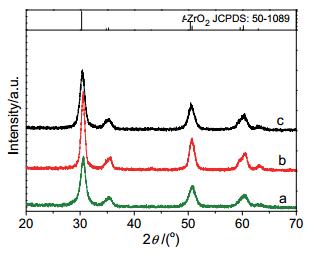
 图 1
(a) ZrO2(3.2)、(b) ZrO2(10.2) 及 (c) ZrO2(11.7) 的XRD谱
Figure 1.
XRD patterns of the (a) ZrO2(3.2), (b) ZrO2(10.2) and (c) ZrO2(11.7)
图 1
(a) ZrO2(3.2)、(b) ZrO2(10.2) 及 (c) ZrO2(11.7) 的XRD谱
Figure 1.
XRD patterns of the (a) ZrO2(3.2), (b) ZrO2(10.2) and (c) ZrO2(11.7)
引用本文:
周功兵, 王浩, 裴燕, 乔明华, 孙斌, 宗保宁. Ru-Zn/ZrO2催化剂在苯部分加氢反应中的孔径效应[J]. 化学学报,
2017, 75(3): 321-328.
doi:
10.6023/A16100569

Citation: Zhou Gongbing, Wang Hao, Pei Yan, Qiao Minghua, Sun Bin, Zong Baoning. Pore Size Effect of Ru-Zn/ZrO2 Catalyst on Partial Hydrogenation of Benzene to Cyclohexene[J]. Acta Chimica Sinica, 2017, 75(3): 321-328. doi: 10.6023/A16100569
Citation: Zhou Gongbing, Wang Hao, Pei Yan, Qiao Minghua, Sun Bin, Zong Baoning. Pore Size Effect of Ru-Zn/ZrO2 Catalyst on Partial Hydrogenation of Benzene to Cyclohexene[J]. Acta Chimica Sinica, 2017, 75(3): 321-328. doi: 10.6023/A16100569
Ru-Zn/ZrO2催化剂在苯部分加氢反应中的孔径效应
摘要:
采用沉淀法和溶剂热法合成了三种具有相同晶型但不同孔径的四方ZrO2(t-ZrO2),以此为载体,采用沉积沉淀-硫酸锌溶液中还原的方法制备了Ru-Zn/ZrO2催化剂,考察了Ru-Zn/ZrO2催化剂的孔径对苯部分加氢性能的影响.采用粉末X射线衍射(XRD)、N2物理吸附、电感耦合等离子体原子发射光谱(ICP-AES)、CO化学吸附、X射线光电子能谱(XPS)、X射线吸收近边结构(XANES)、X射线激发俄歇电子能谱(XAES)、H2程序升温还原(H2-TPR)和透射电子显微镜(TEM)等手段对载体和催化剂进行了系统的表征.研究表明,在苯部分加氢反应中,Ru-Zn/ZrO2催化剂的孔径对环己烯的选择性有显著影响.随催化剂孔径的增大,苯的转换频率(TOF)基本不变,环己烯初始选择性(S0)则逐渐升高,孔径为11.7 nm的ZrO2(ZrO2(11.7))负载的Ru-Zn/ZrO2(11.7)催化剂的S0及得率最高,分别可达88%和54%.结合催化剂的表征和加氢结果,讨论了孔径影响苯部分加氢活性和选择性的原因.
-
关键词:
- Ru-Zn/ZrO2
- / 孔径效应
- / 苯
- / 环己烯
- / 加氢
English
Pore Size Effect of Ru-Zn/ZrO2 Catalyst on Partial Hydrogenation of Benzene to Cyclohexene
Abstract:
Partial hydrogenation of benzene to cyclohexene is an important industrial process and features exceptional superiority to processes such as dehydration of cyclohexanol, dehydrogenation of cyclohexane, and the Birch reduction in terms of inexpensive feedstock, succinct reaction route and consequently, improved operational simplicity. In this work, the pore size effect on the partial hydrogenation of benzene to cyclohexene over the Ru-Zn/ZrO2 catalysts was studied for the first time. Three ZrO2 supports with the same tetragonal crystallographic form (t-ZrO2) but different pore sizes were synthesized by the precipitation and the solvothermal methods. Using these ZrO2 samples, the Ru-Zn/ZrO2 catalysts were prepared by the deposition-precipitation method followed by reduction in ZnSO4·7H2O aqueous solution. The supports and catalysts were characterized by powder X-ray diffraction (XRD), N2 physisorption, inductively coupled plasma-atomic emission spectroscopy (ICP-AES), CO chemisorption, X-ray photoelectron spectroscopy (XPS), X-ray absorption near-edge structure (XANES), temperature-programmed reduction of H2 (H2-TPR), and transmission electron microscopy (TEM). It is identified that the Ru nanoparticles (NPs) on these catalysts had similar size and chemical state. In the partial hydrogenation of benzene to cyclohexene, a pronounced pore size effect of the catalyst was identified. With the increase in the pore size, while the turnover frequency (TOF) of benzene was essentially unchanged, the initial selectivity (S0) to cyclohexene increased steadily. The Ru-Zn/ZrO2(11.7) catalyst with the ZrO2 support having the pore size of 11.7 nm exhibited the highest S0 (88%) and yield (54%) of cyclohexene. On the basis of the characterization results, the similarity in the TOFs of benzene on the Ru-Zn/ZrO2 catalysts with different pore sizes is associated with the identical sizes of the Ru NPs. On the other hand, we tentatively propose that the ZrO2 support with large pore size is beneficial for the out-diffusion of the cyclohexene nano-droplets formed in the pore channels, thus avoiding consecutive hydrogenation to cyclohexane and improving the S0.
-
Key words:
- Ru-Zn/ZrO2
- / pore size effect
- / benzene
- / cyclohexene
- / hydrogenation
-
1 引言
环己烯有活泼的C=C双键, 能够很容易地转化为高附加值的环己醇、己内酰胺和己二酸等, 因而是一种重要的有机化学中间体[1].制备环己烯的方法有环己醇脱水、卤代环己烷脱卤化氢和环己烷脱氢等[2].相比而言, 苯部分加氢生产环己烯具有原料来源广泛、原子经济性高、反应路线简单等显著优点[3].然而苯加氢生成环己烷的标准吉布斯自由能变化为-98 kJ•mol-1, 远比生成环己烯 (-23 kJ•mol-1) 为负, 故在热力学上更容易生成前者[4].因此, 为提高环己烯的选择性, 需要设计合适的催化剂, 使环己烯的生成在动力学上变得有利.
在多相催化加氢反应中, 载体的孔径及孔结构对催化剂的性能有很大影响[5~10].徐华龙等[5]发现对于采用浸渍法、吸附沉淀法及醇盐法制备的Cu/SiO2催化剂, 以吸附沉淀法制备的Cu/SiO2催化剂具有较大孔径和孔容, CuO分散更均匀, 易于还原, 因而具有更高的苯甲醛加氢活性和苯甲醇选择性. Kang等[6]报道对于Ru/meso-ZSM-5催化剂上的费托合成反应 (FTS), 具有多级孔结构的meso-ZSM-5能抑制连续裂解反应, 从而提高C5~C11产物的选择性. Liu等[7]发现Co/ZrO2催化剂的FTS性能与ZrO2的孔径密切相关.随ZrO2孔径增大, FTS活性、C5+产物选择性逐渐升高, 而C1产物的选择性降低, 归因于大孔径的ZrO2对Co还原的促进作用以及对α-烯烃再吸附和链增长的控制作用. Zuo等[8]发现对于Al/Ce柱撑粘土负载的Pd催化剂, 载体中的大孔是提高苯催化氧化反应活性的关键因素. Gelesky等[9]报道具有更大孔径的SiO2负载的Rh纳米粒子的烯烃加氢活性更高. Xia等[10]报道Pt/MCM-41催化剂的芳烃氧化活性比Pt/ZSM-5催化剂高, 归因于前者的载体有更大孔径.
在苯加氢反应中, 载体的孔径同样会影响催化剂的性能.王建强等[11]报道在苯部分加氢反应中, 采用共沉淀法制备的Ru/AlOOH催化剂上的环己烯选择性高于原位焙烧上述催化剂或浸渍法制得的Ru/g-Al2O3催化剂, 归结为薄水铝石表面羟基的亲水性和存在较大的堆积孔.较大的孔有利于环己烯脱附, 抑制环己烯深度加氢. Job等[12]报道Pt/炭干凝胶催化剂上的苯加氢活性比Pt/活性炭催化剂高4~10倍, 归因于干凝胶上存在的大孔使得Pt的分散度提高. Preising等[13]发现对于多孔玻璃负载的Ni催化剂, 苯的扩散系数与载体的平均孔径之间为线性关系, 载体孔径越大, 苯加氢活性越高.
我们[14]的前期研究表明, ZrO2的晶型 (无定形、四方、单斜) 会影响Ru-B/ZrO2催化剂的环己烯选择性, 其中四方ZrO2作为载体选择性最高.在本文中, 我们采用沉淀法和溶剂热法合成了三种具有相同晶型但不同孔径的四方ZrO2.以其为载体, 采用沉积沉淀-硫酸锌溶液中还原的方法制备了Ru-Zn/ZrO2催化剂, 系统考察了载体的孔径对催化剂物化性质和苯部分加氢催化性能的影响.
2 结果与讨论
2.1 载体及催化剂表征
图 1为ZrO2(3.2)、ZrO2(10.2) 及ZrO2(11.7) 的XRD谱.三个ZrO2样品均在30.3°、34.8°、35.3°、50.4°、50.7°、59.6°、60.2°及63.0°处出现四方ZrO2的 (011)、(002)、(110)、(112)、(020)、(013)、(121) 及 (202) 衍射峰 (JCPDS 50-1089), 说明样品具有良好且单一的晶型.由Scherrer公式及 (011) 最强衍射峰算得的ZrO2(3.2)、ZrO2(10.2) 及ZrO2(11.7) 的晶粒尺寸分别约为7.5、11.0及7.6 nm.
图 1
(a) ZrO2(3.2)、(b) ZrO2(10.2) 及 (c) ZrO2(11.7) 的XRD谱
Figure 1.
XRD patterns of the (a) ZrO2(3.2), (b) ZrO2(10.2) and (c) ZrO2(11.7)
从表 1可知, 比表面积 (SBET)、平均孔径 (dpore) 和孔容 (Vpore) 均按ZrO2(3.2)、ZrO2(10.2) 及ZrO2(11.7) 的顺序逐渐变大.三个ZrO2样品的N2吸脱附等温线如图S1(左) 所示, 均为IUPAC定义的IV型吸脱附等温线. ZrO2(3.2) 的吸脱附等温线中出现H2型滞后环, 而ZrO2(10.2) 及ZrO2(11.7) 的吸脱附等温线中则出现H3型滞后环, 说明由溶剂热法和沉淀法制备的ZrO2的孔形状不同.图S1(右) 为由脱附支得到的BJH孔径分布曲线.由图可见, 三个ZrO2样品的孔径分布相似, 但最可几孔径不同.其中, ZrO2(3.2) 的最可几孔径最小, 为3.7 nm, 且由吸附支得到的ZrO2(3.2) 的最可几孔径 (3.8 nm) 与由脱附支得到的结果接近. ZrO2(11.7) 的最可几孔径最大, 为12.7 nm.
 表 1
ZrO2的织构性质
Table 1.
Textural properties of ZrO2
表 1
ZrO2的织构性质
Table 1.
Textural properties of ZrO2
Sample SBET/(m2•g-1) dpore/nm Vpore/(cm3•g-1) ZrO2(3.2) 60 3.2 0.06 ZrO2(10.2) 88 10.2 0.28 ZrO2(11.7) 128 11.7 0.48 表 1 ZrO2的织构性质
Table 1. Textural properties of ZrO2图 2为Ru-Zn/ZrO2催化剂的XRD谱.负载Ru后, 载体晶型保持不变.由Scherrer公式及最强 (011) 衍射峰算得的Ru-Zn/ZrO2(3.2)、Ru-Zn/ZrO2(10.2) 及Ru-Zn/ZrO2(11.7) 催化剂中ZrO2的晶粒尺寸分别约为7.4、10.4及7.1 nm, 表明负载Ru对载体ZrO2的晶粒尺寸影响较小.此外, 催化剂在约44°处存在一个较弱的宽峰, 归属于六方密堆积 (hcp) Ru的最强 (101) 衍射峰 (JCPDS 06-0663).三个催化剂的XRD谱中均未观察到Zn的衍射峰, 表明Zn在催化剂中高度分散.
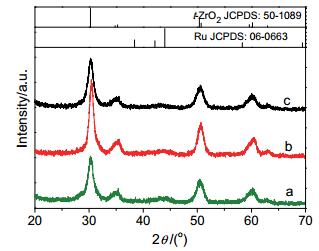 图 2
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的XRD谱
Figure 2.
XRD patterns of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
图 2
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的XRD谱
Figure 2.
XRD patterns of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
Ru-Zn/ZrO2催化剂的物化参数列于表 2.由ICP-AES测得的Ru-Zn/ZrO2(3.2)、Ru-Zn/ZrO2(10.2) 及Ru-Zn/ZrO2(11.7) 催化剂上的Ru负载量均接近理论值.三个催化剂中Zn的负载量也相近, 且大于理论值, 多出的Zn应来源于催化剂还原过程中Zn的进一步沉积. CO化学吸附结果表明, 催化剂上Ru的分散度顺序为Ru-Zn/ZrO2(3.2)<Ru-Zn/ZrO2(10.2)<Ru-Zn/ZrO2(11.7), 计算得到的相应的Ru的粒径顺序则相反. Zhao等[15]发现对于Ru/Al2O3-ZrO2/堇青石催化剂, 载体比表面积越大, 金属越容易分散, 故分散度越高. Wang等[16]也报道比表面积较大的g-Al2O3负载的Ru催化剂的分散度高于比表面积较小的ZrO2负载的Ru催化剂.三个催化剂的N2吸脱附等温线类型、滞后环类型及孔径分布 (图S2) 均与载体相似, 但Ru-Zn/ZrO2(10.2) 和Ru-Zn/ZrO2(11.7) 催化剂的最可几孔径有所减小.由表 2可见, 与载体相比, Ru-Zn/ZrO2(3.2) 催化剂的SBET降低, 但dpore及Vpore均增大, 这是因为ZrO2(3.2) 自身的dpore及Vpore不高, 使得Ru-Zn的贡献得以体现.对于Ru-Zn/ZrO2(10.2) 和Ru-Zn/ZrO2(11.7) 催化剂, 负载Ru-Zn后, SBET、dpore及Vpore均有所降低, 表明金属颗粒进入了ZrO2的部分孔中.
表 2
Ru-Zn/ZrO2催化剂的物化性质
Table 2.
Physicochemical properties of the Ru-Zn/ZrO2 catalysts
Catalyst Ru loadinga/wt% Zn loadinga/wt% Dispersionb/% dRuc/nm SBET/(m2•g−1) dpore/nm Vpore/(cm3•g-1) Ru-Zn/ZrO2(3.2) 10.15 3.88 15.1 8.9 48 5.6 0.07 Ru-Zn/ZrO2(10.2) 10.43 3.84 25.6 5.3 64 9.2 0.25 Ru-Zn/ZrO2(11.7) 10.55 4.12 26.2 5.1 97 9.8 0.30 aDetermined by ICP-AES; bDispersion of Ru determined by CO chemisorption; cParticle size of Ru determined by CO chemisorption. 表 2 Ru-Zn/ZrO2催化剂的物化性质
Table 2. Physicochemical properties of the Ru-Zn/ZrO2 catalysts由于Ru 3d峰与C 1s峰发生部分重叠, 因此我们采用Ru 3p峰确定催化剂中Ru的化学态. 图 3(左) 表明, 三个催化剂中Ru 3p3/2峰的结合能均为461.4 eV, 且3p3/2峰与3p1/2峰的结合能相差22.1 eV, 证明Ru为金属态[17].这些催化剂的Ru K边XANES谱 (图 4) 表明, 催化剂与Ru箔标样的Ru K边的能量接近, 进一步证明Ru为金属态.由图 3(右) 可知, 三个催化剂中Zn 2p3/2峰的结合能均为1021.8 eV.由于金属Zn的2p3/2峰的结合能为1021.8 eV[18], 而ZnO和Zn (OH)2的Zn 2p3/2峰的结合能为1021.8~1022.7 eV[18, 19], 因此Zn的价态难以直接确定, 故我们进一步采集了这三个催化剂的Zn LMM XAES谱.如图 5所示, 三个催化剂中Zn LMM峰的动能均为988.1 eV, 说明Zn均以ZnO或Zn (OH)2的形式存在[2, 20].
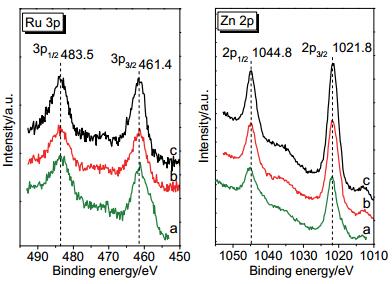 图 3
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的Ru 3p (左) 及Zn 2p (右) XPS谱
Figure 3.
Ru 3p (left) and Zn 2p (right) XPS spectra of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
图 3
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的Ru 3p (左) 及Zn 2p (右) XPS谱
Figure 3.
Ru 3p (left) and Zn 2p (right) XPS spectra of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
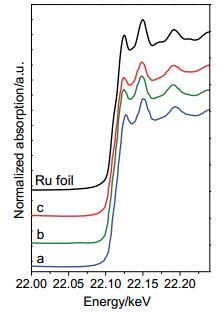 图 4
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2)、(c) Ru-Zn/ZrO2(11.7) 催化剂及Ru箔标样归一化后的Ru K边XANES谱
Figure 4.
Normalized Ru K-edge XANES spectra of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2), (c) Ru-Zn/ZrO2(11.7) catalysts and the Ru foil standard
图 4
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2)、(c) Ru-Zn/ZrO2(11.7) 催化剂及Ru箔标样归一化后的Ru K边XANES谱
Figure 4.
Normalized Ru K-edge XANES spectra of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2), (c) Ru-Zn/ZrO2(11.7) catalysts and the Ru foil standard
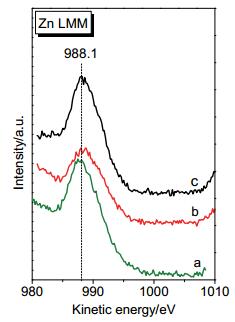 图 5
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的Zn LMM XAES谱
Figure 5.
Zn LMM XAES spectra of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
图 5
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的Zn LMM XAES谱
Figure 5.
Zn LMM XAES spectra of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
金属前驱体与载体之间的相互作用采用H2-TPR进行了研究.如图 6所示, Ru-Zn/ZrO2(3.2)、Ru-Zn/ZrO2(10.2) 及Ru-Zn/ZrO2(11.7) 催化剂均在348 K和361 K处出现两个还原峰.尽管还原峰温度受测试条件如催化剂质量、气体流速和组成以及升温速率等的影响, 但一般认为低温还原峰 (<523 K) 为高分散Ru的氧化物或氢氧化物的还原[21].三个催化剂中相同的还原温度表明金属前驱体与载体之间存在相似的相互作用.此外, 制备催化剂时加入的ZnSO4•7H2O会形成Zn (OH)2.为考察Zn (OH)2对Ru前驱体与载体之间相互作用的影响, 我们以与Ru-Zn/ZrO2(3.2) 催化剂相同的步骤制备了Ru/ZrO2(3.2) 催化剂, 但沉积沉淀时未加ZnSO4•7H2O. 图 6显示Ru/ZrO2(3.2) 催化剂的还原温度 (336 K) 低于Ru-Zn/ZrO2(3.2) 催化剂的还原温度, 表明催化剂中的Zn (OH)2增强了Ru前驱体与载体之间的相互作用.
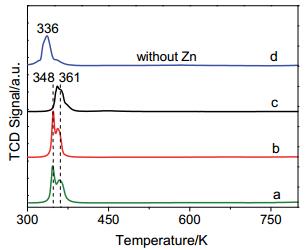 图 6
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2)、(c) Ru-Zn/ZrO2(11.7) 及 (d) Ru/ZrO2(3.2) 催化剂的H2-TPR谱
Figure 6.
H2-TPR profiles of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2), (c) Ru-Zn/ZrO2(11.7) and (d) Ru/ZrO2(3.2) catalysts
图 6
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2)、(c) Ru-Zn/ZrO2(11.7) 及 (d) Ru/ZrO2(3.2) 催化剂的H2-TPR谱
Figure 6.
H2-TPR profiles of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2), (c) Ru-Zn/ZrO2(11.7) and (d) Ru/ZrO2(3.2) catalysts
图 7为Ru-Zn/ZrO2催化剂的TEM照片及含高斯拟合曲线的粒径分布图. Ru-Zn/ZrO2(3.2)、Ru-Zn/ZrO2(10.2) 及Ru-Zn/ZrO2(11.7) 催化剂上Ru粒子 (如图中箭头所示) 的平均粒径相近, 约为4.2 nm, 表明载体孔径对金属粒子的尺寸基本无影响.但Ru-Zn/ZrO2(3.2) 催化剂上金属粒子存在聚集现象 (如图 7a圆圈所示), 这使得Ru的活性比表面积降低, 吸附的CO分子数减少.这可能是Ru-Zn/ZrO2(3.2) 催化剂上Ru的分散度低于Ru-Zn/ZrO2(10.2) 和Ru-Zn/ZrO2(11.7) 催化剂的原因.
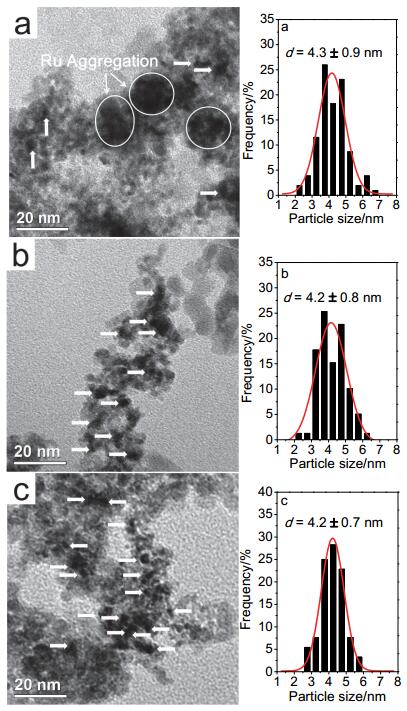 图 7
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的TEM照片及含高斯拟合曲线的粒径分布图
Figure 7.
TEM images and particle size distribution histograms with Gaussian analysis fittings of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
图 7
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的TEM照片及含高斯拟合曲线的粒径分布图
Figure 7.
TEM images and particle size distribution histograms with Gaussian analysis fittings of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
我们进一步采用高分辨TEM (HRTEM) 对Ru-Zn/ZrO2(3.2)、Ru-Zn/ZrO2(10.2) 及Ru-Zn/ZrO2(11.7) 催化剂的微观结构进行了表征.如图 8所示, 三个催化剂上Ru纳米粒子 (如图中圆圈所示) 的尺寸相近.另外, HRTEM照片上均可观察到明显的晶格条纹, 其间距约为2.94 ,归属于t-ZrO2的 (011) 晶面. Ru-Zn/ZrO2(3.2) 和Ru-Zn/ZrO2(10.2) 催化剂的HRTEM照片上还出现了间距为2.50 的晶格条纹, 归属于ZnO的 (101) 晶面, 进一步证实了Zn LMM XAES谱的结论.由于载体与金属纳米粒子的晶格条纹之间可能相互干扰, 因此我们对图 8中的正方形区域进行了傅立叶变换 (FFT). FFT图片进一步证实上述t-ZrO2和ZnO晶面的归属.同时, Ru-Zn/ZrO2(3.2) 和Ru-Zn/ZrO2(11.7) 催化剂的FFT图片上还出现了晶面间距约为1.58 的衍射点, 归属于hcpRu的 (102) 面.
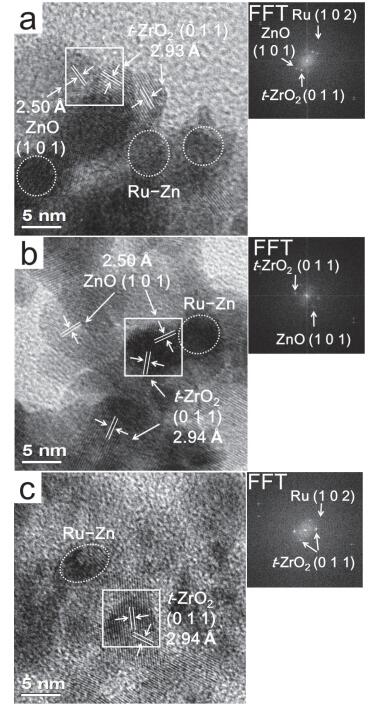 图 8
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的含FFT图片的HRTEM照片
Figure 8.
HRTEM images with FFT patterns of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
图 8
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的含FFT图片的HRTEM照片
Figure 8.
HRTEM images with FFT patterns of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
2.2 苯部分加氢制环己烯性能
Ru-Zn/ZrO2(3.2)、Ru-Zn/ZrO2(10.2) 及Ru-Zn/ZrO2(11.7) 催化剂的苯加氢反应历程如图 9所示.由图可见, 由于反应温度较低, 产物中仅含环己烯和环己烷, 没有出现异构化产物甲基环戊烷.随着反应的进行, 苯的量逐渐降低, 环己烷的量逐渐增加, 而环己烯的量先升高后降低, 在一定时间出现最大值, 体现了苯加氢的连续反应特征.对于Ru-Zn/ZrO2(3.2) 催化剂, 在反应过程中环己烯的量始终低于环己烷.而对于Ru-Zn/ZrO2(10.2) 和Ru-Zn/ZrO2(11.7) 催化剂, 环己烯的量始终高于环己烷.由图 9和表 3可知, 在这三个催化剂中, Ru-Zn/ZrO2(3.2) 催化剂的环己烯得率最低, 为21%, 相应苯的转化率为83%. Ru-Zn/ZrO2(11.7) 上的环己烯得率最高, 为54%, 高于Wang等[22]和Yuan等[23]报道的结果. Wang等[22]采用RuCl3•3H2O、ZrOCl2•8H2O和NH3•H2O共沉淀法制备了催化剂前驱体, 然后在ZnSO4•7H2O水溶液中还原得到Ru-Zn/ZrO2催化剂, 其环己烯得率为43.4%. Yuan等[23]采用相同方法制备了Ru-Zn/ZrO2催化剂, 但用KOH代替氨水, 其环己烯得率为44%.
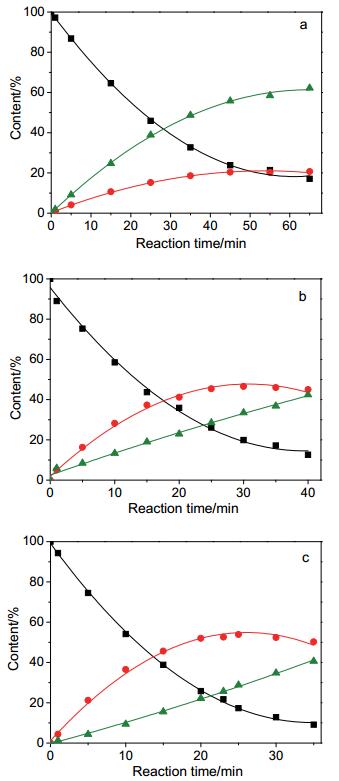 图 9
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的苯加氢反应历程
Figure 9.
The time courses of benzene hydrogenation over the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts. Reaction conditions: 1.0 g catalyst, 50 mL benzene, 100 mL H2O, 15.0 g ZnSO4•7H2O, temperature of 413 K, H2 pressure of 5.0 MPa, and stirring rate of 1200 r/min. (■) Benzene, (●) cyclohexene, and (▲) cyclohexane
表 3
Ru-Zn/ZrO2催化剂的苯部分加氢结果a
Table 3.
Results of the partial hydrogenation of benzene over the Ru-Zn/ZrO2 catalystsa
图 9
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的苯加氢反应历程
Figure 9.
The time courses of benzene hydrogenation over the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts. Reaction conditions: 1.0 g catalyst, 50 mL benzene, 100 mL H2O, 15.0 g ZnSO4•7H2O, temperature of 413 K, H2 pressure of 5.0 MPa, and stirring rate of 1200 r/min. (■) Benzene, (●) cyclohexene, and (▲) cyclohexane
表 3
Ru-Zn/ZrO2催化剂的苯部分加氢结果a
Table 3.
Results of the partial hydrogenation of benzene over the Ru-Zn/ZrO2 catalystsa
Entry Catalyst Conv.b/ % SCHEb/ % YCHEb/ % tb/ min S0c/ % r0d TOF/ s-1 1 Ru-Zn/ZrO2(3.2) 83 25 21 65 32 14.9 1.6 2 Ru-Zn/ZrO2(10.2) 87 51 44 40 74 21.3 1.3 3 Ru-Zn/ZrO2(11.7) 83 65 54 25 88 28.3 1.7 aReaction conditions: 1.0 g catalyst, 50 mL benzene, 100 mL H2O, 15.0 g ZnSO4•7H2O, temperature of 413 K, H2 pressure of 5.0 MPa, and stirring rate of 1200 r/min; bValues recorded at the maximum yield of cyclohexene; cInitial selectivity to cyclohexene; dWeight-specific activity, unit in mmol•gcat-1• min-1. 表 3 Ru-Zn/ZrO2催化剂的苯部分加氢结果a
Table 3. Results of the partial hydrogenation of benzene over the Ru-Zn/ZrO2 catalystsa表 3亦列出了各个Ru-Zn/ZrO2催化剂的苯加氢活性.其中, Ru-Zn/ZrO2(3.2) 催化剂的r0最低, 为14.9 mmol•gcat-1•min-1, 而Ru-Zn/ZrO2(11.7) 催化剂的r0最高, 为28.3 mmol•gcat-1•min-1.根据r0和分散度算得的催化剂上苯的TOF值亦列于表 3, 显示Ru-Zn/ZrO2(3.2)、Ru-Zn/ZrO2(10.2) 和Ru-Zn/ZrO2(11.7) 催化剂有相近的TOF值, 与三个催化剂上的金属纳米粒子粒径相近且化学态相同的表征结果一致.
从表 3可知, Ru-Zn/ZrO2(3.2) 催化剂的S0最低, 为32%. Ru-Zn/ZrO2(11.7) 催化剂的S0最高, 为88%, 高于Schwab等[24, 25]报道的Ru/Al2O3和Ru/La2O3催化剂的S0 (分别约为60%和70%), 也高于Ru-Zn/ZrO2(11.7)-H2催化剂的S0 (74%), 说明在ZnSO4•7H2O水溶液中还原比在H2/Ar混合气中还原更好. 图 10为催化剂的环己烯选择性与转化率的关系, 由图可见在不同转化率下选择性顺序均为Ru-Zn/ZrO2(3.2)<Ru-Zn/ZrO2(10.2)<Ru-Zn/ZrO2(11.7).由于这三个催化剂上Ru的粒径和电子性质相近, 因此粒径效应和电子效应不应是引起选择性显著差异的根本原因. Zhao等[26]发现在苯部分加氢反应中, Ru/Al2O3/堇青石催化剂中较大孔径的Al2O3有利于环己烯传质, 使生成的环己烯容易从催化剂上脱附, 避免深度加氢, 因而环己烯的选择性较高. Liu等[27]发现Ru-Mn-Zn催化剂的环己烯选择性高于二元Ru-Mn或Ru-Zn催化剂, 归因于前者较大的孔径, 使环己烯更易扩散.据此我们推测, Ru-Zn/ZrO2(3.2)、Ru-Zn/ZrO2(10.2) 及Ru-Zn/ZrO2(11.7) 催化剂的环己烯选择性差异可能由载体孔径的不同所引起.苯部分加氢是氢气-苯-水-催化剂四相共存的复杂催化体系, 反应中生成的环己烯由于与水互不相溶, 故以有机液滴的形式分散在水相中[28].当环己烯在催化剂活性位上生成并脱附后, 应该首先以纳米液滴的形式悬浮于催化剂孔道中的水相里.催化剂的孔径越大, 这些环己烯纳米液滴越容易及时离开催化剂, 而不在孔中发生进一步加氢. 表 2表明催化剂的dpore有如下顺序: Ru-Zn/ZrO2(3.2)<Ru-Zn/ZrO2(10.2)<Ru-Zn/ZrO2(11.7), 故Ru-Zn/ZrO2(11.7) 催化剂上的环己烯选择性和得率最高.需要说明的是, 大的催化剂孔径并不意味着水相中的环己烯也会更容易进入催化剂的孔道.这是因为环己烯纳米液滴离开催化剂后会聚集为更大的液滴, 从而不容易再次进入催化剂孔道发生反应.
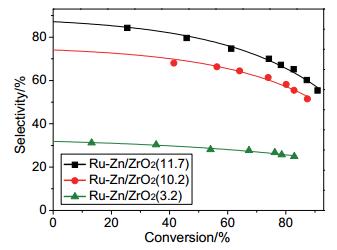 图 10
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂上环己烯选择性与苯转化率之间的关系
Figure 10.
Plots of the selectivity to cyclohexene versus the conversion of benzene over the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
图 10
(a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂上环己烯选择性与苯转化率之间的关系
Figure 10.
Plots of the selectivity to cyclohexene versus the conversion of benzene over the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
3 结论
制备了具有不同孔径的Ru-Zn/ZrO2催化剂, 考察了催化剂在苯部分加氢反应中的孔径效应.结果表明, 虽然这些Ru-Zn/ZrO2催化剂上苯加氢的TOF值基本相同, 但环己烯选择性随催化剂孔径的增大而升高.孔径最大的Ru-Zn/ZrO2(11.7) 催化剂上的S0及环己烯得率最高, 为88%和54%.我们推测催化剂较大的孔径有利于环己烯纳米液滴及时离开催化剂孔道, 避免深度加氢, 因而有利于环己烯选择性的提高.
4 实验部分
4.1 试剂与仪器 (见supporting information)
4.2 催化剂制备
根据文献, 采用溶剂热法[29]和沉淀法[30]制备具有相同晶型但不同孔径的四方ZrO2.溶剂热法的具体步骤为:将ZrOCl2•8H2O和尿素溶解于210 mL甲醇中, 浓度分别为0.60 mol•L−1和1.20 mol•L-1.转移至250 mL水热釜中后, 于473 K下溶剂热20 h.用去离子水将所得固体洗涤至中性, 373 K下干燥24 h.研磨后, 在673 K下焙烧4 h, 升温速率2 K•min-1.由N2物理吸附测得上述ZrO2的孔径为3.2 nm, 将该ZrO2记为ZrO2(3.2).
沉淀法的具体步骤为:在室温及搅拌条件下, 将25 wt%的氨水逐滴加入0.50 mol•L-1的ZrOCl2•8H2O水溶液中, 至pH为10.搅拌3 h后, 于373 K及常压下回流48 h.然后用去离子水洗涤沉淀至中性, 373 K下干燥24 h.所得固体经研磨后, 分别在1073 K及873 K下焙烧5 h, 升温速率10 K•min-1.由N2物理吸附测得上述ZrO2的孔径分别为10.2 nm及11.7 nm, 分别记为ZrO2(10.2) 及ZrO2(11.7).
以上述ZrO2为载体, 采用沉积沉淀-硫酸锌溶液中还原的方法制备Ru-Zn/ZrO2催化剂.具体步骤如下:在室温及搅拌条件下, 将19 mL水加入1.0 g上述ZrO2中, 再依次逐滴加入3.0 mL的RuCl3水溶液 (0.40 mol•L-1) 和1.5 mL的ZnSO4•7H2O水溶液 (62 mmol•L-1), 然后逐滴加入3.2 mL的NaOH水溶液 (30 wt%) 作为沉淀剂, 于353 K下回流3 h. Ru和Zn的理论负载量分别为10.65 wt%和0.54 wt%.待沉淀冷却后, 水洗至中性, 即得到催化剂前驱体.将该催化剂前驱体、90 mL的H2O、15.0 g的ZnSO4•7H2O置于500 mL高压反应釜中.密闭后, 用H2置换4次以排除釜内空气, 于3.0 MPa H2下加热.升温至423 K后, 调节H2压力至5.0 MPa, 开启搅拌至转速为1200 r/min, 并开始计时, 还原3 h.还原结束后, 停止搅拌.待高压釜冷却至室温后, 取出催化剂并水洗至滤液中不含硫酸锌 (采用BaCl2•2H2O水溶液检测), 保存在乙醇中以备表征.根据所用ZrO2的不同, 制备的催化剂记为Ru-Zn/ZrO2(3.2)、Ru-Zn/ZrO2(10.2) 及Ru-Zn/ZrO2(11.7).
4.3 催化剂表征
XRD谱在德国Bruker公司AXS D8 Advance型X射线粉末衍射仪上测定. Cu Kα线为激发源 (λ=0.15418 nm), 管电压40 kV, 管电流40 mA, 扫描范围10°~70°, 扫描速率4 (°)•min-1. ZrO2及Ru的晶粒尺寸根据Scherrer公式采用最强衍射峰的宽度进行计算, 并采用Warren规则扣除仪器宽化的影响[31]. Scherrer公式为d=0.89λ/bcosq[b=(B2-b2)1/2], 其中d为晶粒尺寸, q为衍射角, B和b分别为q处衍射峰及仪器产生的半高宽 (FWHM), b为扣除仪器宽化影响后q处衍射峰的半高宽.采用在相同条件下测量高结晶度的石英的衍射峰半高宽作为仪器造成的增宽[32].
样品的N2吸脱附等温线于77 K下在美国Micromeritics Tristar 3000型物理吸附仪上测定.测试前, 样品在N2(99.9%) 气氛中于473 K下预处理4 h.样品的孔径分布曲线根据吸脱附等温线的脱附支采用Barrett-Joyner-Halenda (BJH) 等效圆柱模型计算.催化剂体相组成采用电感耦合等离子体原子发射光谱 (ICP-AES, 美国Thermo Elemental公司IRIS Intrepid) 测定.
催化剂上Ru的分散度 (dispersion) 和粒径 (dRu) 采用CO化学吸附在美国Micromeritics 2750型化学吸附仪上测定.将约100 mg样品装入样品管中, 在Ar (99.999%) 气氛中于473 K下预处理1 h, 然后降温至298 K.在该温度下CO (99.999%) 脉冲进样[24], 至热导检测器 (TCD) 检测的峰面积恒定为止, 即达到饱和吸附.根据CO吸附面积, 采用文献提供的方法计算SRu和Ru的分散度, 假定吸附CO与金属Ru的化学计量比 (CO/Ru) 为0.5, 且Ru的表面原子密度为1.63×1019 atoms•m-2[33].
XPS和XAES谱在美国Perkin-Elmer PHI5000C型X射线光电子能谱仪上测定. Mg Kα线 (hν=1253.6 eV) 为激发源, 电压为14.0 kV, 功率为250 W, 通能为93.90 eV, X射线与样品夹角为54°.将乙醇保护的样品转移至预处理室, 室温下真空脱气过夜, 然后转移至分析室, 其真空度优于2×10-9 Torr.由于Ru 3d峰与污染碳的C 1s峰部分重叠, 因此采用载体ZrO2的Zr 3d5/2峰的结合能 (182.2 eV) 进行荷电校正, 结合能测量误差±0.2 eV.
Ru的K边X射线吸收光谱采用荧光模式在北京同步辐射中心 (BSRF) 1W1B线站上测定, 吸收边能量为22117 eV, 电子束能量为2.5 GeV, 电流为200 mA.将乙醇保护的催化剂涂于胶带上, 压入Al窗, 置于样品台上.采用IFEFFIT软件包中的Athena软件拟合吸收边前的吸收曲线, 将其外推至吸收边以后, 作为本底部分扣除, 即获得X射线吸收近边结构 (XANES)[34].
催化剂中金属前驱体与载体之间的相互作用采用H2-TPR在美国Micromeritics 2750型化学吸附仪上测定.取约100 mg样品置于样品管中, 在Ar (99.999%) 气氛中于473 K下预处理1 h, 冷却至室温, 将Ar切换为5 vol% H2/Ar混合气, 气体流速为25 mL•min-1.待基线稳定后, 从室温升温至973 K, 升温速率10 K•min-1, 得到H2-TPR曲线.
催化剂的形貌、Ru纳米粒子的粒径及微观结构采用日本JEOL公司JEM2011型透射电子显微镜观察, 工作电压为200 kV.将样品分散在无水乙醇中, 超声10 min后滴到表面覆有碳膜的铜网上.在TEM照片中量取约300个粒子的粒径, 绘制粒径分布图, 进行Gauss拟合, 并计算平均粒径和标准偏差.
4.4 催化性能评价
苯加氢反应在容积为500 mL的机械搅拌式高压反应釜中进行, 于典型的苯部分加氢反应条件下考察催化剂的性能[1~4, 35~37].在釜中加入1.0 g催化剂, 100 mL的H2O及15.0 g的ZnSO4•7H2O.密闭后, 用H2置换4次以除去釜内空气, 于3.0 MPa H2下加热.升温至413 K后, 用柱塞泵将50 mL苯打入高压釜中, 速率为10 mL•min-1.调节H2压力至5.0 MPa, 开启搅拌至转速为1200 r/min, 以消除扩散效应[22], 并开始计时.反应过程中定时取样, 每次取样约0.3 mL.同一个催化剂的催化性能至少重复考察两次, 反应结果的误差在2%以内.采用GC122型气相色谱仪分析产物组成.色谱分析条件为PEG-20M填充柱, TCD检测器, H2载气, 色谱柱、检测器及进样器温度均为403 K, 桥电流为100 mA.
苯转化率 (Conv.), 环己烯选择性 (SCHE) 及得率 (YCHE) 由以下公式算得:
Conv.=([C6H6]0-[C6H6]t)/[C6H6]0
SCHE=[C6H10]t/([C6H6]0-[C6H6]t)
YCHE=[C6H10]t/[C6H6]0
其中, 下标“0”和“t”分别表示苯的初始浓度及时间为t时苯或环己烯的浓度.
催化活性用初始反应速率 (r0) 及苯的转换频率TOF来表示. r0通过外推苯的浓度-时间曲线至反应时间为零时得到, 表示反应初始单位时间单位质量催化剂上转化的苯的量, 单位为mmol/(min•gcat). TOF=r0×MRu/(dispersion×W), 其中MRu和W分别为Ru的摩尔质量及负载量.环己烯的初始选择性 (S0) 通过外推环己烯选择性-时间曲线至反应时间为零时得到.
-
-
[1]
窦镕飞, 谭晓荷, 范义秋, 裴燕, 乔明华, 范康年, 孙斌, 宗保宁, 化学学报, 2016, 74, 503. doi: 10.6023/A16020074Dou, R. F.; Tan, X. H.; Fan, Y. Q.; Pei, Y.; Qiao, M. H.; Fan, K. N.; Sun, B.; Zong, B. N. Acta Chim. Sinica 2016, 74, 503. doi: 10.6023/A16020074
-
[2]
Sun, H. J.; Jiang, H. B.; Li, S. H.; Dong, Y. Y.; Wang, H. X.; Pan, Y. J.; Liu, S. C.; Tang, M. S.; Liu, Z. Y. Chem. Eng. J. 2013, 218, 415. doi: 10.1016/j.cej.2012.12.041
-
[3]
Sun, H. J.; Wang, H. X.; Jiang, H. B.; Li, S. H.; Liu, S. C.; Liu, Z. Y.; Yuan, X. M.; Yang, K. J. Appl. Catal. A 2013, 450, 160. doi: 10.1016/j.apcata.2012.10.016
-
[4]
Zhang, P.; Wu, T. B.; Jiang, T.; Wang, W. T.; Liu, H. Z.; Fan, H. L.; Zhang, Z. F.; Han, B. X. Green Chem. 2013, 15, 152. doi: 10.1039/C2GC36596K
-
[5]
徐华龙, 黄静静, 杨新艳, 杜俊明, 沈江, 沈伟, 化学学报, 2006, 64, 1615. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract336873.shtmlXu, H. L.; Huang, J. J.; Yang, X. Y.; Du, J. M.; Shen, J.; Shen, W. Acta Chim. Sinica 2006, 64, 1615. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract336873.shtml
-
[6]
Kang, J. C.; Cheng, K.; Zhang, L.; Zhang, Q. H.; Ding, J. S.; Hua, W. Q.; Lou, Y. C.; Zhai, Q. G.; Wang, Y. Angew. Chem. Int. Ed. 2011, 50, 5200. doi: 10.1002/anie.v50.22
-
[7]
Liu, Y. C.; Fang, K. G.; Chen, J. G.; Sun, Y. H. Green Chem. 2007, 9, 611. doi: 10.1039/B614266D
-
[8]
Zuo, S. F.; Huang, Q. Q.; Zhou, R. X. Catal. Today 2008, 139, 88. doi: 10.1016/j.cattod.2008.08.026
-
[9]
Gelesky, M. A.; Chiaro, S. S. X.; Pavan, F. A.; dos Santos, J. H. Z.; Dupont, J. Dalton Trans. 2007, 5549.
-
[10]
Xia, Q. H.; Hidajat, K.; Kawi, S. Catal. Today 2001, 68, 255. doi: 10.1016/S0920-5861(01)00285-1
-
[11]
王建强, 郭平均, 乔明华, 闫世润, 范康年, 化学学报, 2004, 62, 1765. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344136.shtmlWang, J. Q.; Guo, P. J.; Qiao, M. H.; Yan, S. R.; Fan, K. N. Acta Chim. Sinica 2004, 62, 1765. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344136.shtml
-
[12]
Job, N.; Pereira, M. F. R.; Lambert, S.; Cabiac, A.; Delahay, G.; Colomer, J. F.; Marien, J.; Figueiredo, J. L.; Pirard, J. P. J. Catal. 2006, 240, 160. doi: 10.1016/j.jcat.2006.03.016
-
[13]
Preising, H.; Enke, D. Colloids Surf. A 2007, 300, 21. doi: 10.1016/j.colsurfa.2006.12.036
-
[14]
Zhou, G. B.; Liu, J. L.; Tan, X. H.; Pei, Y.; Qiao, M. H.; Fan, K. N.; Zong, B. N. Ind. Eng. Chem. Res. 2012, 51, 12205.
-
[15]
Zhao, Y. J.; Zhou, J.; Zhang, J. G.; Wang, S. D. J. Mol. Catal. A 2009, 309, 35. doi: 10.1016/j.molcata.2009.04.012
-
[16]
Wang, Z. Q.; Ma, Y. C.; Lin, J. X. J. Mol. Catal. A 2013, 378, 307. doi: 10.1016/j.molcata.2013.07.003
-
[17]
Campbell, P. S.; Santini, C. C.; Bayard, F.; Chauvin, Y.; Collière, V.; Podgoršek, A.; Costa Gomes, M. F.; Sá, J. J. Catal. 2010, 275, 99. doi: 10.1016/j.jcat.2010.07.018
-
[18]
Moulder, J. F.; Stickle, W. F.; Sobol, P. E.; Bomben, K. D. In Handbook of X-ray Photoelectron Spectroscopy, Ed.:Chastain, J., Perkin-Elmer, Minnesota, 1992, p. 89.
-
[19]
Deroubaix, G.; Marcus, P. Surf. Interface Anal. 1992, 18, 39. doi: 10.1002/(ISSN)1096-9918
-
[20]
Silvestre-Albero, J.; Serrano-Ruiz, J. C.; Sepúlveda-Escribano, A.; Rodríguez-Reinoso, F. Appl. Catal. A 2005, 292, 244. doi: 10.1016/j.apcata.2005.06.005
-
[21]
Lorenzut, B.; Montini, T.; Pavel, C. C.; Comotti, M.; Vizza, F.; Bianchini, C.; Fornasiero, P. Chem Cat Chem 2010, 2, 1096.
-
[22]
Wang, J. Q.; Wang, Y. Z.; Xie, S. H.; Qiao, M. H.; Li, H. X.; Fan, K. N. Appl. Catal. A 2004, 272, 29. doi: 10.1016/j.apcata.2004.04.038
-
[23]
Yuan, P. Q.; Wang, B. Q.; Ma, Y. M.; He, H. M.; Cheng, Z. M.; Yuan, W. K. J. Mol. Catal. A 2009, 309, 124. doi: 10.1016/j.molcata.2009.05.006
-
[24]
Schwab, F.; Lucas, M.; Claus, P. Angew. Chem. Int. Ed. 2011, 50, 10453. doi: 10.1002/anie.201104959
-
[25]
Schwab, F.; Lucas, M.; Claus, P. Green Chem. 2013, 15, 646. doi: 10.1039/c3gc36615d
-
[26]
Zhao, Y. J.; Zhou, J.; Zhang, J. G.; Wang, S. D. Catal. Lett. 2009, 131, 597. doi: 10.1007/s10562-009-0025-9
-
[27]
Zhou, X. L.; Sun, H. J.; Guo, W.; Liu, Z. Y.; Liu, S. C. J. Nat. Gas Chem. 2011, 20, 53. doi: 10.1016/S1003-9953(10)60152-1
-
[28]
Foppa, L.; Dupont, J. Chem. Soc. Rev. 2015, 44, 1886. doi: 10.1039/C4CS00324A
-
[29]
Li, W. Z.; Huang, H.; Li, H. J.; Zhang, W.; Liu, H. C. Langmuir 2008, 24, 8358. doi: 10.1021/la800370r
-
[30]
Jung, K. T.; Bell, A. T. J. Mol. Catal. A 2000, 163, 27. doi: 10.1016/S1381-1169(00)00397-6
-
[31]
Warren, B. E. J. Appl. Phys. 1941, 12, 375. doi: 10.1063/1.1712915
-
[32]
Robertson, S. D.; Anderson, R. B. J. Catal. 1971, 23, 286. doi: 10.1016/0021-9517(71)90051-0
-
[33]
Elmasides, C.; Kondarides, D. I.; Grünert, W.; Verykios, X. E. J. Phys. Chem. B 1999, 103, 5227.
-
[34]
Ravel, B.; Newville, M. J. Synchrotron Rad. 2005, 12, 537. doi: 10.1107/S0909049505012719
-
[35]
孙海杰, 李永宇, 李帅辉, 张元馨, 刘寿长, 刘仲毅, 任保增, 物理化学学报, 2014, 30, 1332.Sun, H. J.; Li, Y. Y.; Li, S. H.; Zhang, Y. X.; Liu, S. C.; Liu, Z. Y.; Ren, B. Z. Acta Phys.-Chim. Sin. 2014, 30, 1332.
-
[36]
卜娟, 王建强, 乔明华, 闫世润, 李和兴, 范康年, 化学学报, 2007, 65, 1338. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract328945.shtmlBu, J.; Wang, J. Q.; Qiao, M. H.; Yan, S. R.; Li, H. X.; Fan, K. N. Acta Chim. Sinica 2007, 65, 1338. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract328945.shtml
-
[37]
王丽娟, 张爱清, 李琳, 刘汉范, 刘书正, 化学学报, 2012, 70, 1021. doi: 10.6023/A1110173Wang, L. J.; Zhang, A. Q.; Li, L.; Liu, H. F.; Liu, S. Z. Acta Chim. Sinica 2012, 70, 1021. doi: 10.6023/A1110173
-
[1]
-

图 1 (a) ZrO2(3.2)、(b) ZrO2(10.2) 及 (c) ZrO2(11.7) 的XRD谱
Figure 1 XRD patterns of the (a) ZrO2(3.2), (b) ZrO2(10.2) and (c) ZrO2(11.7)

图 2 (a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的XRD谱
Figure 2 XRD patterns of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts

图 3 (a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的Ru 3p (左) 及Zn 2p (右) XPS谱
Figure 3 Ru 3p (left) and Zn 2p (right) XPS spectra of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts

图 4 (a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2)、(c) Ru-Zn/ZrO2(11.7) 催化剂及Ru箔标样归一化后的Ru K边XANES谱
Figure 4 Normalized Ru K-edge XANES spectra of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2), (c) Ru-Zn/ZrO2(11.7) catalysts and the Ru foil standard

图 5 (a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的Zn LMM XAES谱
Figure 5 Zn LMM XAES spectra of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts

图 6 (a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2)、(c) Ru-Zn/ZrO2(11.7) 及 (d) Ru/ZrO2(3.2) 催化剂的H2-TPR谱
Figure 6 H2-TPR profiles of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2), (c) Ru-Zn/ZrO2(11.7) and (d) Ru/ZrO2(3.2) catalysts

图 7 (a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的TEM照片及含高斯拟合曲线的粒径分布图
Figure 7 TEM images and particle size distribution histograms with Gaussian analysis fittings of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts

图 8 (a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的含FFT图片的HRTEM照片
Figure 8 HRTEM images with FFT patterns of the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts

图 9 (a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂的苯加氢反应历程
Figure 9 The time courses of benzene hydrogenation over the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts. Reaction conditions: 1.0 g catalyst, 50 mL benzene, 100 mL H2O, 15.0 g ZnSO4•7H2O, temperature of 413 K, H2 pressure of 5.0 MPa, and stirring rate of 1200 r/min. (■) Benzene, (●) cyclohexene, and (▲) cyclohexane

图 10 (a) Ru-Zn/ZrO2(3.2)、(b) Ru-Zn/ZrO2(10.2) 及 (c) Ru-Zn/ZrO2(11.7) 催化剂上环己烯选择性与苯转化率之间的关系
Figure 10 Plots of the selectivity to cyclohexene versus the conversion of benzene over the (a) Ru-Zn/ZrO2(3.2), (b) Ru-Zn/ZrO2(10.2) and (c) Ru-Zn/ZrO2(11.7) catalysts
表 1 ZrO2的织构性质
Table 1. Textural properties of ZrO2
Sample SBET/(m2•g-1) dpore/nm Vpore/(cm3•g-1) ZrO2(3.2) 60 3.2 0.06 ZrO2(10.2) 88 10.2 0.28 ZrO2(11.7) 128 11.7 0.48  下载: 导出CSV
下载: 导出CSV
表 2 Ru-Zn/ZrO2催化剂的物化性质
Table 2. Physicochemical properties of the Ru-Zn/ZrO2 catalysts
Catalyst Ru loadinga/wt% Zn loadinga/wt% Dispersionb/% dRuc/nm SBET/(m2•g−1) dpore/nm Vpore/(cm3•g-1) Ru-Zn/ZrO2(3.2) 10.15 3.88 15.1 8.9 48 5.6 0.07 Ru-Zn/ZrO2(10.2) 10.43 3.84 25.6 5.3 64 9.2 0.25 Ru-Zn/ZrO2(11.7) 10.55 4.12 26.2 5.1 97 9.8 0.30 aDetermined by ICP-AES; bDispersion of Ru determined by CO chemisorption; cParticle size of Ru determined by CO chemisorption.
下载: 导出CSV
表 3 Ru-Zn/ZrO2催化剂的苯部分加氢结果a
Table 3. Results of the partial hydrogenation of benzene over the Ru-Zn/ZrO2 catalystsa
Entry Catalyst Conv.b/ % SCHEb/ % YCHEb/ % tb/ min S0c/ % r0d TOF/ s-1 1 Ru-Zn/ZrO2(3.2) 83 25 21 65 32 14.9 1.6 2 Ru-Zn/ZrO2(10.2) 87 51 44 40 74 21.3 1.3 3 Ru-Zn/ZrO2(11.7) 83 65 54 25 88 28.3 1.7 aReaction conditions: 1.0 g catalyst, 50 mL benzene, 100 mL H2O, 15.0 g ZnSO4•7H2O, temperature of 413 K, H2 pressure of 5.0 MPa, and stirring rate of 1200 r/min; bValues recorded at the maximum yield of cyclohexene; cInitial selectivity to cyclohexene; dWeight-specific activity, unit in mmol•gcat-1• min-1.
下载: 导出CSV
-











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 9
- 文章访问数: 1338
- HTML全文浏览量: 132
 下载:
下载:
