引用本文:
向兴德, 卢艳莹, 陈军. 钠离子电池先进功能材料的研究进展[J]. 化学学报,
2017, 75(2): 154-162.
doi:
10.6023/A16060275

Citation: Xiang Xingde, Lu Yanying, Chen Jun. Advance and Prospect of Functional Materials for Sodium Ion Batteries[J]. Acta Chimica Sinica, 2017, 75(2): 154-162. doi: 10.6023/A16060275
Citation: Xiang Xingde, Lu Yanying, Chen Jun. Advance and Prospect of Functional Materials for Sodium Ion Batteries[J]. Acta Chimica Sinica, 2017, 75(2): 154-162. doi: 10.6023/A16060275
钠离子电池先进功能材料的研究进展
摘要:
钠离子电池作为一种新型的化学电源,因钠资源储量丰富、成本低廉等优势,在规模储能领域具有应用前景,近年来受到了人们的广泛关注.为了获得比能量高、循环寿命长和快速充放电能力强的先进钠离子电池,人们正致力于开发比容量高、循环性能好和倍率性能佳的储钠电极材料和离子电导率高、电化学窗口宽的功能电解液,并取得了重要进展.目前,有前景的正极材料主要有高容量的层状氧化物、高电位的氟磷酸盐和长寿命的磷酸盐;可用的负极材料主要包括循环稳定性强的钛基层状氧化物和碳材料、比容量大的金属/非金属单质和低成本的金属化合物;有效的功能电解液有酯类电解液和醚类电解液.本综述详细总结了上述几类电极材料和电解液的最新研究进展,重点介绍了它们的电化学性质、科学难题及解决策略.
English
Advance and Prospect of Functional Materials for Sodium Ion Batteries
Abstract:
Sodium ion batteries (SIBs) as a new chemical power source have recently attracted a great attention for large-scale energy storage owing to the abundance and low cost of sodium resources. In order to achieve advanced SIBs with high specific energy, long cycling lifetime and fast charge/discharge ability, efforts have been devoted to developing advanced electrode materials with large specific capacity, robust cycling stability and good rate capability, as well as functional electrolytes with high ion-conductivity and wide electrochemical window. Promising cathode materials include high-capacity layered oxides, high-potential fluorophosphates and long-lifetime phosphates. Available anode materials consist of highly stable Ti-based layered oxides and carbon materials, high-capacity elemental metals/non-metals and low-cost metal-based compounds. Effective electrolytes involve ester-based electrolytes and ether-based electrolytes. This review summarizes the recent advance of electrode materials and electrolytes for SIBs, mainly focusing on their electrochemical properties, existing challenges and resolution strategies.
-
Key words:
- sodium ion batteries
- / cathode materials
- / anode materials
- / electrolytes
-
1 引言
钠离子电池作为一种新型的二次电池, 近年来在新能源储存与转换领域引起了广泛关注[1~4]. 相比于已经商业化使用的锂离子电池, 它具有以下特点: (1) 相类似的充放电机制. 充电过程中, 正极材料发生氧化反应, 失去电子, 并脱出钠离子; 电子通过外电路到达负极, 同时钠离子也经过电解液迁移到负极; 负极材料得到电子, 并嵌入钠离子, 发生还原反应. 放电过程与充电过程相反. (2) 相似的嵌插化学性质. 钠与锂属于同主族元素, 物理与化学性质相近. 锂离子和钠离子可以在相似的材料结构中进行可逆的嵌入与脱出. (3) 更低的资源成本. 钠资源在地壳中的储量约1%, 广泛存在于海水中; 而锂资源储量只有约10 ppm[2], 且分布不均, 70%集中在南美国家. 此外, 为了更好地满足实用化要求, 钠离子电池还需要提升比能量、循环寿命、倍率性能等[1].
电池的比能量主要由正负极材料的比容量和电位差决定[5]. 比容量与材料的分子量和电荷转移数有关(见表 1). 当分子量相同时, 电荷转移数越大, 比容量越大; 当电荷转移数相同时, 分子量越小, 比容量越大. 工作电位与活性材料的氧化还原能相对应. 氧化还原能越高, 电位越低; 反之则越高. 通常, 电负性强的阴离子可以通过诱导效应降低材料的氧化还原能, 从而提高其工作电位[6]. 因此, 高比能量要求电池具有比容量大、工作电位高的正极材料和比容量大、工作电位低的负极材料.
 表 1
代表性正极材料[NaCrO2、Na3(VO)2(PO4)2F、Na3V2(PO4)2F3、NaFePO4和Na3V2(PO4)3]和负极材料(石墨、硬碳、Sn、P和Fe2O3)的分子量、反应机理、理论容量和平均工作电位
Table 1.
Molecular weights, reaction mechanism, theoretical capacities, and average operating potentials of representative cathode materials [NaCrO2, Na3(VO)2(PO4)2F, Na3V2(PO4)2F3, NaFePO4, and Na3V2(PO4)3] and anode materials (graphite, hard carbon, Sn, P, and Fe2O3)
表 1
代表性正极材料[NaCrO2、Na3(VO)2(PO4)2F、Na3V2(PO4)2F3、NaFePO4和Na3V2(PO4)3]和负极材料(石墨、硬碳、Sn、P和Fe2O3)的分子量、反应机理、理论容量和平均工作电位
Table 1.
Molecular weights, reaction mechanism, theoretical capacities, and average operating potentials of representative cathode materials [NaCrO2, Na3(VO)2(PO4)2F, Na3V2(PO4)2F3, NaFePO4, and Na3V2(PO4)3] and anode materials (graphite, hard carbon, Sn, P, and Fe2O3)
Na3(VO)2(PO4)2F 411.9 Na3(VO)2(PO4)2F↔Na(VO)2(PO4)2F+2Na++2e- 130 ca. 3.8 Na3V2(PO4)2F3 417.9 Na3V2(PO4)2F3↔NaV2(PO4)2F3+2Na++2e- 128 ca. 3.9 NaFePO4 173.8 NaFePO4↔FePO4+Na++e- 154 ca. 2.6 Na3V2(PO4)3 455.9 Na3V2(PO4)3↔NaV2(PO4)3+2Na++2e- 117 ca. 3.4 负极材料 石墨(C) 12 20C+Na++e-↔NaC20 (醚类溶剂) 112 ca. 0.8 硬碳(C) 12 6C+Na++e-↔NaC6 375 ca. 0.2 P 31.0 P+3Na++3e-↔Na3P 2594 ca. 0.5 Sn 118.7 4Sn+15Na++15e-↔Na15Sn4 847 ca. 0.3 表 1 代表性正极材料[NaCrO2、Na3(VO)2(PO4)2F、Na3V2(PO4)2F3、NaFePO4和Na3V2(PO4)3]和负极材料(石墨、硬碳、Sn、P和Fe2O3)的分子量、反应机理、理论容量和平均工作电位
Table 1. Molecular weights, reaction mechanism, theoretical capacities, and average operating potentials of representative cathode materials [NaCrO2, Na3(VO)2(PO4)2F, Na3V2(PO4)2F3, NaFePO4, and Na3V2(PO4)3] and anode materials (graphite, hard carbon, Sn, P, and Fe2O3)电池的循环寿命主要受电极材料的结构稳定性和电解液的电化学稳定性影响. 由于Na+离子半径比较大(1.02 Å), 大量的嵌入或脱出Na+离子会使材料发生很大的体积膨胀或收缩, 容易诱导电极材料发生不可逆的结构变化, 从而使高容量钠离子电池表现出不足的循环寿命[7]. 加强材料结构稳定性的常见策略主要有: (1) 控制正极材料的充电截止电位或负极材料的放电截止电位, 限制钠离子的嵌脱量; (2) 引入键合能力强、非活性的过渡金属离子, 提高结构框架的稳定性. 另外, 电解液分解也是影响电池循环寿命的重要原因. 当电解液的最高占据轨道(HOMO)能高于正极材料的氧化还原能时, 电子容易从电解液HOMO轨道转移到正极, 导致电解液发生氧化分解反应; 当电解液的最低空轨道(LUMO)能低于负极材料的氧化还原能时, 电子容易从负极转移到LUMO轨道, 导致电解液发生还原分解反应[8]. 电解液持续的不可逆分解不仅会增加电池的内阻, 还会带来安全性隐患. 减少电解液副反应的路途有两种: (1) 优化电解液组分. 拓宽它的电化学稳定窗口, 使其HOMO能低于正极材料的氧化还原能, LUMO能高于负极材料的氧化还原能. (2) 开发功能成膜添加剂[9]. 这种添加剂可以在电解液分解之前发生反应, 在电极表面生成一薄层能够传输离子、绝缘电子、性质稳定的固态电解质界面膜(SEI), 防止电解液分解. 因此, 长循环寿命要求电池具有结构稳定的电极材料和电化学稳定的电解液.
电池的倍率性能与电池反应动力学有关, 主要受电极材料的电子传导性和离子传输能力影响[10]. 碳包覆可以加强材料的电子传导性; 纳米结构可以缩短离子的扩散路径、增加离子的扩散界面, 从而提高材料的离子传输能力; 多孔形貌有利于电解液对材料的浸润. 因此, 设计具有碳包覆、纳米结构和多孔形貌的电极材料, 可以优化电极材料的反应动力学, 显著提高电极材料的倍率性能.
本文将详细综述几类有前景的正极材料(金属氧化物、氟磷酸盐和磷酸盐)、负极材料(钛基层状氧化物、碳材料、金属/非金属单质和金属化合物)和电解液(酯类电解液和醚类电解液), 重点介绍它们的电化学性质、科学难题以及解决策略.
2 正极材料
正极材料是钠离子电池的关键要素之一, 负责提供活性钠离子和高电位氧化还原电对, 对电池的比容量和工作电压有重要影响. 可用的正极材料主要有低成本的金属氧化物(NaxMO2, M=Fe, Mn, Ni, Co等)、高电位的氟磷酸盐[Na3(VOx)2(PO4)2F3-2x, 0≤x≤1]和长循环的磷酸盐[NaFePO4和Na3V2(PO4)3].
2.1 金属氧化物
金属氧化物具有毒性小、成本低、合成工艺简单等特点, 被认为是一类有前景的低成本钠离子电池正极材料[7], 主要包括层状氧化物和隧道型氧化物.
层状氧化物的理论容量很高(约240 mAh·g-1), 在高容量钠离子电池中具有诱人的应用前景. 但是, 由于单金属层状氧化物NaMO2(M=Fe, Ni, Mn, Cr等)在深度脱钠状态下容易发生不可逆的结构变化, 表现出低的库仑效率和快速的容量衰退[11~15]. 为了提高循环稳定性, 往往需要通过限制充电截止电位来降低材料的脱钠深度, 以致于它们的实际可用容量只有100~120 mAh·g-1左右[14~16]. 形成多金属氧化物可以适当提高材料的比容量和循环性能[17~21]. 据报道, NaNi1/3Co1/3Mn1/3O2在2.0~3.75 V范围内可以释放出120 mAh·g-1的可逆容量, 循环50圈后无容量衰减[18]; NaNi1/4Fe1/2Mn1/4O2在0.1 C (2.1~3.9 V)条件下的可逆容量达到140 mAh·g-1, 在1 C循环50圈后的容量保持率为90.4%[19]; Na[Ni0.4-Fe0.2Mn0.2Ti0.2]O2在0.1 C (2.0~4.2 V)条件下的可逆容量达到145 mAh·g-1, 循环200圈后还能保持121 mAh· g-1 [20]. 通常, 富镍氧化物的比容量较高, 但循环性能较差, 而富锰氧化物的比容量较低, 但循环性能较好. 构建内部富镍、外部富锰的多级结构可以制备出循环性能好、比容量高的氧化物正极材料.比如, 具有多级结构的Na[Ni0.60Co0.05Mn0.35]O2在0.1 C (1.5~4.1 V)条件下的可逆容量达到157 mAh·g-1, 循环100圈后的容量保持率为84%[21]. 此外, 设计P2/O3堆积结构的复合层状氧化物也可以加强氧化物的比容量和循环稳定性. 具有P2/O3堆积结构的Na0.66Li0.18Mn0.71Ni0.21Co0.08O2+d在0.2 C条件下的可逆容量达到185 mAh·g-1, 循环50圈后的容量保持率大于80%[22]. 另外, 表面包覆不仅有利于增强循环稳定性, 还可以提高倍率性能. 有研究表明[23], 碳包覆的NaCrO2在20 mA·g-1 (2.0~3.6 V)条件下的可逆容量为121 mAh·g-1, 循环300圈后的容量保持率高达90%. 即使电流密度增大到5.5 A·g-1, 可逆容量还能保持在106 mAh·g-1. 这主要是因为碳包覆不仅可以减少活性材料与电解液间的副反应, 提高材料的结构稳定性,还可以增加材料的电子传导性, 优化材料的反应动力学.
相对于层状氧化物, 隧道型氧化物的比容量较低(理论容量121 mAh·g-1, 实际容量约100 mAh·g-1)[24], 在高容量钠离子电池中没有明显的竞争力. 但是, 常见的隧道型氧化物大多数是Mn-Ti基化合物(如Na0.44MnO2、Na0.44TixMn1-xO2、Na0.61Ti0.48Mn0.52O2)[24~26], 具有资源丰富、价格低廉、工作电位较低(2.6~3.0 V)、循环稳定性好等优点, 适合用于构建低成本、长寿命的水系钠离子电池.
2.2 氟磷酸盐
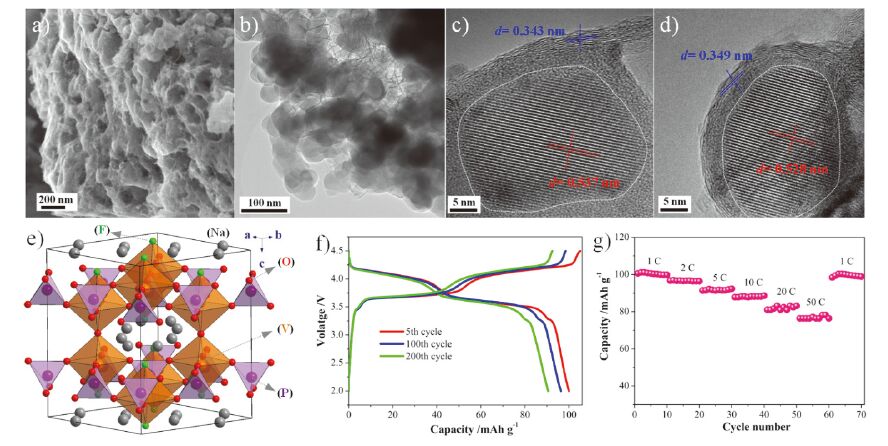
氟磷酸钒钠[Na3(VOx)2(PO4)2F3-2x, 0≤x≤1]是Na3(VO)2(PO4)2F和Na3V2(PO4)2F3的固溶体, 是一类结构框架稳定、比容量适中(约130 mAh·g-1)、工作电位高(约3.8 V)的聚阴离子化合物[27~29], 适合用于构建高电压钠离子电池. 这类材料在嵌脱钠过程中表现出两个高电位平台, 分别位于约3.7和约4.0 V. 但是, 它在反应过程中的相变机制很复杂, 目前还存在争议. 有人认为, 氟磷酸钒钠在整个充放电过程中发生单相反应[30, 31]. 也有人认为, 它在低电位平台主要发生两相反应, 在高电位平台主要发生单相反应[32~34]. 这可能是由于合成方法不同或组分不同引起的. 此外, 这类材料的电子传导性低、离子扩散能力不足, 反应动力学慢, 在高倍率条件下容量衰退很快. 通过改进材料合成方法, 构建电子传导性高的表面包覆层和纳米尺寸的材料颗粒, 可以提高氟磷酸钒钠在高倍率条件下的可逆容量和循环稳定性[31, 35~38]. 本课题组通过简单的固相反应路线制备了兼有薄层碳包覆、Na3(VO0.5)2(PO4)2F2嵌入多孔石墨烯的Na3(VO0.5)2(PO4)2F2/C材料(见图 1)[37]. 这种材料表现出了优异的高倍率性能: 在1、10、20和50 C条件下的放电容量分别达到100、88、82和77 mAh·g-1; 在50 C循环1000圈后的容量保持率为73%.
2.3 磷酸盐
NaFePO4存在无定形、磷铁钠矿型和橄榄石型三种结构形态. 不同结构形态的NaFePO4具有不同的电化学特性. 无定形NaFePO4是一种高容量、长寿命的钠离子电池正极材料. 平均工作电位为2.6 V, 可逆容量约150 mAh·g-1, 循环300圈后容量保持率可达95%[39]. 磷铁钠矿型NaFePO4是热力学稳定相, 可以由高温固相法直接合成. 但是, 由于缺乏离子扩散通道, 它本身并没有明显的电化学储钠活性. 不过, 纳米结构的磷铁钠矿型NaFePO4在首次充电过程中可以向无定形相转变, 从而表现出优异的电化学性能(可逆容量约142 mAh·g-1, 循环200圈后容量保持率为95%)[40]. 橄榄石型NaFePO4可以通过离子交换法、电化学转换法制备得到. 其工作电压约2.7 V、可逆容量为125 mAh·g-1, 循环50圈后还能保持90%左右[41]. 相比而言, 无定形NaFePO4的制备工艺简单、比容量大、能量效率高、循环稳定性好, 展示出了更好的应用前景.
Na3V2(PO4)3属于经典的NASICON(超快钠离子导体)结构[42]: 2个[VO6]八面体与3个[PO4]四面体通过共享“角”组成一个“灯笼”单元; 每个“灯笼”单元与其它6个单元相联, 形成可以容纳Na+离子的隙位空间. 由于有利的三维离子扩散通道, 钠离子可以在Na3V2(PO4)3中进行快速地扩散. 在充电过程中可以脱出2个Na+离子, 发生两相反应, 展现出3.4 V的电位平台和117 mAh·g-1的理论容量[43]. 但是, 它的电子传导性低, 极化严重, 以致于实际容量和循环稳定性不足. 制备具有纳米结构的Na3V2(PO4)3/C复合材料, 可以获得高倍率、长寿命的钠离子电池正极材料[44~49]. 据报道[48], 利用软化学快速处理法制备的多孔Na3V2(PO4)3/C在1、10、50、100和200 C的放电容量分别达到104、103、91、74和44 mAh·g-1, 在100 C循环1000圈后容量还能保持在50 mAh·g-1.
目前, 磷酸盐和氟磷酸盐在循环性能和倍率性能方面已经取得了一定进展. 但是, 它们的比容量较低, 限制了其在高能量钠离子电池中的应用. 开发循环性能好、倍率性能佳的高容量正极材料(如层状氧化物)将是钠离子电池研究的重点方向.
3 负极材料
负极材料也是钠离子电池的关键要素, 负责提供钠离子储存位点和低电位氧化还原电对, 对电池的比容量和工作电压有直接影响. 可用的负极材料主要有长寿命的钛基层状氧化物(如Na2/3Co1/3Ti2/3O2和Na0.66[Li0.22Ti0.78]O2)和碳材料(石墨和硬碳)、低电位的金属/非金属单质(如P和Sn)和低成本的金属化合物(如Fe2O3和MnFe2O4).
3.1 钛基层状氧化物
钛基层状氧化物是一类以Ti4+/Ti3+为低电位氧化还原电对的储钠负极材料, 允许钠离子在其层间进行可逆的嵌入与脱出[50~53]. 这类材料不仅循环性能好, 而且倍率性能佳, 适合用于构建长寿命、大功率的钠离子电池. 代表性的钛基层状氧化物主要有NaTiO2、Na0.66[Li0.22Ti0.78]O2和Na2/3Co1/3Ti2/3O2. 据报道, NaTiO2在0.1 C的可逆容量为152 mAh·g-1, 循环60圈后的容量保持率为98%[50]. Na0.66[Li0.22Ti0.78]O2在0.1 C的可逆容量为116 mAh·g-1, 循环200圈后的容量保持率大于86%[51]. Na2/3Co1/3Ti2/3O2在2 C的可逆容量约为60 mAh·g-1, 循环1000圈后的容量保持率接近96%[52].
3.2 碳材料
碳材料由于原料丰富、成本低廉、合成简单、易回收处理、工作电位低、性质稳定等特点, 适合用于构建高能量、长寿命的钠离子电池. 具有储钠活性的碳材料主要包括结构有序的石墨[54~57]和结构无序的硬碳[58~60]. 石墨在传统的碳酸酯类电解液中几乎没有电化学活性, 这可能是因为石墨烯层间距太小(约0.34 nm)、溶剂化钠离子太大的缘故. 增大石墨的层间距可以提高它的储钠性能[61]. 有趣的是, 在醚类电解液中, 石墨不仅可以释放出约120 mAh·g-1的可逆容量, 还可以表现出优异的循环稳定性(在0.2 A·g-1循环6000圈后, 容量保持率高达95%)和倍率性能(在10 A·g-1能保持102 mAh·g-1的容量)[57]. 相对于石墨, 硬碳的层间距较大(约0.4 Å)且结构无序. Na+离子可以在硬碳的表面或缺陷处发生可逆吸脱附, 在纳米孔内进行可逆填充, 在石墨烯层间进行可逆脱嵌. 充放电曲线包括一个位于0.1~1.0 V之间的电位斜区和一个处于0.1 V的电位平台. 最新研究表明[62], 电位斜区对应于钠离子在纳米孔内的填充; 电位平台归因于钠离子在石墨烯层间的嵌插以及在表面或缺陷处的吸脱附. 在低电流密度下, 硬碳的可逆容量可以达到约300 mAh·g-1, 循环50圈几乎无容量衰减[63].
3.3 金属/非金属单质
金属/非金属单质(P、Sn、Sb等)可以通过合金化反应机制进行电化学储钠[64~67], 具有工作电位低、理论容量高等优点, 是一类高能量钠离子电池负极材料. 代表性的金属/非金属单质材料有P和Sn. P是目前理论比容量最高的储钠负极材料(2594 mAh·g-1), 具有储量丰富、环境友好等优点. 但是, 它的循环性能和倍率性能很低, 主要是因为: (1) 它是电子绝缘体, 导电性差, 反应动力学缓慢; (2) 它在充放电过程中体积变化严重, 电极材料容易粉化脱落. 引入多孔结构的导电碳不仅可以加强材料的电子传导性, 还可以缓解体积变化产生的形变应力, 从而大大改善P的电化学性能. 据报道[68], 封装在石墨烯卷中的P纳米颗粒可以表现出优异的循环性能和倍率性能: 在电流密度为250、1000和4000 mA· g-1时的可逆容量分别达到2355、1705和1084 mAh· g-1; 在250 mA·g-1循环150圈后容量保持率为92%. Sn具有成本低、工作电位低(约0.3 V)、比容量高(847 mA· g-1)等优点, 是一种重要的金属单质类储钠材料. 与P相比, 它的电子传导性较好, 但在充放电过程中同样遭受了大的体积变化. 为了适应这种变化, 需要将其均匀分散在多孔碳材料中以制备高性能的Sn/C复合材料. 本课题组通过静电纺丝技术制备了具有多孔结构的Sn/C纳米纤维(见图 2)[69]. Sn颗粒大小为1~2 nm, 均匀分散在直径为80~120 nm的碳纤维中; 初始态的Sn属于四方晶系(空间群为I41/amd, 晶胞体积为约108 Å3), 嵌钠后变成立方结构的Na15Sn4 (空间群为I-43d, 约2269 Å3). 由于其单一的多孔纳米结构, Sn/C在0.2和10 A·g-1条件下的可逆容量分别达到633和450 mAh·g-1, 循环1300圈后的容量保持率大于90%.
 图 2
多孔Sn/C纳米纤维的SEM (a)、TEM (b~c)和充放电曲线(d插图是放电产物Na15Sn4与充电产物Sn的HRTEM)[69], 以及Sn (e)和Na15Sn4 (f)的晶胞结构
Figure 2.
SEM (a), TEM (b~c) and charge/discharge curves (d, the inset is the HRTEM images of fully discharged product Na15Sn4 and fully charged product Sn) of porous Sn/C nanofibers[69], and crystal structures of Sn (e) and Na15Sn4 (f)
图 2
多孔Sn/C纳米纤维的SEM (a)、TEM (b~c)和充放电曲线(d插图是放电产物Na15Sn4与充电产物Sn的HRTEM)[69], 以及Sn (e)和Na15Sn4 (f)的晶胞结构
Figure 2.
SEM (a), TEM (b~c) and charge/discharge curves (d, the inset is the HRTEM images of fully discharged product Na15Sn4 and fully charged product Sn) of porous Sn/C nanofibers[69], and crystal structures of Sn (e) and Na15Sn4 (f)
3.4 金属化合物
金属化合物主要有氧化物(如CuO、Fe2O3、MnFe2O4)[70~72]、硫化物(如MoS2和SnS2)[73~77]、硒化物(如FeSe2)[78]和磷化物(如SnP3和Sn4P3)[79, 80]. 在放电过程中, 氧化物通常发生转换反应; 硫化物和硒化物一般先发生嵌插反应, 然后进行转换反应; 磷化物主要发生转换反应和合金化反应. 在金属化合物材料中, 铁基氧化物由于铁资源丰富、比容量较高等特点, 在低成本、高容量钠离子电池中具有应用前景. 制备碳/铁基氧化物纳米材料是当前的研究重点. 这种材料不仅可以缓解体积变化, 还可以加强反应动力学, 从而提高材料的循环性能和倍率性能. 据报道, Fe2O3/C纳米球在0.2和8 A·g-1条件下的可逆容量分别为740和317 mAh·g-1, 在2 A·g-1循环1400圈后的可逆容量保持在358 mAh· g-1 [71]. MnFe2O4/C纳米纤维在电流密度为0.1和10 A· g-1时的可逆容量分别为504和305 mAh·g-1, 循环4200圈后的容量保持率接近90%[72].
总的来说, 钠离子电池负极材料的比容量很高, 在嵌脱钠过程中体积变化严重, 循环稳定性较差. 制备多孔的碳基纳米复合物是负极材料领域的研究热点. 常见的碳基纳米复合物结构有由喷雾热解法制备的多孔纳米球和由静电纺丝技术合成的多孔纳米纤维.
4 电解液
电解液由钠盐、有机溶剂和功能添加剂组成, 在钠离子电池中起着传输离子的作用. 可用的钠盐主要有高氯酸钠(NaClO4)、六氟磷酸钠(NaPF6)、三氟甲基磺酸钠(NaCF3SO3)和双三氟甲基磺酰亚胺钠[NaN(SO2CF3)2, NaTFSI]. 常见的有机溶剂(图 3)包括酯类小分子化合物如碳酸乙烯酯(EC)、碳酸丙烯酯(PC)、碳酸二甲酯(DMC)、碳酸甲乙酯(EMC)、碳酸二乙酯(DEC)和甲基磺酸乙酯(EMS), 醚类小分子化合物如四甘醇二甲醚(TGM)、二甘醇二甲醚(DGM)和乙二醇二甲醚(DME);功能添加剂主要有氟代碳酸乙烯酯(FEC). 根据有机溶剂的类型不同, 可以将电解液分为酯类电解液和醚类电解液两部分.
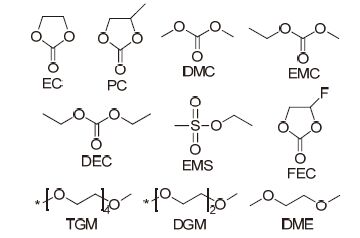 图 3
常见有机溶剂和功能添加剂的分子结构
Figure 3.
Molecular structures of selected organic solvents and functional additives
图 3
常见有机溶剂和功能添加剂的分子结构
Figure 3.
Molecular structures of selected organic solvents and functional additives
4.1 酯类电解液
酯类电解液具有种类多、热稳定性好、离子电导率高(约10-2mS·cm-1)、电化学窗口宽(0.2~5.0 V vs. Na+/Na)等特点[81], 广泛应用于钠离子电池研究. 对于碳酸酯类电解液, 溶剂组分对其离子导电性和电化学稳定性有重要影响, 优化溶剂组分是改善电解液性能最重要的手段之一. 增大介电常数、降低粘度和提高离子迁移率都有利于加强电解液的导电性. 但是, 电化学稳定性与溶剂分子没有严格的依赖关系, 是电解液各组分协同作用的结果. 相对来说, 钠盐类型对电解液性能的影响较小. EC:PC基电解液由于离子电导率高、电化学窗口宽, 被认为是目前最优的碳酸酯类电解液. 另外, 磺酸酯类电解液(如NaClO4/EMS)由于起始氧化电位高(5.6 V vs. Na+/Na), 近年来在正极材料研究中也逐渐得到使用[19, 21]. 值得注意的是, 电解液容易在高电位正极发生氧化分解反应、在低电位负极发生还原分解反应. 分解产物覆盖在电极材料表面会增加电池的电化学阻抗, 降低电池寿命. 使用功能添加剂在电极表面构建一薄层能传导离子和绝缘电子的、性质稳定的固态电解质膜(SEI), 可以有效抑制电解液副反应的发生. FEC是目前钠离子电池研究中最常见的添加剂, 使用量一般为2%~5%(质量分数).
4.2 醚类电解液
醚类电解液的电化学窗口比较窄, 起始氧化电位比较低, 在高电位正极材料和高电压钠离子电池中应用很少. 但是, 它对改善有机正极材料、硫族化合物以及石墨负极材料的电化学性能有明显效果. 有研究表明: 石墨类电极材料在碳酸酯电解液中几乎没有储钠活性, 但是在醚类电解液中却能表现出100~120 mAh·g-1的储钠容量, 并且可以稳定循环1000圈以上[55~57]. 这主要是因为醚类小分子可以与Na+离子在石墨烯层间进行可逆共嵌[54]. 醚类分子越小, 共嵌电位越低. 本课题组报道了蒽醌化合物在醚类电解液中的可逆电化学行为, 发现: 优化电解液浓度、醚类分子类型和添加剂可以显著提高有机电极材料的可逆容量、循环稳定性和倍率性能[82, 83]. 另外, 硫化物和硒化物也能在醚类电解液中展现出稳定的储钠性能[78, 84].
尽管酯类电解液和醚类电解液在优化材料储钠性能方面取得了一定进展, 但是缺乏系统地研究, 作用机理尚不明确. 通过理论计算研究溶剂分子与Na+离子在不同材料体系的“共嵌”行为和利用实验技术分析电解液体系对电极界面性质的影响, 对于电解液优化和电池性能提高至关重要.
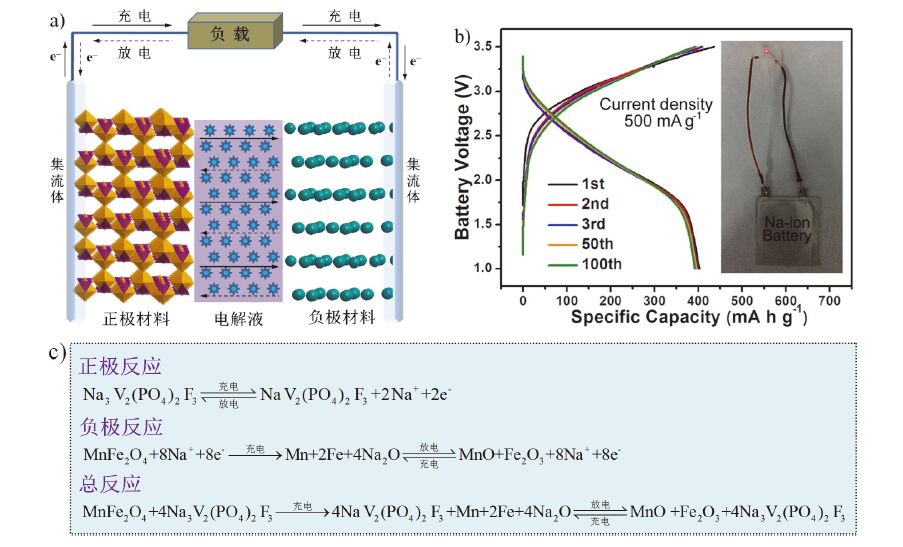
在全电池方面, 随着储钠电极材料和功能电解液的发展(见表 2), 钠离子电池已经取得了一定进展. 据报道, 以Na0.66Ni0.17Co0.17Ti0.66O2为对称电极、1 mol/L NaClO4/PC+2%(体积分数) FEC为电解液组装的钠离子电池可以在电流密度为20 mA·g-1时输出84 mAh·g-1的容量(按负极材料质量计算)和3.1 V的电压, 循环100圈后的容量保持率为95%[53]. 以NaNi1/4Fe1/2Mn1/4O2为正极、Fe3O4为负极、1 mol/L NaClO4/EMS+2%(体积分数) FEC为电解液组装的钠离子电池可以在电流密度为13 mA·g-1时输出130 mAh·g-1的容量(按正极材料质量计算)和2.4 V的电压, 循环150圈后的容量保持率大于76%[19]. 以Na[Ni0.60Co0.05Mn0.35]O2为正极、硬碳为负极、0.5 mol/L NaPF6/EMS+2%(体积分数) FEC为电解液组装的钠离子电池可以在电流密度为75 mA·g-1时输出143 mAh·g-1的容量(按正极材料质量算)和2.7 V的电压, 循环300圈后放电容量还能保持80%[21]. 最近, 本课题组以Na3V2(PO4)2F3为正极、MnFe2O4/C为负极、1 mol/L NaClO4/PC+5%(质量分数) FEC为电解液组装出了能量密度为77.8 Wh·kg-1的软包电池(见图 4). 循环100圈后能量保持率高达96.5%[72]. 目前, 比能量低是限制钠离子电池实用化进程的主要问题. 优化电池组态及材料配比可以有效提高电池的比能量.
表 2
代表性储钠电极材料的实际可逆容量和循环稳定性
Table 2.
Practical capacities and cycling stability of representative Na-storage materials
Na[Ni0.4Fe0.2Mn0.2Ti0.2]O2[20] 1 mol/L NaClO4/PC 145 (2.0~4.2 V) 83% (0.1 C, 200圈) Na[Ni0.60Co0.05Mn0.35]O2[21] 0.5 mol/L NaPF6/EMS 157 (1.5~4.1 V) 84% (0.1 C, 100圈) Na3(VO)2(PO4)2F[38] 1 mol/L NaPF6/EC-DEC 120 (2.5~4.6 V) 91% (0.1 C, 200圈) Na3(VO0.5)2(PO4)2F2[37] 1 mol/L NaClO4/PC-EC 100 (2.0~4.5 V) 92% (1 C, 200圈) NaFePO4[39] 1 mol/L NaPF6/EC-DEC 152 (1.5~4.0 V) 95% (0.1 C, 300圈) Na3V2(PO4)3[49] 1 mol/L NaPF6/EC-DEC 110 (2.7~4.0 V) 93% (1 C, 300圈) 负极材料 硬碳[63] 1 mol/L NaClO4/PC 290 (0.01~2.0 V) 92% (25 mA·g-1, 50圈) P/C[68] 1 mol/L NaClO4/EC-DEC 1229 (0.0~2.0 V) 92.3% (0.25 A·g-1, 150圈) Sn/C[69] 1 mol/L NaClO4/PC 1002 (0.01~2.0 V) 90% (2 A·g-1, 1300圈) Fe2O3/C[71] 1 mol/L NaClO4/EC-DEC 993 (0.04~3.0 V) 74.5% (0.2 A·g-1, 200圈) MnFe2O4/C[72] 1 mol/L NaClO4/PC 755 (0.01~3.0 V) 90% (2 A·g-1, 4200圈) MoS2/C[76] 1 mol/L NaClO4/EC-DMC 716 (0.05~3.0 V) 79.0% (1 A·g-1, 2500圈) SnS2/C[73] 1 mol/L NaClO4/EC-DEC 630 (0.05~3.0 V) 79.3% (1 A·g-1, 400圈) 表 2 代表性储钠电极材料的实际可逆容量和循环稳定性
Table 2. Practical capacities and cycling stability of representative Na-storage materials5 结论与展望
钠离子电池因钠资源储量丰富、成本低廉等优势已在新型二次电池领域受到越来越多的关注. 为了开发比能量大、循环寿命长、倍率性能佳的钠离子电池, 人们致力于研究比容量大、工作电位高、结构稳定的储钠正极材料和比容量大、工作电位低、循环稳定的储钠负极材料以及离子电导率高、电化学窗口宽的功能电解液.目前, 比能量低是制约钠离子电池实用化进程的主要因素. 亟需解决的问题有: 一、正极材料的比容量低, 限制了电池的比容量. 通过本体掺杂和表面改性技术制备结构稳定的高容量氧化物正极材料是提高电池比能量的有效途径; 二、负极材料的首次库仑效率低、电压滞后现象严重, 降低了电池的能量效率. 通过人造SEI膜或预钠化可以有效抑制副反应的发生, 从而提高首次库仑效率. 通过构建碳基纳米复合材料改善材料的反应动力学, 提高反应的可逆性, 从而缓解库电压滞后问题; 三、电解液的电化学窗口比较窄, 限制了电池的工作电压.通过优化电解液组分和设计添加剂功能分子, 拓宽电解液的电化学窗口, 构建高电压钠离子电池. 随着比能量的提高, 钠离子电池有望在新能源储存与转换领域得到广泛应用.
-
-
[1]
Xiang, X. D.; Zhang, K.; Chen, J. Adv. Mater. 2015, 27, 5343.
-
[2]
Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Chem. Rev. 2014, 114, 11636.
-
[3]
Li, H.; Wu, C.; Wu, F.; Bai, Y. Acta Chim. Sinica 2014, 72, 21. (李慧, 吴川, 吴锋, 白莹, 化学学报, 2014, 72, 21.)
-
[4]
Pan, H. L.; Hu, Y. S.; Chen, L. Q. Energy Environ. Sci. 2013, 6, 2338. doi: 10.1039/c3ee40847g
-
[5]
Zhang, K.; Han, X.; Hu, Z.; Zhang, X.; Tao, Z.; Chen, J. Chem. Soc. Rev. 2015, 44, 699. doi: 10.1039/C4CS00218K
-
[6]
Gutierrez, A.; Benedek, N. A.; Manthiram, A. Chem. Mater. 2013, 25, 4010.
-
[7]
Han, M. H.; Gonzalo, E.; Singh, G.; Rojo, T. Energy Environ. Sci. 2015, 8, 81.
-
[8]
Goodenough, J. B.; Kim, Y. Chem. Mater. 2010, 22, 587.
-
[9]
Xu, K. Chem. Rev. 2014, 114, 11503.
-
[10]
Zhang, K.; Hu, Z.; Tao, Z.; Chen, J. Sci. Chin. Mater. 2014, 57, 42. doi: 10.1007/s40843-014-0006-0
-
[11]
Ma, X. H.; Chen, H. L.; Ceder, G. J. Electrochem. Soc. 2011, 158, A1307.
-
[12]
Yabuuchi, N.; Yoshida, H.; Komaba, S. Electrochemistry 2012, 80, 716.
-
[13]
Han, M. H.; Gonzalo, E.; Casas-Cabanas, M.; Rojo, T. J. Power Sources 2014, 258, 266. doi: 10.1016/j.jpowsour.2014.02.048
-
[14]
Vassilaras, P.; Ma, X.; Li, X.; Ceder, G. J. Electrochem. Soc. 2012, 160, A207.
-
[15]
Kubota, K.; Ikeuchi, I.; Nakayama, T.; Takei, C.; Yabuuchi, N.; Shiiba, H.; Nakayama, M.; Komaba, S. J. Phys. Chem. C 2015, 119, 166. doi: 10.1021/jp5105888
-
[16]
Zhao, J.; Zhao, L.; Dimov, N.; Okada, S.; Nishida, T. J. Electrochem. Soc. 2013, 160, A3077.
-
[17]
Yuan, D. D.; Liang, X. M.; Wu, L.; Cao, Y.; Ai, X. C.; Feng, J.; Yang, H. Adv. Mater. 2014, 26, 6301.
-
[18]
Sathiya, M.; Hemalatha, K.; Ramesha, K.; Tarascon, J. M.; Prakash, A. S. Chem. Mater. 2012, 24, 1846. doi: 10.1021/cm300466b
-
[19]
Oh, S. M.; Myung, S. T.; Yoon, C. S.; Lu, J.; Hassoun, J.; Scrosati, B.; Amine, K.; Sun, Y. K. Nano Lett. 2014, 14, 1620. doi: 10.1021/nl500077v
-
[20]
Sun, X.; Jin, Y.; Zhang, C. Y.; Wen, J. W.; Shao, Y.; Zang, Y.; Chen, C. H. J. Mater. Chem. A 2014, 2, 17268. doi: 10.1039/C4TA03828B
-
[21]
Hwang, J. Y.; Oh, S. M.; Myung, S. T.; Chung, K. Y.; Belharouak, I.; Sun, Y. K. Nat. Commun. 2015, 6, 6865. doi: 10.1038/ncomms7865
-
[22]
Guo, S. H.; Liu, P.; Yu, H. J.; Zhu, Y. B.; Chen, M. W.; Ishida, M.; Zhou, H. S. Angew. Chem. Int. Ed. 2015, 54, 5894. doi: 10.1002/anie.201411788
-
[23]
Yu, C. Y.; Park, J. S.; Jung, H. G.; Chung, K. Y.; Aurbach, D.; Sun, Y. K.; Myung, S. T. Energy Environ. Sci. 2015, 8, 2019. doi: 10.1039/C5EE00695C
-
[24]
Zhan, P.; Wang, S.; Yuan, Y.; Jiao, K. L.; Jiao, S. Q. J. Electrochem. Soc. 2015, 162, A1028.
-
[25]
Wang, Y.; Liu, J.; Lee, B.; Qiao, R.; Yang, Z.; Xu, S.; Yu, X.; Gu, L.; Hu, Y. S.; Yang, W.; Kang, K.; Li, H.; Yang, X. Q.; Chen, L.; Huang, X. Nat. Commun. 2015, 6, 6401.
-
[26]
Guo, S. H.; Yu, H. J.; Liu, D. Q.; Tian, W.; Liu, X. Z.; Hanada, N.; Ishida, M.; Zhou, H. S. Chem. Commun. 2014, 50, 7998. doi: 10.1039/c4cc02362e
-
[27]
Park, Y. U.; Seo, D. H.; Kim, H.; Kim, J.; Lee, S.; Kim, B.; Kang, K. Adv. Funct. Mater. 2014, 24, 4603. doi: 10.1002/adfm.201400561
-
[28]
Zhao, J. M.; Mu, L. Q.; Qi, Y. R.; Hu, Y. S.; Liu, H. Z.; Dai, S. Chem. Commun. 2015, 51, 7160.
-
[29]
Qi, Y.; Mu, L.; Zhao, J.; Hu, Y. S.; Liu, H.; Dai, S. Angew. Chem. Int. Ed. 2015, 54, 9911. doi: 10.1002/anie.201503188
-
[30]
Shakoor, R. A.; Seo, D. H.; Kim, H.; Park, Y. U.; Kim, J.; Kim, S. W.; Gwon, H.; Lee, S.; Kang, K. J. Mater. Chem. 2012, 22, 20535. doi: 10.1039/c2jm33862a
-
[31]
Peng, M. H.; Li, B. A.; Yan, H. J.; Zhang, D. T.; Wang, X. Y.; Xia, D. G.; Guo, G. S. Angew. Chem. Int. Ed. 2015, 54, 6452. doi: 10.1002/anie.201411917
-
[32]
Sharma, N.; Serras, P.; Palomares, V.; Brand, H. E. A.; Alonso, J.; Kubiak, P.; Fdez-Gubieda, M. L.; Rojo, T. Chem. Mater. 2014, 26, 3391.
-
[33]
Serras, P.; Palomares, V.; Rojo, T.; Brand, H. E. A.; Sharma, N. J. Mater. Chem. A 2014, 2, 7766. doi: 10.1039/c4ta00773e
-
[34]
Park, Y. U.; Seo, D. H.; Kwon, H. S.; Kim, B.; Kim, J.; Kim, H.; Kim, I.; Yoo, H. I.; Kang, K. J. Am. Chem. Soc. 2013, 135, 13870. doi: 10.1021/ja406016j
-
[35]
Liu, Q.; Wang, D. X.; Yang, X.; Chen, N.; Wang, C. Z.; Bie, X. F.; Wei, Y. J.; Chen, G.; Du, F. J. Mater. Chem. A 2015, 3, 21478. doi: 10.1039/C5TA05939A
-
[36]
Jin, H.; Dong, J.; Uchaker, E.; Zhang, Q.; Zhou, X.; Hou, S.; Li, J.; Cao, G. J. Mater. Chem. A 2015, 3, 17563. doi: 10.1039/C5TA03164H
-
[37]
Xiang, X. D.; Lu, Q. Q.; Han, M.; Chen, J. Chem. Commun. 2016, 52, 3653.
-
[38]
Xu, M. W.; Wang, L.; Zhao, X.; Song, J.; Xie, H.; Lu, Y. H.; Goodenough, J. B. Phys. Chem. Chem. Phys. 2013, 15, 13032. doi: 10.1039/c3cp52408f
-
[39]
Li, C.; Miao, X.; Chu, W.; Wu, P.; Tong, D. G. J. Mater. Chem. A 2015, 3, 8265. doi: 10.1039/C5TA01191D
-
[40]
Kim, J.; Seo, D. H.; Kim, H.; Park, I.; Yoo, J. K.; Jung, S. K.; Park, Y. U.; Goddard, W. A.; Kang, K. Energy Environ. Sci. 2015, 8, 540.
-
[41]
Oh, S. M.; Myung, S. T.; Hassoun, J.; Scrosati, B.; Sun, Y. K. Electrochem. Commun. 2012, 22, 149. doi: 10.1016/j.elecom.2012.06.014
-
[42]
Jian, Z. L.; Yuan, C. C.; Han, W. Z.; Lu, X.; Gu, L.; Xi, X. K.; Hu, Y. S.; Li, H.; Chen, W.; Chen, D. F.; Ikuhara, Y.; Chen, L. Q. Adv. Funct. Mater. 2014, 24, 4265. doi: 10.1002/adfm.v24.27
-
[43]
Jian, Z. L.; Zhao, L.; Pan, H. L.; Hu, Y. S.; Li, H.; Chen, W.; Chen, L. Q. Electrochem. Commun. 2012, 14, 86. doi: 10.1016/j.elecom.2011.11.009
-
[44]
Xu, Y.; Wei, Q.; Xu, C.; Li, Q.; An, Q.; Zhang, P.; Sheng, J.; Zhou, L.; Mai, L. Adv. Energy Mater. 2016, 6, 1600389. doi: 10.1002/aenm.201600389
-
[45]
Li, S.; Dong, Y. F.; Xu, L.; Xu, X.; He, L.; Mai, L. Q. Adv. Mater. 2014, 26, 3545. doi: 10.1002/adma.v26.21
-
[46]
Jian, Z.; Han, W.; Lu, X.; Yang, H.; Hu, Y. S.; Zhou, J.; Zhou, Z.; Li, J.; Chen, W.; Chen, D.; Chen, L. Adv. Energy Mater. 2013, 3, 156. doi: 10.1002/aenm.v3.2
-
[47]
Duan, W.; Zhu, Z.; Li, H.; Hu, Z.; Zhang, K.; Cheng, F.; Chen, J. J. Mater. Chem. A 2014, 2, 8668. doi: 10.1039/c4ta00106k
-
[48]
Zhu, C.; Song, K. P.; van Aken, P. A.; Maier, J.; Yu, Y. Nano Lett. 2014, 14, 2175. doi: 10.1021/nl500548a
-
[49]
Yang, J.; Han, D. W.; Jo, M. R.; Song, K.; Kim, Y. I.; Chou, S. L.; Liu, H. K.; Kang, Y. M. J. Mater. Chem. A 2015, 3, 1005. doi: 10.1039/C4TA06001F
-
[50]
Wu, D.; Li, X.; Xu, B.; Twu, N.; Liu, L.; Ceder, G. Energy Environ. Sci. 2015, 8, 195.
-
[51]
Wang, Y.; Yu, X.; Xu, S.; Bai, J.; Xiao, R.; Hu, Y. S.; Li, H.; Yang, X. Q.; Chen, L.; Huang, X. Nat. Commun. 2013, 6. 6401
-
[52]
Yu, H. J.; Ren, Y.; Xiao, D. D.; Guo, S. H.; Zhu, Y. B.; Qian, Y. M.; Gu, L.; Zhou, H. S. Angew. Chem. Int. Ed. 2014, 53, 8963. doi: 10.1002/anie.201404549
-
[53]
Guo, S. S.; Liu, P.; Sun, Y.; Zhu, K.; Yi, J.; Chen, M. W.; Ishida, M.; Zhou, H. S. Angew. Chem. Int. Ed. 2015, 54, 11701. doi: 10.1002/anie.201505215
-
[54]
Kim, H.; Hong, J.; Yoon, G.; Kim, H.; Park, K. Y.; Park, M. S.; Yoon, W. S.; Kang, K. Energy Environ. Sci. 2015, 8, 2963.
-
[55]
Kim, H.; Hong, J.; Park, Y. U.; Kim, J.; Hwang, I.; Kang, K. Adv. Funct. Mater. 2015, 25, 534. doi: 10.1002/adfm.v25.4
-
[56]
Jache, B.; Adelhelm, P. Angew. Chem. Int. Ed. 2014, 53, 10169. doi: 10.1002/anie.201403734
-
[57]
Zhu, Z. Q.; Cheng, F. Y.; Hu, Z.; Niu, Z. Q.; Chen, J. J. Power Sources 2015, 293, 626. doi: 10.1016/j.jpowsour.2015.05.116
-
[58]
Ponrouch, A.; Goñi, A. R.; Palacín, M. R. Electrochem. Commun. 2013, 27, 85. doi: 10.1016/j.elecom.2012.10.038
-
[59]
Bommier, C.; Luo, W.; Gao, W.-Y.; Greaney, A.; Ma, S.; Ji, X. Carbon 2014, 76, 165.
-
[60]
Zhou, X. S.; Guo, Y. G. ChemElectroChem 2014, 1, 83. doi: 10.1002/celc.201300071
-
[61]
Wen, Y.; He, K.; Zhu, Y. J.; Han, F. D.; Xu, Y. H.; Matsuda, I.; Ishii, Y.; Cumings, J.; Wang, C. S. Nat. Commun. 2014, 5, 4033.
-
[62]
Bommier, C.; Surta, T. W.; Dolgos, M.; Ji, X. L. Nano Lett. 2015, 15, 5888. doi: 10.1021/acs.nanolett.5b01969
-
[63]
Dahbi, M.; Yabuuchi, N.; Kubota, K.; Tokiwa, K.; Komaba, S. Phys. Chem. Chem. Phys. 2014, 16, 15007. doi: 10.1039/c4cp00826j
-
[64]
Qian, J. F.; Wu, X. Y.; Cao, Y. L.; Ai, X. P.; Yang, H. X. Angew. Chem. Int. Ed. 2013, 52, 4633. doi: 10.1002/anie.201209689
-
[65]
Liu, Y. C.; Zhang, N.; Jiao, L. F.; Tao, Z. L.; Chen, J. Adv. Funct. Mater. 2015, 25, 214. doi: 10.1002/adfm.v25.2
-
[66]
Zhang, N.; Liu, Y. C.; Lu, Y. Y.; Han, X. P.; Cheng, F. Y.; Chen, J. Nano Res. 2015, 8, 3384.
-
[67]
Wu, L.; Lu, H. Y.; Xiao, L. F.; Ai, X. P.; Yang, H. X.; Cao, Y. L. J. Mater. Chem. A 2015, 3, 5708. doi: 10.1039/C4TA06086E
-
[68]
Pei, L. K.; Zhao, Q.; Chen, C. C.; Liang, J.; Chen, J. ChemElectroChem 2015, 2, 1652.
-
[69]
Liu, Y.; Zhang, N.; Jiao, L.; Chen, J. Adv. Mater. 2015, 27, 6702.
-
[70]
Lu, Y. Y.; Zhang, N.; Zhao, Q.; Liang, J.; Chen, J. Nanoscale 2015, 7, 2770.
-
[71]
Zhang, N.; Han, X.; Liu, Y.; Hu, X.; Zhao, Q.; Chen, J. Adv. Energy Mater. 2015, 5, 1401123. doi: 10.1002/aenm.201401123
-
[72]
Liu, Y.; Zhang, N.; Yu, C.; Jiao, L.; Chen, J. Nano Lett. 2016, 16, 3321.
-
[73]
Qu, B. H.; Ma, C. Z.; Ji, G.; Xu, C. H.; Xu, J.; Meng, Y. S.; Wang, T. H.; Lee, J. Y. Adv. Mater. 2014, 26, 3854. doi: 10.1002/adma.201306314
-
[74]
Zhang, Y. D.; Zhu, P. Y.; Huang, L. L.; Xie, J.; Zhang, S. C.; Cao, G. S.; Zhao, X. B. Adv. Funct. Mater. 2015, 25, 481. doi: 10.1002/adfm.201402833
-
[75]
Zhu, C.; Mu, X.; van Aken, P. A.; Yu, Y.; Maier, J. Angew. Chem. Int. Ed. 2014, 53, 2152. doi: 10.1002/anie.201308354
-
[76]
Lu, Y.; Zhao, Q.; Zhang, N.; Lei, K.; Li, F.; Chen, J. Adv. Funct. Mater. 2016, 26, 911. doi: 10.1002/adfm.v26.6
-
[77]
Yu, D. Y. W.; Prikhodchenko, P. V.; Mason, C. W.; Batabyal, S. K.; Gun, J.; Sladkevich, S.; Medvedev, A. G.; Lev, O. Nat. Commun. 2013, 4, 2922.
-
[78]
Zhang, K.; Hu, Z.; Liu, X.; Tao, Z.; Chen, J. Adv. Mater. 2015, 27, 3305.
-
[79]
Fan, X.; Mao, J.; Zhu, Y.; Luo, C.; Suo, L.; Gao, T.; Han, F.; Liou, S. C.; Wang, C. Adv. Energy Mater. 2015, 5, 1500174. doi: 10.1002/aenm.201500174
-
[80]
Qian, J.; Xiong, Y.; Cao, Y.; Ai, X.; Yang, H. Nano Lett. 2014, 14, 1865.
-
[81]
Ponrouch, A.; Marchante, E.; Courty, M.; Tarascon, J. M.; Palacín, M. R. Energy Environ. Sci. 2012, 5, 8572. doi: 10.1039/c2ee22258b
-
[82]
Guo, C.; Zhang, K.; Zhao, Q.; Pei, L.; Chen, J. Chem. Commun. 2015, 51, 10244.
-
[83]
Zhang, K.; Guo, C.; Zhao, Q.; Niu, Z.; Chen, J. Adv. Sci. 2015, 2, 1500018.
-
[84]
Hu, Z.; Zhu, Z. Q.; Cheng, F. Y.; Zhang, K.; Wang, J. B.; Chen, C. C.; Chen, J. Energy Environ. Sci. 2015, 8, 1309.
-
[1]
-


图 2 多孔Sn/C纳米纤维的SEM (a)、TEM (b~c)和充放电曲线(d插图是放电产物Na15Sn4与充电产物Sn的HRTEM)[69], 以及Sn (e)和Na15Sn4 (f)的晶胞结构
Figure 2 SEM (a), TEM (b~c) and charge/discharge curves (d, the inset is the HRTEM images of fully discharged product Na15Sn4 and fully charged product Sn) of porous Sn/C nanofibers[69], and crystal structures of Sn (e) and Na15Sn4 (f)

图 3 常见有机溶剂和功能添加剂的分子结构
Figure 3 Molecular structures of selected organic solvents and functional additives

表 1 代表性正极材料[NaCrO2、Na3(VO)2(PO4)2F、Na3V2(PO4)2F3、NaFePO4和Na3V2(PO4)3]和负极材料(石墨、硬碳、Sn、P和Fe2O3)的分子量、反应机理、理论容量和平均工作电位
Table 1. Molecular weights, reaction mechanism, theoretical capacities, and average operating potentials of representative cathode materials [NaCrO2, Na3(VO)2(PO4)2F, Na3V2(PO4)2F3, NaFePO4, and Na3V2(PO4)3] and anode materials (graphite, hard carbon, Sn, P, and Fe2O3)
Na3(VO)2(PO4)2F 411.9 Na3(VO)2(PO4)2F↔Na(VO)2(PO4)2F+2Na++2e- 130 ca. 3.8 Na3V2(PO4)2F3 417.9 Na3V2(PO4)2F3↔NaV2(PO4)2F3+2Na++2e- 128 ca. 3.9 NaFePO4 173.8 NaFePO4↔FePO4+Na++e- 154 ca. 2.6 Na3V2(PO4)3 455.9 Na3V2(PO4)3↔NaV2(PO4)3+2Na++2e- 117 ca. 3.4 负极材料 石墨(C) 12 20C+Na++e-↔NaC20 (醚类溶剂) 112 ca. 0.8 硬碳(C) 12 6C+Na++e-↔NaC6 375 ca. 0.2 P 31.0 P+3Na++3e-↔Na3P 2594 ca. 0.5 Sn 118.7 4Sn+15Na++15e-↔Na15Sn4 847 ca. 0.3  下载: 导出CSV
下载: 导出CSV
表 2 代表性储钠电极材料的实际可逆容量和循环稳定性
Table 2. Practical capacities and cycling stability of representative Na-storage materials
Na[Ni0.4Fe0.2Mn0.2Ti0.2]O2[20] 1 mol/L NaClO4/PC 145 (2.0~4.2 V) 83% (0.1 C, 200圈) Na[Ni0.60Co0.05Mn0.35]O2[21] 0.5 mol/L NaPF6/EMS 157 (1.5~4.1 V) 84% (0.1 C, 100圈) Na3(VO)2(PO4)2F[38] 1 mol/L NaPF6/EC-DEC 120 (2.5~4.6 V) 91% (0.1 C, 200圈) Na3(VO0.5)2(PO4)2F2[37] 1 mol/L NaClO4/PC-EC 100 (2.0~4.5 V) 92% (1 C, 200圈) NaFePO4[39] 1 mol/L NaPF6/EC-DEC 152 (1.5~4.0 V) 95% (0.1 C, 300圈) Na3V2(PO4)3[49] 1 mol/L NaPF6/EC-DEC 110 (2.7~4.0 V) 93% (1 C, 300圈) 负极材料 硬碳[63] 1 mol/L NaClO4/PC 290 (0.01~2.0 V) 92% (25 mA·g-1, 50圈) P/C[68] 1 mol/L NaClO4/EC-DEC 1229 (0.0~2.0 V) 92.3% (0.25 A·g-1, 150圈) Sn/C[69] 1 mol/L NaClO4/PC 1002 (0.01~2.0 V) 90% (2 A·g-1, 1300圈) Fe2O3/C[71] 1 mol/L NaClO4/EC-DEC 993 (0.04~3.0 V) 74.5% (0.2 A·g-1, 200圈) MnFe2O4/C[72] 1 mol/L NaClO4/PC 755 (0.01~3.0 V) 90% (2 A·g-1, 4200圈) MoS2/C[76] 1 mol/L NaClO4/EC-DMC 716 (0.05~3.0 V) 79.0% (1 A·g-1, 2500圈) SnS2/C[73] 1 mol/L NaClO4/EC-DEC 630 (0.05~3.0 V) 79.3% (1 A·g-1, 400圈)
下载: 导出CSV
-





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 141
- 文章访问数: 7142
- HTML全文浏览量: 2384
 下载:
下载:
