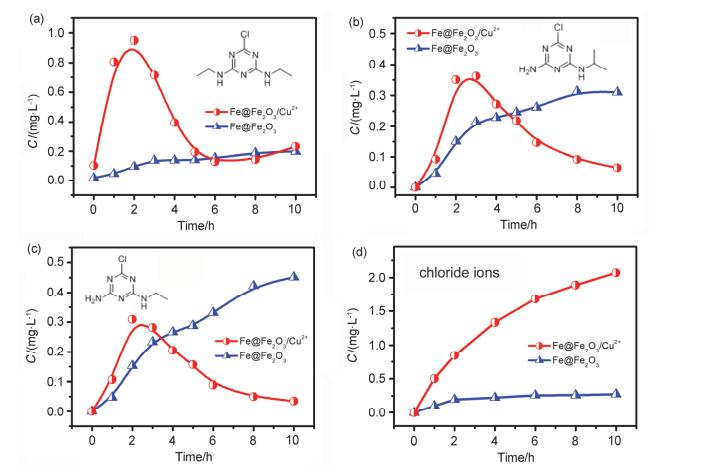 图 5
不同体系(Fe@Fe2O3蓝色, Fe@Fe2O3/Cu2+红色)中阿特拉津降解产物浓度随时间的变化图: (a) 2-氯-4, 6-二乙氨基-1, 3, 5-三嗪; (b)脱乙基阿特拉津; (c)脱异丙基阿特拉津; (d)氯离子
Figure 5.
Time profiles of concentrations of species during the atrazine degradation in Fe@Fe2O3 (blue) and Fe@Fe2O3/Cu2+ (red) systems: (a) 4, 6-bis(ethylamino)-2-chloro-1, 3, 5-triazine; (b) deethylatrazine; (c) deisopropylatrazine; (d) chloride ions
图 5
不同体系(Fe@Fe2O3蓝色, Fe@Fe2O3/Cu2+红色)中阿特拉津降解产物浓度随时间的变化图: (a) 2-氯-4, 6-二乙氨基-1, 3, 5-三嗪; (b)脱乙基阿特拉津; (c)脱异丙基阿特拉津; (d)氯离子
Figure 5.
Time profiles of concentrations of species during the atrazine degradation in Fe@Fe2O3 (blue) and Fe@Fe2O3/Cu2+ (red) systems: (a) 4, 6-bis(ethylamino)-2-chloro-1, 3, 5-triazine; (b) deethylatrazine; (c) deisopropylatrazine; (d) chloride ions
由于脱侧链烷基中间产物继续上羟基脱氯的过程会产生游离氯离子, 我们也检测了溶液中的氯离子含量, 结果如图 5d所示. Fe@Fe2O3与Fe@Fe2O3/Cu2+体系中10 h时的氯离子积累量分别为0.25和2.0 mg•L-1, 而实验体系中20 mg•L-1阿特拉津理论上含3.3 mg•L-1氯离子, 因此这两个体系对应的脱氯率分别为7.6%和60.6%, Fe@Fe2O3/Cu2+体系中氯离子产生量明显高于Fe@Fe2O3体系.
在阿特拉津降解过程中, 氨氮的产生量是评价阿特拉津能否开环降解的重要依据.本实验中使用阿特拉津浓度为20 mg•L-1, 如果其中的氮全部转变为氨氮, 那么对应的氨氮浓度将达到6.5 mg•L-1.经过检测, 我们发现10 h降解后Fe@Fe2O3体系与Fe@Fe2O3/Cu2+体系中的氨氮含量分别为0.27 mg•L-1和0.37 mg•L-1, 对应的氨氮转化率仅为4.1%和5.7%.如果仅按阿特拉津的支链脱氮计算, 理论上氨氮含量会达到1.3 mg•L-1, 而在Fe@Fe2O3体系与Fe@Fe2O3/Cu2+体系中的氨氮含量也明显低于此数值.这说明Fe@Fe2O3与Fe@Fe2O3/ Cu2+体系均难以使阿特拉津开环, 阿特拉津的降解途径并没有明显差异, 只是发生脱氯氧化及侧链烷基氧化.但是从最终阿特拉津的降解和脱氯效果来看, Fe@Fe2O3/Cu2+体系具有明显优势.
2.4 Cu2+离子的促进机制
我们接着探讨了Cu2+对Fe@Fe2O3释放溶解态亚铁离子的影响.从图 6a可以看出Fe@Fe2O3/Cu2+体系中的二价铁含量远远高于Fe@Fe2O3体系, 这说明Cu2+的加入促进了铁纳米线中二价铁的释放.另外, 溶液中游离Fe3+的浓度相对而言很低(图S3), 这是由于产生的Fe3+主要以氧化物或氢氧化物的沉积物形式存在.我们同时也监测了Fe@Fe2O3/Cu2+体系中溶液Cu2+的浓度变化(图S4), 反应1 h后铜离子浓度从初始的145 mg•L-1迅速降低至5 mg•L-1, 随后基本维持在4 mg•L-1左右, 这意味着铜离子在Fe@Fe2O3表面还原聚集而导致溶液中的铜离子浓度较低.
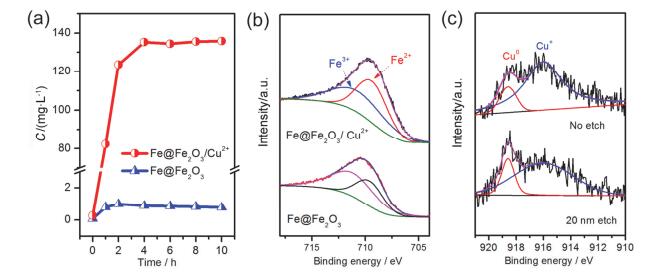 图 6
(a)溶液中亚铁浓度随时间的变化曲线; (b) Fe@Fe2O3和Fe@Fe2O3/Cu2+反应10 h后Fe 2p的XPS表征; (c) Fe@Fe2O3/Cu2+反应10 h后Cu的俄歇电子能谱表征
Figure 6.
(a) Time profiles of the Fe2+ concentration; (b) XPS spectra of Fe 2p in Fe@Fe2O3 and Fe@Fe2O3/Cu2+ after 10 h degradation; (c) AES spectra of Cu in Fe@Fe2O3/Cu2+ after 10 h degradation
图 6
(a)溶液中亚铁浓度随时间的变化曲线; (b) Fe@Fe2O3和Fe@Fe2O3/Cu2+反应10 h后Fe 2p的XPS表征; (c) Fe@Fe2O3/Cu2+反应10 h后Cu的俄歇电子能谱表征
Figure 6.
(a) Time profiles of the Fe2+ concentration; (b) XPS spectra of Fe 2p in Fe@Fe2O3 and Fe@Fe2O3/Cu2+ after 10 h degradation; (c) AES spectra of Cu in Fe@Fe2O3/Cu2+ after 10 h degradation
为了研究铁纳米线反应前后的表面结构变化以及存在物种的形态, 我们测试了样品的X射线光电子能谱(XPS).位于结合能709.37和710.64 eV处的峰分别对应Fe2+和Fe3+的特征峰(图 6b)[15, 19], 计算结果表明反应后Fe@Fe2O3纳米线中Fe2+和Fe3+物种的峰面积比为0.63, 而在有Cu2+存在时峰面积比增加至1.08.这说明Cu2+存在时Fe@Fe2O3纳米线更容易释放出Fe2+而导致表面Fe2+的比例较高, 这与图 6a的结果也是一致的.根据相关文献及化学手册, 铁、铜相关物种以及分子氧活化的氧化还原电势大小排序为: E(Fe3+/Fe2+)>E(O2/H2O2)>E(Cu2+/Cu)>E(Fe2+/Fe)[20]. Cu2+会被铁纳米线还原为Cu0而在表面附着, 俄歇电子能谱中918.6 eV处的特征峰证实了这一点(图 6c)[21~23], 而且样品经过20 nm刻蚀后也能观察到明显的Cu0信号.至于能谱中出现的Cu+特征峰, 很可能是由于样品接触空气后部分氧化而导致.
结合上述实验结果, 我们提出了Cu2+促进Fe@Fe2O3活化分子氧降解阿特拉津的机理(图 7).首先, 铜离子在铁纳米线表面被还原成Cu0颗粒(Cu2++2Fe0 → 2Fe2++Cu), Cu0与Fe0之间的原电池效应促使Fe0的电子向Cu颗粒转移并发生氧还原[24], 同时Fe2+的释放速度也得以加快, 并与水溶液中的溶解氧进一步发生分子氧活化反应, 最终经历Fenton过程产生•OH而降解阿特拉津.
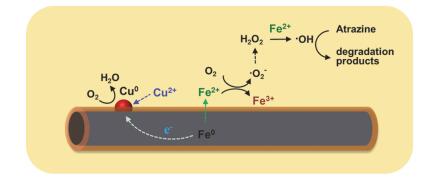 图 7
Cu2+促进Fe@Fe2O3活化分子氧降解阿特拉津示意图
Figure 7.
Schematic illustration for the enhanced molecular oxygen activation with Fe@Fe2O3 nanowires in the presence of Cu2+ ions
图 7
Cu2+促进Fe@Fe2O3活化分子氧降解阿特拉津示意图
Figure 7.
Schematic illustration for the enhanced molecular oxygen activation with Fe@Fe2O3 nanowires in the presence of Cu2+ ions
3 结论
本文研究了Cu2+离子存在时Fe@Fe2O3纳米线活化分子氧降解阿特拉津的过程, 发现Cu2+离子可以有效促进Fe@Fe2O3纳米线降解阿特拉津的效率.结果表明Cu2+在Fe@Fe2O3纳米线表面被还原成Cu0并促进了铁的溶出, 释放的亚铁活化分子氧进而产生•OH等活性物种降解阿特拉津.在降解过程中, 阿特拉津发生脱氯氧化及侧链烷基氧化, 降解后的产物毒性大大降低.本论文不仅开发了一种增强型Fe@Fe2O3纳米线活化分子氧的新途径, 也提供了一种同时去除环境中可能存在的重金属铜污染及农药阿特拉津污染的新方法, 为环境中阿特拉津的去除提供了新的思路.
4 实验部分
本研究所使用的测试仪器及试剂见支持信息S2; Fe@Fe2O3纳米线的合成和各种溶液的配置见支持信息S2;详细的测试过程见支持信息S3~S4;部分测试结果见支持信息S4~S5.
阿特拉津降解过程均在室温(25±2 ℃)和常压下进行, 以Fe@Fe2O3/Cu2+体系降解阿特拉津为例, 具体操作过程如下:取20 mL的20 mg•L-1 Atrazine降解溶液于50 mL锥形瓶中, 加入0.056 g Fe@Fe2O3纳米线以及200 μL Cu2+贮备液, 随后持续鼓入空气并计时.如果没有特别说明, 降解过程均在鼓入空气条件下进行.在隔绝氧气的对照实验中, 通入氩气而其它条件保持不变.降解过程中按间隔时间取1 mL液体样, 经由滤膜过滤后使用高效液相色谱定量分析Atrazine, 使用离子色谱分析仪测定降解过程中氯离子的生成情况, 以及用原子吸收仪测定溶液中金属离子的浓度变化. Fe@Fe2O3体系降解阿特拉津的实验过程与上述基本相同, 只是溶液中没有加入Cu2+贮备液.
-
-
[1]
Hayes, T. B.; Collins, A.; Lee, M.; Mendoza, M.; Noriega, N.; Stuart, A. A.; Vonk, A. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 5476. doi: 10.1073/pnas.082121499
-
[2]
Dombek, T.; Dolan, E.; Schultz, J.; Klarup, D. Environ. Pollut. 2001, 111, 21. doi: 10.1016/S0269-7491(00)00033-6
-
[3]
Rangsivek, R.; Jekel, M. Water Res. 2005, 39, 4153. doi: 10.1016/j.watres.2005.07.040
-
[4]
Sun, Y.; Li, J.; Huang, T.; Guan, X. Water Res. 2016, 100, 277. doi: 10.1016/j.watres.2016.05.031
-
[5]
刘娜, 赵勇胜, 张兰英, 刘红, 刘鹏, 中国环境科学, 2006, 26, 116. doi: 10.3321/j.issn:1000-6923.2006.01.025Liu, N.; Zhao, Y. S.; Zhang, L. Y.; Liu, H.; Liu, P. China Environ. Sci. 2006, 26, 116. doi: 10.3321/j.issn:1000-6923.2006.01.025
-
[6]
Chen, J. L.; Alabed, S. R.; Ryan, J. A.; Li, Z. J. Hazard. Mater. 2001, 83, 243. doi: 10.1016/S0304-3894(01)00193-5
-
[7]
Boussahel, R.; Harik, D.; Mammar, M.; Lamara-Mohamed, S. Desalination 2007, 206, 369. doi: 10.1016/j.desal.2006.04.059
-
[8]
Kim, G.; Jeong, W.; Choe, S. J. Hazard. Mater. 2008, 155, 502. doi: 10.1016/j.jhazmat.2007.11.092
-
[9]
Liang, L.; Guan, X.; Shi, Z.; Li, J.; Wu, Y.; Tratnyek, P. G. Environ. Sci. Technol. 2014, 48, 6326. doi: 10.1021/es500958b
-
[10]
程荣, 王建龙, 张伟贤, 化学进展, 2006, 18, 93. doi: 10.3321/j.issn:1005-281X.2006.01.014Cheng, R.; Wang, J. L.; Zhang, W. X. Prog. Chem. 2006, 18, 93. doi: 10.3321/j.issn:1005-281X.2006.01.014
-
[11]
Nagpal, V.; Bokare, A. D.; Chikate, R. C.; Rode, C. V.; Paknikar, K. M. J. Hazard. Mater. 2010, 175, 680. doi: 10.1016/j.jhazmat.2009.10.063
-
[12]
徐新华, 卫建军, 汪大翚, 中国环境科学, 2004, 24, 76. doi: 10.3321/j.issn:1000-6923.2004.01.018Xu, X. H.; Wei, J. J.; Wang, D. H. China Environ. Sci. 2004, 24, 76. doi: 10.3321/j.issn:1000-6923.2004.01.018
-
[13]
Zhang, W. X. J. Nanopart. Res. 2003, 5, 323. doi: 10.1023/A:1025520116015
-
[14]
Li, Y.; Zhang, Y.; Li, J.; Zheng, X. Environ. Pollut. 2011, 159, 3744. doi: 10.1016/j.envpol.2011.07.016
-
[15]
Ai, Z.; Gao, Z.; Zhang, L.; He, W.; Yin, J. J. Environ. Sci. Technol. 2013, 47, 5344. doi: 10.1021/es4005202
-
[16]
Huang, Q.; Cao, M.; Ai, Z.; Zhang, L. Appl. Catal. B-Environ. 2015, 162, 319. doi: 10.1016/j.apcatb.2014.06.046
-
[17]
Kettle, A. J.; Anderson, R. F.; Hampton, M. B.; Winterbourn, C. C. Biochemistry 2007, 46, 4888. doi: 10.1021/bi602587k
-
[18]
Wang, L.; Cao, M.; Ai, Z.; Zhang, L. Environ. Sci. Technol. 2014, 48, 3354. doi: 10.1021/es404741x
-
[19]
Mills, P.; Sullivan, J. J. Phys. D:Appl. Phys. 1983, 16, 723. doi: 10.1088/0022-3727/16/5/005
-
[20]
Dean, J. A., Lange's Handbook of Chemistry, 13th ed., McGraw-Hill Book, New York, 1991.
-
[21]
Dong, G.; Ai, Z.; Zhang, L. Water Res. 2014, 66C, 22. http://www.sciencedirect.com/science/article/pii/S0043135414005788
-
[22]
Yang, L.; He, J.; Zhang, Q.; Wang, Y. J. Catal. 2010, 276, 76. doi: 10.1016/j.jcat.2010.09.002
-
[23]
Mudunkotuwa, I. A.; Pettibone, J. M.; Grassian, V. H. Environ. Sci. Technol. 2012, 46, 7001. doi: 10.1021/es203851d
-
[24]
杜诚, 高小惠, 陈卫, 催化学报, 2016, 37, 1049. http://www.chxb.cn/CN/abstract/abstract21833.shtmlDu, C.; Gao, X.; Chen, W. Chin. J. Catal. 2016, 37, 1049. http://www.chxb.cn/CN/abstract/abstract21833.shtml
-
[1]
-

图 1 (a)不同体系中阿特拉津降解的效率图; (b) -ln (C/C0)随时间的变化曲线
Figure 1 (a) Time profiles of the aerobic atrazine degradation in Cu2+, Fe@Fe2O3 and Fe@Fe2O3/Cu2+ systems; (b) plots of ln(C/C0) versus time for the aerobic atrazine degradation in different systems

图 2 (a)不同体系pH值的变化; (b) Fe@Fe2O3/Cu2+体系调节不同pH值后降解阿特拉津的效率图
Figure 2 (a) Time profiles of the aerobic atrazine degradation in Cu2+, Fe@Fe2O3 and Fe@Fe2O3/Cu2+systems; (b) Time profiles of atrazine degradation in Fe@Fe2O3/Cu2+ systems at different initial pH values

图 3 Fe@Fe2O3/Cu2+/Air体系加入不同捕获剂后阿特拉津的降解图
Figure 3 Degradation of atrazine in the Fe@Fe2O3/Cu2+/Air system after adding different scavengers

图 4 不同体系中活性氧物种的测定: (a)产生•OH量随时间变化曲线; (b) •OH的ESR图谱; (c)产生H2O2量随时间变化曲线; (d) 5 mmol•L-1 H2O2的分解速度
Figure 4 Detection of reactive oxygen species under different systems: (a) amount of generated •OH radical versus time; (b) ESR spectra for •OH species; (c) generated H2O2 versus time; (d) Time profiles of the H2O2 decomposition

图 5 不同体系(Fe@Fe2O3蓝色, Fe@Fe2O3/Cu2+红色)中阿特拉津降解产物浓度随时间的变化图: (a) 2-氯-4, 6-二乙氨基-1, 3, 5-三嗪; (b)脱乙基阿特拉津; (c)脱异丙基阿特拉津; (d)氯离子
Figure 5 Time profiles of concentrations of species during the atrazine degradation in Fe@Fe2O3 (blue) and Fe@Fe2O3/Cu2+ (red) systems: (a) 4, 6-bis(ethylamino)-2-chloro-1, 3, 5-triazine; (b) deethylatrazine; (c) deisopropylatrazine; (d) chloride ions

图 6 (a)溶液中亚铁浓度随时间的变化曲线; (b) Fe@Fe2O3和Fe@Fe2O3/Cu2+反应10 h后Fe 2p的XPS表征; (c) Fe@Fe2O3/Cu2+反应10 h后Cu的俄歇电子能谱表征
Figure 6 (a) Time profiles of the Fe2+ concentration; (b) XPS spectra of Fe 2p in Fe@Fe2O3 and Fe@Fe2O3/Cu2+ after 10 h degradation; (c) AES spectra of Cu in Fe@Fe2O3/Cu2+ after 10 h degradation
-


 下载:
下载:






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 1601
- HTML全文浏览量: 239
 下载:
下载:
