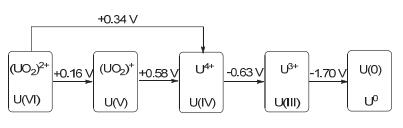
 图 1
铀的氧化还原电位(vs.标准氢电极pH=1的HClO4溶液, 25 ℃)
Figure 1.
Oxidation-reduction potentials of uranium (vs. SHE in pH=1 HClO4 at 298 K)
图 1
铀的氧化还原电位(vs.标准氢电极pH=1的HClO4溶液, 25 ℃)
Figure 1.
Oxidation-reduction potentials of uranium (vs. SHE in pH=1 HClO4 at 298 K)
引用本文:
岳国宗, 高瑞, 赵鹏翔, 褚明福, 帅茂兵. 三价铀有机络合物对小分子气体的活化研究[J]. 化学学报,
2016, 74(8): 657-663.
doi:
10.6023/A16050260

Citation: Yue Guozong, Gao Rui, Zhao Pengxiang, Chu Mingfu, Shuai Maobing. Trivalent Uranium Complex in Small Molecules Activation[J]. Acta Chimica Sinica, 2016, 74(8): 657-663. doi: 10.6023/A16050260
Citation: Yue Guozong, Gao Rui, Zhao Pengxiang, Chu Mingfu, Shuai Maobing. Trivalent Uranium Complex in Small Molecules Activation[J]. Acta Chimica Sinica, 2016, 74(8): 657-663. doi: 10.6023/A16050260
三价铀有机络合物对小分子气体的活化研究
摘要:
铀是典型的锕系元素,具有高度极化性,可以利用5f轨道与相应配体进行配位.由三价铀化合物参与的典型反应类型主要有:迁移插入、σ键复分解反应和氧化还原反应等.这为研究和发展一系列结构新颖、反应独特的有机铀络合物和材料提供了重要的基础.在过去的20年中,化学家们发现三价铀有机络合物具有可以活化气体小分子的反应特点,由于其重要的科学研究意义及工业应用价值,该领域迅速得到了科学界的广泛关注和发展.工作主要针对三价铀有机络合物对一些小分子气体(N2,CO,CO2)的活化研究做一简要概述.
-
关键词:
- 锕系化学
- / 三价铀络合物
- / 配位化学
- / N2、CO、CO2的活化
English
Trivalent Uranium Complex in Small Molecules Activation
Abstract:
Uranium, one of typical actinide elements, has strong polarizing property. Using 5f orbitals for bonding with ligands, uranium(III) compounds have some unique reactivities including: migratory insertion, σ-bond metathesis and redox activity etc., which provides researchers with good opportunities to obtain organic uranium complexes or materials with unique structures and reactional properties. In the last 20 years, it has been found that trivalent uranium organic complexes exhibit a wide variety of activation towards small molecules. Due to the significant research and potential industrial value, this field has been well developed in recent years. Some research results for small molecules (such as N2, CO, CO2) activation promoted by trivalent uranium complexes were summarized in the paper.
-
1 引言
得益于锕系元素蕴藏着的丰富宝藏, 越来越多的科学家投入到核科学技术领域并开展相关的研究工作[1, 2].对含铀化合物物理化学性质的深入研究不仅可以为开发新型核材料、发展先进的乏燃料储存及处理技术提供帮助, 而且有助于深入认识铀的物理化学行为以及预测其他锕系元素性质.因此, 开展对含铀元素化合物的性质研究具有非常重要的意义.
铀相比于其他金属元素具有许多独特的性质.首先, 铀原子具有较大的原子半径, 其f轨道也可以参与到共价键的形成, 配位数目多, 例如四价铀的配位数可以达到12个甚至更高.其次, 一些含铀化合物具有亲电特征, 涉及的反应包括:迁移插入、σ键复分解反应、氧化还原反应等, 对部分小分子气体如N2、CO、CO2等表现出很高的活化能力[3].这些对小分子气体所表现出来的活化性质与镧系金属化合物所表现出的性质形成了鲜明的对比.其原因是镧系元素的4f价键轨道高度收缩, 相应的镧系金属配位化合物中以离子键相互作用为主; 而由于相对论效应导致锕系元素5f轨道能级更易受到晶体场劈裂效应的影响, 相对来说其复合物中共价键作用成分更高; 同时, 6d轨道的贡献也不可忽略.
铀元素价电子层结构为5f36d17s2, 根据电子丢失程度的不同, 可呈现从三价到六价一系列的氧化态.铀的氧化还原电位数据如图 1所示, 根据吉布斯自由能方程, 电极电位大于零, 化学反应的吉布斯自由能变ΔG小于零, 说明反应可以自发进行[4].这也说明了三价铀不稳定, 具有较强的还原性; 而铀(V)和铀(VI)具有较强的氧化性; 同时铀(V)很容易发生歧化反应生成稳定的铀(IV)和铀(VI), 而这两种价态也是铀元素中最为常见的两种氧化态.
图 1
铀的氧化还原电位(vs.标准氢电极pH=1的HClO4溶液, 25 ℃)
Figure 1.
Oxidation-reduction potentials of uranium (vs. SHE in pH=1 HClO4 at 298 K)
铀元素拥有三价到六价一系列的氧化态也为实现含铀化合物的催化循环提供了必备条件[5~7].目前, 科学家对六价铀酰化合物开展的研究较多, 也取得了相当大的进展[8~10].而由于低价铀化合物易被氧化等原因, 低价铀有机络合物的研究起步较晚, 相关的文献报道资料相对较少.近年来, 随着科学家们对锕系元素配位化学研究的日益重视, 低价铀有机配位化合物的研究也越来越受到化学家们的关注[11~13].例如, 四价铀有机配位化合物已被报道出具有一定的催化活性, 对醛、内酯等底物的聚合反应具有一定的催化性能[14, 15]; 同时, 由于铀原子5f轨道的扩展以及+3价氧化态较高的还原能力, 三价铀有机络合物被认为是最有可能实现以催化活化小分子气体为功能导向的候选化合物[16~18], 特别是三价铀有机配位化合物对CO、N2、CO2等小分子气体的活化研究.对这些小分子气体的活化与利用具有重要的科学研究意义及工业应用价值.低价铀配位化合物领域也逐渐成为锕系化学领域的研究热点之一.
2 三价铀有机络合物对小分子气体的活化
2.1 三价铀有机络合物对氮气的配位及活化作用
氨是化肥工业、现代农业和有机化工业的主要原料, 氮气活化制氨为现代社会发展起到了重要的推动作用.由于N≡N键的键能较强, 所以目前使用N2和H2制备氨气需要在比较高的温度和压力条件下进行, 条件较为苛刻, 耗能巨大.因此, 如何在温和的条件下实现对氮气的活化固氮一直是科学家们研究的重点.早期, 科学家们就发现了铀元素化合物对惰性气体分子N2具有一定的还原活化能力, Haber-Bosch工艺流程中所用到的催化剂就含有部分铀元素化合物[19].
首例含N2配体的锕系络合物的合成是由Scott团队[20]于1996年报道的.他们发现将化合物1[U(N3N)] (N3N=N(CH2CH2NSi(t-Bu)Me2)3)暴露于氮气的气氛下可以得到化合物2[{(N3N)U}2(μ, η2, η2-N2)], N2作为配体处于两个三价铀有机络合物中间.经过单晶X射线衍射验证了其结构, 并测得N—N键长为1.109±0.007 Å, 这与自由的N2键长基本一致, 说明该配位物中的N2配体并没有发生实质的活化反应(见图 2).
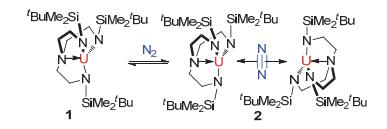 图 2
三价铀有机络合物1与N2形成复合物2
Figure 2.
Synthesis of compound 2 from compound 1
图 2
三价铀有机络合物1与N2形成复合物2
Figure 2.
Synthesis of compound 2 from compound 1
1998年, Cummins教授[21]首次报道了与N2配位的含铀异核双核复合物5[(Ar[t-Bu]N)3U(μ, η1, η1-N2)Mo(N[t-Bu]Ph)3].在甲苯溶液中, 他们使用等量的化合物[(Ar[t-Bu]N)3U(THF)](3)和[Mo(N[t-Bu]Ph)3](4)在N2气氛下反应得到化合物5.由于化合物5中的N—N键长已经变为1.232±0.011 Å, 表明N2分子已经被活化. Mo—N的原子间距为1.773±0.008 Å, 与Mo—N双键键长相一致, 而U—N的距离为2.220±0.009 Å, 表明元素铀才是对N2活化的关键, 见图 3.
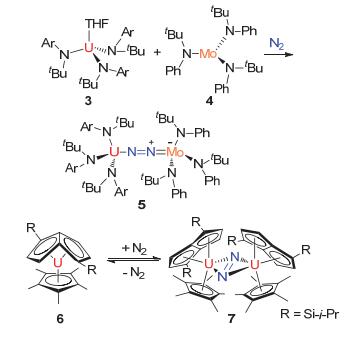 图 3
三价铀有机络合物3和6对N2的活化
Figure 3.
N2activation by trivalent uranium complex 3/6 to obtain compounds 5and7
图 3
三价铀有机络合物3和6对N2的活化
Figure 3.
N2activation by trivalent uranium complex 3/6 to obtain compounds 5and7
2002年, Hitchcock教授[22]制备出了化合物7 [{(Cp*)(η8-1, 4-(Si-i-Pr3)2C8H4)U}2(μ, η2, η2-N2)] (Cp*=η5-C5Me5, i-Pr=-CH(CH3)2).在该化合物中, N—N键长为1.232±0.010 Å, 这与N=N双键的键长基本一致, 表明铀核中心对N2配体有还原作用(见图 3).
化学家们还发现N2也可以以端位的模式进行配位, Evans教授[23]发现金属茂合物[(Cp*)3U] (8)可以与N2分子反应(见图 4), 产物中N2以端位方式进行可逆性配位.形成的化合物[(Cp*)3U(η1-N2)] (9)经过X单晶衍射实验和红外光谱测定, 发现其N—N键长为1.120±0.014 Å, 振动光谱为2207 cm-1, 配体N2并没有发生活化反应.
Arnold教授[24]于2011年发现化合物10uranium(III) tris(aryloxide)可以在一个大气压下与氮气发生配位作用, 如图 4所示, 其配位方式为桥联式的侧面配位, 将两个铀核连接.其N—N键长为1.189(18) Å, 比正常的N2键稍长, 验证了铀核中心对氮气的还原活化.在此前关于N2端位配位方式的研究中, 由于N2配体较弱的σ-donor和π-acceptor性质, 导致金属与N2配体的相互作用较弱, 而Arnold教授的研究表明:化合物11中金属核心的π对称轨道电子密度反馈进入了N2的π*反键轨道, 导致配体N2侧面配位方式中铀核中心对N2具有更强的活化作用, 这也使得形成的化合物更加稳定.
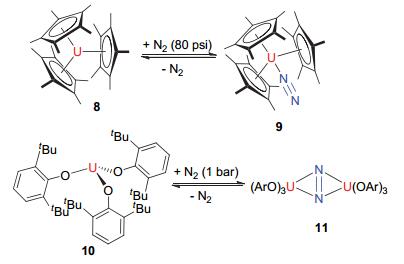 图 4
三价铀有机络合物8和10对N2的活化
Figure 4.
Reaction of trivalent uranium complex 8 and 10 with N2 to give the compounds 9and 11
图 4
三价铀有机络合物8和10对N2的活化
Figure 4.
Reaction of trivalent uranium complex 8 and 10 with N2 to give the compounds 9and 11
2.2 三价铀有机络合物与CO的配位及羰基化作用
自19世纪30年代Fischer-Tropsch流程的发现和使用以来, 科学家们一直在探索通过各种过渡金属化合物活化CO制备石油等化工产品的可行性.目前, 过渡金属化合物活化CO的研究取得了一定的进展[25], 而锕系元素化合物对CO活化反应研究开展较晚; 尽管如此, 目前该领域已经出现了许多优秀的工作.
最早报道含有CO的铀复合物来自于Andersen教授的工作[26].他将三价铀复合物[(C5Me4H)3U]暴露于CO的气氛中即可得到化合物12[(C5Me4H)3U(η1-CO)].该产物的产生可以通过CO在1976 cm-1的红外吸收峰所证实, 当使用13CO气体时, 峰值为1935 cm-1, 使用C18O时, 吸收峰为1932 cm-1.在减压条件下, 该化合物又重新回到了起始原料.在这之后, 与之相似的铀羰基复合物13[(Me5C5)3U(η1-CO)]也被制备分离, 并经过了光谱和X单晶衍射分析.其红外光谱特征明显, 1900 cm-1的C—O振动光谱表明了明显的U—CO π-反馈键. X-单晶衍射显示U—C—O角度为175.2°±0.6°, U—C键长为2.383±0.006 Å, 表明CO与铀核中心有很强的成键作用[27](见图 5). 2005年, Meyer教授[28]的研究证明, 一分子的CO可以被两分子的三价铀化合物还原得到化合物14, 该化合物是通过CO的端位配位的方式将两个铀核中心链接, 通过光谱表征可以判断作为配体的CO已经被还原活化(见图 5).
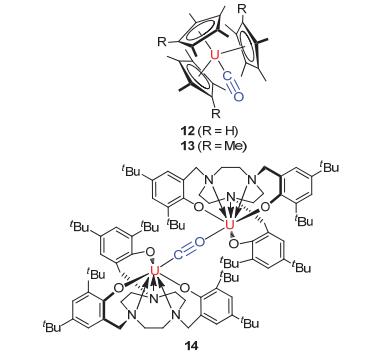 图 5
几种铀有机络合物与CO配位(化合物12、13和14)的结构
Figure 5.
The structure of uranium compounds 12, 13and 14
图 5
几种铀有机络合物与CO配位(化合物12、13和14)的结构
Figure 5.
The structure of uranium compounds 12, 13and 14
紧接着, Cloke等[29, 30]报道了关于三价铀化合物活化CO的重要进展, 见图 6.他们发现, 将暗紫色的化合物15[(η8-COT)(Cp*)U(THF)]在一个大气压下与CO反应可以得到暗红色的双核化合物16[{(η8-COT)(Cp*)U}2(μ, η1, η2-C3O3)], 两分子的三价铀化合物15促使三分子CO之间形成C—C键从而得到平面三元环△核心结构, 新形成的C—O键长处于正常C—O单键和双键键长之间; 三个C—C键中有两个较短[1.379(6) Å]和一个稍长的C—C键[1.436(7) Å], 较长的C—C键与其中一个铀核中心有着明显的相互作用.理论计算表明, 铀核中心不仅可以作为配体CO的还原剂, 还可以作为Lewis酸, 在反应历程中起到了至关重要的作用.同时, 作者通过对有机配体的修饰改变有机配体和含铀络合物的空间位阻, 并进一步制备得到了新颖的平面四方形的双核化合物18, 如图 6所示, 新形成的(C4O4)2-双离子与两个四价铀核中心配位; 这也说明了有机配体对该类反应的选择性具有一定的调控能力.
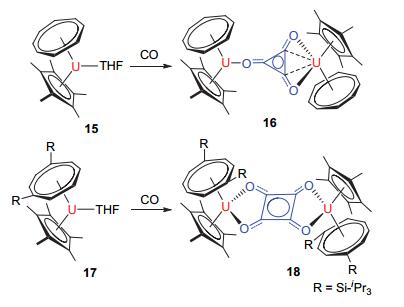 图 6
三价铀有机络合物15和17对CO的活化
Figure 6.
Reaction of 15/17 with CO to give the compounds 16 and 18
图 6
三价铀有机络合物15和17对CO的活化
Figure 6.
Reaction of 15/17 with CO to give the compounds 16 and 18
Arnold教授[31]于2011年发现, 使用简单的三价铀化合物与一个大气压下的CO反应, 就可以获得金黄色的晶体化合物20, 见图 7.作者通过X-单晶衍射确证了其结构, 发现其为乙炔基连接的四价铀复合物.化合物20中的OCCO基团是线性的, 新生成的C—C键长为1.183(7) Å, 确认其为C≡C三键.由于OCCO基团被周围的大位阻基团包围, 阻止了其与额外CO的进一步反应.然而, 通过加热化合物20可以分离得到金黄色的晶体化合物21, 作者通过X-单晶衍射确认了其结构.该化合物的形成是化合物20中的一个三甲基硅基中的C—H键对OCCO基团发生分子内加成而得到的七元铀环产物.
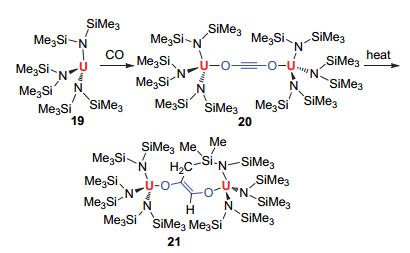 图 7
化合物20和21的合成
Figure 7.
Synthesis of compounds 20 and 21
图 7
化合物20和21的合成
Figure 7.
Synthesis of compounds 20 and 21
2013年, Liddle教授[32]报道了化合物1[U(trenDMBS)]可以选择性地实现常温常压下两分子CO自身的还原偶联得到双核化合物22[{U(trenDMBS)}2(μ, η1, η1-OCCO)] (图 8).化合物22可以与有机硅试剂反应, 以高产率得到官能化产物RMe2SiOCCOSiMe2R (23; R=Ph, Me)和前体化合物24.化合物24可以被回收和重新利用. DFT理论计算结果表明:独特的trenDMBS配体导致了这一不寻常的化学反应.而且, 值得一提的是反应产物23, 25都是工业兆吨级制备二醇和呋喃的重要前体, 这对CO的催化活化循环反应的设计以及实际应用具有重要的指导意义.
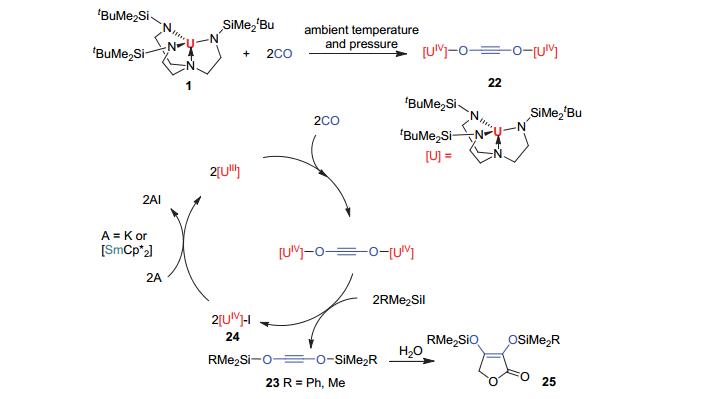 图 8
化合物1对CO的活化和相关官能化反应
Figure 8.
Activation of CO promoted by compound 1
图 8
化合物1对CO的活化和相关官能化反应
Figure 8.
Activation of CO promoted by compound 1
综上可以看到, CO与铀化合物的反应模式是多种多样的.三价铀有机络合物不仅可以活化配位的CO, 还可以在一定程度上对其实现官能化, 表明了三价铀复合物很可能发展成为一类具有CO官能化的功能催化剂.
2.3 三价铀有机络合物对CO2的配位及活化
温室效应不仅是全球气候、环境的问题, 也是涉及到人类社会的生产、消费和生活方式以及社会和经济发展各个领域的重大问题. CO2是温室气体中一种排放量最大的气体, 它主要来源于化石性燃料的燃烧和工业生产排放的废气.由于CO2含量日益增多, 国际上提出了相应的对策以减少CO2的排放量, 科学家们也一直致力于CO2的活化和功能化, 以废物资源化的原则, 开发其潜在的应用价值.
CO2中稳定的C=O双键需要一个高度活化的金属中心来克服其热力学和动力学上的稳定性, 这就增加了CO2的利用难度[33~36].目前, 尽管多数对CO2进行活化的报道采用的是过渡金属[37, 38], 但使用低价铀有机络合物作为催化剂, 通过新颖的反应模式来固定CO2的研究也越来越受到科学家们的关注.
2004年, Meyer团队[39]在Science上发表了关于低价铀有机络合物活化CO2的重要进展.三价铀有机络合物26可以将CO2还原得到四价铀氧桥化合物27[{((t-BuArO)3tacn)U}2(μ-O)], 并释放出CO.使用adamantyl基团替换化合物26中的叔丁基, 即化合物28[((AdArO)3tacn)U] (Ad=1-adamantyl), 将其暴露于CO2气氛下, 可以生成CO2以端位方式配位的含铀复合物29[((AdArO)3tacn)U(η1-OCO)] (见图 9).在该化合物中, CO2以接近线性的形式配位, U—O—C和O—C—O键角分别为171.1°±0.2°和178.0°±0.3°.化合物29中配体CO2的振动谱带出现在2188 cm-1, 相比正常CO2的2349 cm-1出现了明显的偏移, 这表明CO2已被活化.通过单晶X射线衍射实验发现, 配位端的O—C与末端的O—C键长有着明显的差别.配位端的O—C键长仍在C—O双键的正常范围, 为1.122±0.004 Å;而末端的O—C键长较长, 为1.277±0.004 Å, 这进一步验证了CO2的还原.晶体学数据以及光谱数据证实了化合物29是一种电荷分离的铀(IV)物种, U(iv)L·- (L=ligand).这种稳定的电荷分离物种与d区元素的化学性质有着显著的区别, 这对于催化反应中生成的中间体具有非常重要的稳定作用.
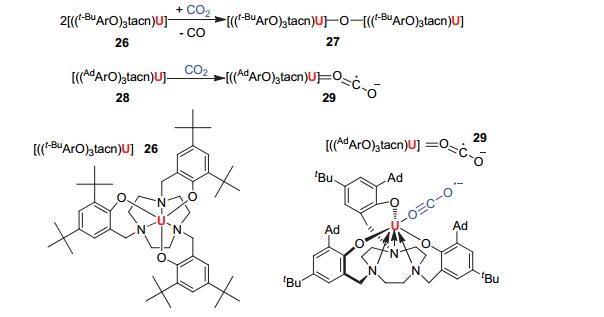 图 9
三价铀有机络合物26和28对CO2的活化
Figure 9.
Reaction of 26/28 with CO2 to give the compounds 27 and 29
图 9
三价铀有机络合物26和28对CO2的活化
Figure 9.
Reaction of 26/28 with CO2 to give the compounds 27 and 29
Meyer团队[40~42]相继报道了一系列的相关研究, 得到了三价铀有机配合物与CO2反应生成化合物30和31(见图 10).对比化合物29与30、31的结构可以看到, 配体在空间位阻上的差别可以改变三价铀化合物与CO2的反应性, 这也就产生了选择性.化合物29中体积庞大的大位阻配体可以稳定端位配位的CO2·-; 而相对较小的配体, 可以进一步形成稳定的碳酸根配位模式的化合物, 例如化合物30、31.
它们的反应途径可能是:首先, 一分子CO2与两分子三价铀化合物反应释放一分子CO后得到氧桥中间体; 然后, 具有高度活性的氧桥中间体再与另一分子的CO2反应而生成最终产物. 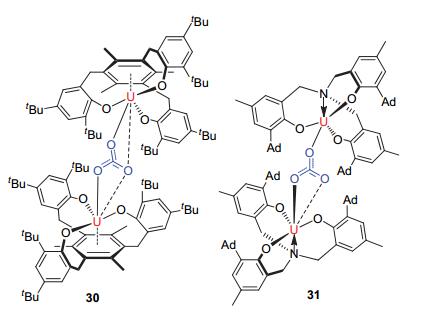 图 10
含铀复合物30和31的结构
Figure 10.
The structures of uranium compounds 30and 31
图 10
含铀复合物30和31的结构
Figure 10.
The structures of uranium compounds 30and 31
此外, 普渡大学的Bart教授[43]报道了三价铀络合物32[U(TpMe)2-(CH2Ph)]可以与CO2反应得到羰基化的四价铀化合物33[U(TpMe)2(a-O2CCH2Ph)](见图 11).向体系加入Me3SiCl或Me3SiI可以得到化合物34Me3Si-OC(O)CH2Ph和化合物31[U(TpMe)2(X)], 而化合物31是制备[U(TpMe)2(CH2Ph)]的前体化合物, 这为实现该类反应的催化循环提供了实验依据, 使得该类活化反应具有一定的工业应用价值.
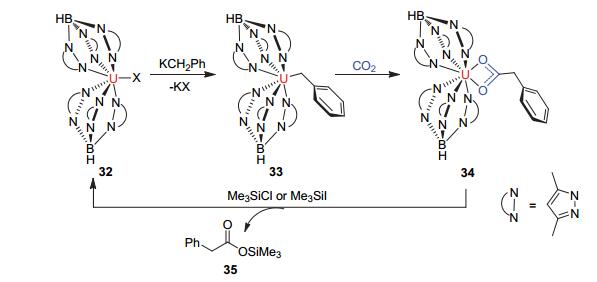 图 11
三价铀有机络合物33对CO2的活化
Figure 11.
Insertion of CO2 into a uranium-benzyl bond to give 34
图 11
三价铀有机络合物33对CO2的活化
Figure 11.
Insertion of CO2 into a uranium-benzyl bond to give 34
最近, Cloke教授[44]制备了一系列三价铀化合物35[U(COTTIPS2)(CpEMe4)] (CpEMe4=EC4Me4, E=N, P, As; COTTIPS2=C8H6{1, 4-SiiPr3}), 并研究了该类化合物的氧化还原性质.该类三价铀化合物可以对CO2进行还原插入反应, 得到氨基甲酸铀类化合物37和膦基甲酸铀类化合物38, 见图 12.这些化合物的结构都得到了单晶X射线衍射的确认.
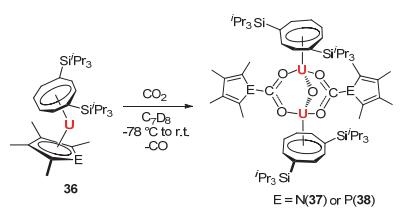 图 12
三价铀有机络合物36对CO2的活化
Figure 12.
Reaction of 36 with CO2 to give the compounds 37 or 38
图 12
三价铀有机络合物36对CO2的活化
Figure 12.
Reaction of 36 with CO2 to give the compounds 37 or 38
3 结论与展望
三价铀化合物的物理化学性质研究是锕系化学领域中重要的课题之一, 其在基础科学、国防科研和商业经济领域中具有重要的价值.本文综述了多种三价铀有机络合物的结构和活化性质:利用元素铀丰富的成键轨道以及多种氧化态的特性, 通过对有机配体位阻和电子效应的微妙修饰, 获取了多种具有催化活化性质的三价铀有机络合物, 并探讨了该类化合物对N2, CO和CO2的活化反应方式.
目前, 国际上在对三价铀有机络合物的制备以及其对小分子气体的活化研究领域已经取得了较大的进展, 国外以实验研究和理论计算并重的方式, 发展和研究了一系列三价铀有机配位化合物.其中大多数的研究工作主要来自美国的国家实验室(洛斯阿拉莫斯国家实验室, 劳伦斯伯克利国家实验室和阿贡国家实验室等)及著名高校(麻省理工大学, 芝加哥大学, 加州大学以及普渡大学等)、英国著名高校(爱丁堡大学, 诺丁汉大学和萨塞克斯大学等)、德国和以色列等一些高校和科研机构.虽然我国的核化学和放射化学研究范围和成果在不断扩展和增加[45], 国内许多课题组在放射化学, 特别是在锕系领域开展了大量有意义的研究工作, 如清华大学的李隽课题组[46], 苏州大学的王殳凹课题组[47], 北京大学的刘春立课题组, 刘文剑课题组[48], 胡少文课题组[49], 北京师范大学的自国甫课题组[50]等, 但是对低价锕系金属有机化学的研究起步较晚.目前国内工作主要以锕系配位化合物的理论计算研究为主.例如, 中国科学院高能物理研究所的柴之芳院士等[51]使用DFT计算方法研究了(Tp*)2UCH2Ph对CO2和CS2的活化机理, 其计算结果表明CO2和CS2对(Tp*)2UCH2Ph的反应机理是分步进行的.首先, CO2或CS2直接插入U—C(benzyl)键, 生成C—C键和U—E(E=O, S).然后, 新生成的Ph2CH2CE2-(E=O, S)通过转动羧基形成第二个U—E键, 进而得到相应的产物. 2014年, 王东琪等[52]研究了CO2对含有NHC卡宾配体的三价铀络合物中U—N键的插入机理.他们通过计算研究提出该反应是通过一个新颖的四步反应机理进行的, 这有效避免了过高的反应能垒, 比前人所提出的机理更加具有说服力; 此外, 近期一篇关于锕系元素计算化学的综述也详细阐述了近年来锕系计算化学的进展[53].
经过二十年的发展, 尽管三价铀有机络合物新颖独特的化学性质已经引起了科学家们的关注, 并取得了较好的进展, 但人们对三价铀有机络合物的物理化学性质的了解还不系统, 不全面, 还有大量的研究工作有待深入开展:如不同配体与三价铀的配位化学行为以及其对活化性质的影响, 三价铀有机络合物对小分子气体活化官能化反应的反应机理研究, 对该类反应的反应活性及选择性规律的总结和完善等.我们认为, 在研究方法上, 应加强实验方面的研究, 并注重实验方法与理论计算相结合, 从分子层次上理解三价铀化合物对小分子的活化根源, 探索反应的机理.
-
-
[1]
石伟群, 赵宇亮, 柴之芳, 化学进展, 2011, 23, 1478.Shi W. Q., Zhao Y. L., Chai Z. F.Prog. Chem., 2011, 23:1478.
-
[2]
袁立永, 石伟群, 蓝建慧, 柴之芳, 科学通报, 2012, 57, 581. doi: 10.1360/972011-2513Yuan Y. L., Shi W. Q., Lan J. H., Chai Z. F.Chin. Sci. Bull., 2012, 57:581. doi: 10.1360/972011-2513
-
[3]
Monreal M. J., Diaconescu P. L.Nat. Chem., 2010, 2:424. doi: 10.1038/nchem.642
-
[4]
Jones C. J.d-and f-Block Chemistry, Polestar Wheatons, Exeter, 2001 p.86.
-
[5]
Ephritikhine M.Dalton Trans. 2006, 2501.
-
[6]
Fox A. R., Bart S. C., Meyer K., Cummins C. C.Nature, 2008, 455:341. doi: 10.1038/nature07372
-
[7]
Liddle S. P.Angew. Chem., Int. Ed., 2015, 54:30.
-
[8]
Fortier S., Hayton T. W.Coord. Chem. Rev., 2010, 254:197. doi: 10.1016/j.ccr.2009.06.003
-
[9]
Altmaier M., Gaona X., Fanghänel T.Chem. Rev., 2013, 113:901. doi: 10.1021/cr300379w
-
[10]
Loiseau. T.; Mihalcea I., Henry N., Volkringer C.Coord. Chem. Rev. 2014, 266~267, 69.
-
[11]
Gardner B. M., Liddle S. T.Eur. J. Inorg. Chem. 2013, 3753.
-
[12]
Jones M. B., Gaunt A. J.Chem. Rev., 2013, 113:1137. doi: 10.1021/cr300198m
-
[13]
Arnold P. L., McMullon M. W., Rieb J., Kuhn F. E.Angew. Chem., Int. Ed., 2014, 53:2. doi: 10.1002/anie.v53.1
-
[14]
Karmel I. S. R., Fridman N., Tamm M., Eisen M. S.J. Am. Chem. Soc., 2014, 136:17180. doi: 10.1021/ja5091436
-
[15]
Karmel I. S. R., Fridman N., Eisen M. S.Organometallics, 2015, 34:636. doi: 10.1021/om501179e
-
[16]
Ossola F., Zanella P., Ugo P., Seeber R.Inorg. Chim. Acta, 1988, 147:123. doi: 10.1016/S0020-1693(00)80640-4
-
[17]
Avens L. R., Barnhart D. M., Burns C. J., McKee S. D., Smith W. H.Inorg. Chem., 1994, 33:4245. doi: 10.1021/ic00097a010
-
[18]
Morris D. E., Re R. E. D., Jantunen K. C., Castro-Rodriguez I., Kiplinger J. L.Organometallics, 2004, 23:5142. doi: 10.1021/om049634v
-
[19]
Haber F.DE 229126, 1909.
-
[20]
Roussel P., Hitchcock P. B., Tinker N., Scott P.Chem. Commun., 1996, 17:2053.
-
[21]
Odom A. L., Arnold P. L., Cummins C. C.J. Am. Chem. Soc., 1998, 120:5836. doi: 10.1021/ja980095t
-
[22]
Cloke F. G. N., Hitchcock P. B. J.Am. Chem. Soc., 2002, 124:9352. doi: 10.1021/ja027000e
-
[23]
Evans W. J., Kozimor S. A., Ziller J. W.J. Am. Chem. Soc., 2003, 125:14264. doi: 10.1021/ja037647e
-
[24]
Mansell S. M., Kaltsoyannis N., Arnold P. L.J. Am. Chem.Soc., 2011, 133:9036. doi: 10.1021/ja2019492
-
[25]
Khodakov A. Y., Chu W., Fongarland P.Chem. Rev., 2007, 107:1692. doi: 10.1021/cr050972v
-
[26]
Brennan J. G., Andersen R. A., Robbins J. L.J. Am. Chem. Soc., 1986, 108:335. doi: 10.1021/ja00262a046
-
[27]
Parry J., Carmona E., Coles S., Hursthouse M. J.Am. Chem. Soc., 1995, 117:2649. doi: 10.1021/ja00114a030
-
[28]
Castro-Rodriguez I., Meyer K. J.Am. Chem. Soc., 2005, 127:11242. doi: 10.1021/ja053497r
-
[29]
Summerscales O. T., Cloke F. G. N., Hitchcock P. B., Green J. C., Hazari N.Science, 2006, 311:829. doi: 10.1126/science.1121784
-
[30]
Summerscales O. T., Cloke F. G. N., Hitchcock P. B., Green J. C., Hazari N. J.Am. Chem. Soc., 2006, 128:9602. doi: 10.1021/ja063222r
-
[31]
Arnold P. L., Turner Z. R., Bellabarba R. M., Tooze R. P.Chem. Sci., 2011, 2:77. doi: 10.1039/C0SC00452A
-
[32]
Gardner B. M., Stewart J. C., Davis A. L., McMaster L. W., Blake A. J., Liddle S. T.Proc. Natl. Acad. Sci., 2012, 109:9265. doi: 10.1073/pnas.1203417109
-
[33]
Gibson D. H.Chem. Rev., 1996, 96:2063. doi: 10.1021/cr940212c
-
[34]
Fujita E.Coord. Chem. Rev. 1999, 185-6, 373.
-
[35]
Keith D. W.Science, 2009, 325:1654. doi: 10.1126/science.1175680
-
[36]
Solomon S., Plattner G. K., Knutti R., Friedlingstein P.Proc. Natl. Acad. Sci., 2009, 106:1704. doi: 10.1073/pnas.0812721106
-
[37]
Natrajan L., Pecaut J., Mazzanti M.Dalton Trans. 2006, 1002.
-
[38]
Andrews P. C., Beck T., Forsyth C. M., Fraser B. H., Junk P. C., Massi M., Roesky P. W.Dalton Trans. 2007, 5651.
-
[39]
Castro-Rodriguez I., Nakai H., Zakharov L. N., Rheingold A. L., Meyer K.Science, 2004, 305:1757. doi: 10.1126/science.1102602
-
[40]
Lam O. P., Bart S. C., Kameo H., Heinemann F. W., Meyer K.Chem. Commun., 2010, 46:3137. doi: 10.1039/b927142b
-
[41]
Castro L., Lam O. P., Bart S. C., Meyer K., Maron L.Organometallics, 2010, 29:5504. doi: 10.1021/om100479r
-
[42]
Schmidt A. C., Nizovtsev A. V., Scheurer A., Heinemann F. W., Meyer K.Chem. Commun., 2012, 48:8634. doi: 10.1039/c2cc34150f
-
[43]
Matson E. M., Forrest W. P., Fanwick P. E., Bart S. C.J. Am. Chem. Soc., 2011, 133:4948. doi: 10.1021/ja110158s
-
[44]
Kahan R. J., Cloke F. G. N., Roea S. M., Niefb F.New J. Chem., 2015, 39:7602. doi: 10.1039/C5NJ00590F
-
[45]
张生栋, 丁有钱, 顾忠茂, 王祥云, 叶国安, 汪小琳, 沈兴海, 秦芝, 赵宇亮, 师全林, 李金英, 化学通报, 2014, 77, 660.Zhang S. D., Ding Y. Q., Gu Z. M., Wang X. Y., Ye G. A., Wang X. L., Shen X. H., Qin Z., Zhao Y. L., Shi Q. L., Li J. Y.Chemistry Online, 2014, 77:660.
-
[46]
Xiao H., Hu H. S., Schwarz W. H. E., Li J. J.Phys. Chem. A, 2010, 114:8837. doi: 10.1021/jp102107n
-
[47]
Wang Y. L., Liu Z. Y., Li Y. X., Bai Z. L., Liu W., Wang Y. X., Xu X. M., Xiao C. L., Sheng D. P., Diwu J., Su J., Chai, Z.F.; Albrecht-Schmitt T. E., Wang S. A.J. Am. Chem. Soc., 2015, 137:6144. doi: 10.1021/jacs.5b02480
-
[48]
Liu W. J.Mol. Phys., 2010, 108:1679. doi: 10.1080/00268971003781571
-
[49]
Hu S. W., Wang X. Y., Chu T. W., Liu X. Q.J. Phys. Chem. A, 2009, 113:9243. doi: 10.1021/jp904655w
-
[50]
Zhang L., Hou G. H., Zi G. F., Ding W. J., Walter M. D.J. Am. Chem. Soc., 2016, 138:5130. doi: 10.1021/jacs.6b01391
-
[51]
Ding W. J., Fang W. H., Chai Z. F., Wang D. Q.J. Chem. Theory Comput., 2012, 8:3605. doi: 10.1021/ct300075n
-
[52]
Ding W. J., Wang D. Q.Organometallics, 2014, 33:7007. doi: 10.1021/om500797h
-
[53]
王东琪, Gunsteren W F V., 化学进展, 2011, 23, 1566.Wang D.-Q., Gunsteren W. F.V. Prog. Chem., 2011, 23:1566.
-
[1]
-

图 1 铀的氧化还原电位(vs.标准氢电极pH=1的HClO4溶液, 25 ℃)
Figure 1 Oxidation-reduction potentials of uranium (vs. SHE in pH=1 HClO4 at 298 K)

图 3 三价铀有机络合物3和6对N2的活化
Figure 3 N2activation by trivalent uranium complex 3/6 to obtain compounds 5and7

图 4 三价铀有机络合物8和10对N2的活化
Figure 4 Reaction of trivalent uranium complex 8 and 10 with N2 to give the compounds 9and 11

图 5 几种铀有机络合物与CO配位(化合物12、13和14)的结构
Figure 5 The structure of uranium compounds 12, 13and 14

图 6 三价铀有机络合物15和17对CO的活化
Figure 6 Reaction of 15/17 with CO to give the compounds 16 and 18

图 9 三价铀有机络合物26和28对CO2的活化
Figure 9 Reaction of 26/28 with CO2 to give the compounds 27 and 29

图 11 三价铀有机络合物33对CO2的活化
Figure 11 Insertion of CO2 into a uranium-benzyl bond to give 34
-


 下载:
下载:











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 4116
- HTML全文浏览量: 915
 下载:
下载:
