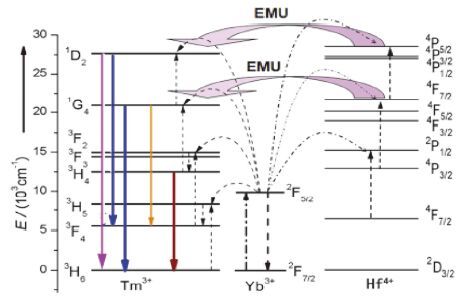
 图 1
离子Yb3+、Tm3+和Hf4+之间的可能能量转移机制图
Figure 1.
Proposed energy transfer mechanism among Yb3+,Hf4+and Tm3+
图 1
离子Yb3+、Tm3+和Hf4+之间的可能能量转移机制图
Figure 1.
Proposed energy transfer mechanism among Yb3+,Hf4+and Tm3+
引用本文:
黄清明, 俞瀚, 张新奇, 曹文兵, 俞建长. Hf4+共掺NaYF4:Yb/Tm提高上转换发光性能研究[J]. 化学学报,
2015, 74(2): 191-198.
doi:
10.6023/A15040257

Citation: Huang Qingming, Yu Han, Zhang Xinqi, Cao Wenbing, Yu Jianchang. Upconversion Performance Enhancement of NaYF4:Yb/Tm by Codoping Hf4+ as Energy Migrator[J]. Acta Chimica Sinica, 2015, 74(2): 191-198. doi: 10.6023/A15040257
Citation: Huang Qingming, Yu Han, Zhang Xinqi, Cao Wenbing, Yu Jianchang. Upconversion Performance Enhancement of NaYF4:Yb/Tm by Codoping Hf4+ as Energy Migrator[J]. Acta Chimica Sinica, 2015, 74(2): 191-198. doi: 10.6023/A15040257
Hf4+共掺NaYF4:Yb/Tm提高上转换发光性能研究
摘要:
利用X射线多晶衍射仪、场发射扫描电镜、场发射透射电镜、X射线光电子能谱和荧光光谱仪对相近半径离子Hf4+和Zr4+共掺六方NaYF4:Yb3+/Tm3+的结构、形貌和上转换发光性能进行研究.结果表明Hf4+和Zr4+离子共掺六方NaYF4:Yb3+/Tm3+可有效调控晶场的不对称性, Hf4+相对于Zr4+是个更好的掺杂离子,它在调控晶场的同时还参与Tm3+离子上转换发光的能量传递过程,明显提高了短波500 nm以下发射带的荧光强度;而Zr4+离子仅扮演晶场调控角色而未能参与稀土离子Tm3+的上转换发光过程, Tm3+离子小于500 nm短波发射带的荧光强度没有得到明显的提高,仅提高802 nm发射带的荧光强度.该研究发现Hf4+可作为蓄能离子参与稀土离子的上转换发光过程,有助于将Hf4+作为蓄能离子和晶格操纵工具用于设计和制备其它高性能的稀土上转换发光材料.
English
Upconversion Performance Enhancement of NaYF4:Yb/Tm by Codoping Hf4+ as Energy Migrator
Abstract:
In this paper we report the infrared to visible upconversion(UC) luminescence properties of Hf4+ and Zr4+ codoped NaYF4:Yb3+/Tm3+. Samples were synthesised by hydrothermal method. Concentration of Tm3+ and Yb3+ ions were fixed to be 2 mol% and 5 mol% for all samples, respectively. NaY0.93-xYb0.05Tm0.02F4 was tridoped with 0, 2, 4, 6, 8 mol% Hf4+ or Zr4+ and corresponding samples were named as Hf0, Hf2, Hf4, Hf6, Hf8 and Zr0, Zr2, Zr4, Zr6, Zr8, respectively. In a typical procedure, trivalent nitrate stock solutions of 0.2 mol/L were prepared at first by dissolving the corresponding metal oxide in concentrated nitric acid or hydrofluoric acid at elevated temperatures. And then, a certain mole percentage of trivalent nitrate solutions were added into 20 mL 0.04 mmol EDTA aqueous solution. After vigorous stirring for 30 min, 25 mL ethanol solution containing 0.2 mmol NaF, 0.2 mmol NH4HF2 and corresponding stoichiometric amount Hf4+ or Zr4+ were dropwise added into the solution, and then pH value of solution was adjusted to 3.0 by addition 1 mol/L HF, and stirring continued for 30 min. Then the emulsion mixture was moved to PTFE-lined high pressure pot and incubated in oven at 180℃ for 12 h. The final products were collected, washed several times with water and ethanol alternately, and gathered by centrifugation, and then dried in oven at 60℃. Crystal microstructure, morphology and UC luminescence properties of samples were investigated by X-ray diffraction, field emission scanning electron microscopy, transmission electron microscopy, X-ray photoelectron spectroscopy and upconversion photoluminescence spectra. Results revealed the bond distance of F1-Y and F2-Y become close with the rising of Hf4+ or Zr4+ codoped concentration, indicating crystal field asymmetry of rare earth ions were tuned effectively by Hf4+ or Zr4+ codoping. Electron hypersensitive transition was promoted, and the intensity of 802 nm emission was enhanced obviously. Hf4+ ion was a better dopant than Zr4+, for the extranuclear electronic structure of Hf4+ was the same with rare earth ions, and Hf4+ ion was involved in UC process as a migrator to improve Tm3+ upper state levels'population. UC luminescence of Hf4+ codoped sample were enhanced obviously, especially the shortwave emissions. The reported work establishes the understanding of Hf4+ as a migrator for Tm3+ ions upconversion luminescence, which may be helpful for design and synthesis of high-performance upconversion materials.
-
Key words:
- upconversion
- / hafnium doping
- / crystal field
- / energy migration
-
1 引言
近年,稀土掺杂无机材料成为光学材料的研究热 点[1~3],其中稀土掺杂六方NaYF4特别引人关注,因为六方NaYF4具有卓越的光物理性能和在众多领域的潜在应用[4]. 其应用主要包括: 生命科学[5~7]、医学成 像[8~10]、药物释放[11, 12]、光动力学治疗[13]、温度传感 器[14, 15]、光催化[16]和太阳能光伏器件[17~19]等. 稀土铥的核外4f电子层具有多重能级结构,这些能级间的电子跃迁可产生从红光到蓝光的多谱带发射[20],它是一个优秀的上转换发光激活离子. 在众多应用领域,短波发射扮演着重要的角色[21],特别是在光催化和太阳能频谱转换领域. 但三光子以上的上转换发光强度通常都很低,弱的短波发射强度成为上转换发光材料应用的主要障碍. 上转换发光的主要途径有激发态的光子吸收(ESA)、两光子同时吸收(TPA)、能量转移上转换(ETU)和能量调谐转移上转换(EMU)[22]等. 在过去几年里,人们通过多种途径试图提高上转换发光强度,比如敏化剂共掺杂[23]、晶粒大小与形貌调控[24]、构建纳米核壳结 构[25, 26]、纳米粒子的表面修饰[27, 28]和晶场调控[29, 30]等. 但大多数研究成果都集中在提高绿光发射强度,而对于更短波长的蓝光发射带依然收效甚微.
众所周知,离子掺杂是制备新型、高性能功能材料的一种有效途径[31~33]. 刘教授团队揭示了能量调谐转移上转换发光模式[25, 27],在该模式中敏化剂被用于大量吸收泵浦光子,然后将能量转移给邻近的蓄能离子,把蓄能离子基态的电子激发至激发态上,而后能量在蓄能离子间逐步转移,最终,把能量转移给激活离子. 在它们的研究中Tm3+离子550 nm发射带的荧光强度得到显著的增强,但450 nm发射带的强度无法通过提高能量传递离子Gd3+的浓度而得以增强,因为Gd3+的能级与Tm3+450 nm发射带的能量不匹配.
基于此,本文主要介绍一种新方法,将Hf4+离子作为蓄能或能量转移离子共掺NaYF4: Yb3+/Tm3+来提高多光子吸收上转换发光. 因为Hf4+离子的核外电子结构4f135d与稀土离子相同,能级4P5/2 (28547 cm-1)和4F7/2 (21638 cm-1)与Tm3+离子的1D2 (27642 cm -1)和1G4 (20882 cm-1)相近. Hf4+可能会参与Tm3+的上转换发光过程,它们之间的可能能量转移机制如图 1所示. 同时将与Hf4+相近半径但不具有4f层电子结构的Zr4+离子作为参照,对比研究Hf4+或Zr4+共掺杂对六方NaYF4: Yb3+/Tm3+发光性能的影响. 另一方面,由于Tm3+离子的3H4到3H6的能级跃迁符合超敏跃迁选择定则[34],较小半径的Hf4+与Zr4+共掺也将导致NaYF4晶场对称性的变化,这有利于符合超敏跃迁激发态能级的电子布居,或将增强Tm3+的上转换发光强度.
图 1
离子Yb3+、Tm3+和Hf4+之间的可能能量转移机制图
Figure 1.
Proposed energy transfer mechanism among Yb3+,Hf4+and Tm3+
2 结果与讨论
2.1 结构与形貌分析
利用ICP对各样品的元素进行定量分析,分析结果如支持信息中表S1、S2所示,样品中元素Y、Yb、Tm和Hf或Zr的含量与实验加入量基本一致.
采用XRD分别对Hf4+和Zr4+掺杂六方NaYF4: Yb/Tm系列样品进行物相分析,各样品的衍射峰都可以归属于六方相NaYF4(JCPDS 16-0344),没有出现其它物相的衍射峰,说明Hf4+或Zr4+共掺杂未导致其它物相的生成,如图 2所示. 利用Rietveld全谱拟合法精修各图谱,求出各样品的精确晶胞参数和键长; 各样品的精修结果评价因子Rp、Rwp都小于5%,GOF都处在1.3~1.7之间,说明结构精修结果可信. 图 3a,3b显示,各样品的晶胞体积和晶胞参数c/a随着Hf4+或Zr4+掺杂量的增加逐渐减小,因Hf4+和Zr4+的离子半径比Y3+小,所以晶胞体积变小说明Hf4+和Zr4+掺入六方相NaYF4的晶格结构中,且晶格不同方向的变形不同步,c轴方向缩小的比较大,a轴方向缩小的比较小. 图 3c,3d显示六方相晶格结构(见支持信息中图S1)中三价阳离子与F-的配位键长r1、r2随着Hf4+和Zr4+掺杂量增加的变化趋势刚好相反,r1随着Hf4+和Zr4+掺杂量增加逐渐减小,减小的趋势先急后缓,而r2反之. Zr4+掺杂系列在Zr4 +掺杂量为6 mol%时出现了一个明显的拐点,而Hf4+掺杂系列中掺杂量为4~8 mol%时键长的变化不是很明显,这可能是由于Hf4+具有4f层未充满电子轨道,当掺杂浓度在4~8 mol%之间时,通过Hf4+离子核外4f层不同轨道上电子的重新分配予以平衡,导致键长的相对稳定. 而Zr4+离子由于没有未充满的4f电子轨道,在同样的畸变晶场中无法实现价键稳定的电子重新平衡分布,所以出现了明显的拐点. 该图也显示随着Hf4+和Zr4+共掺杂量的增加,相邻三价阳离子的间距都逐渐减小,由于Hf4+/Zr4+离子的半径为72 pm左右,比Y3+离子90 pm小,表明Hf4+或Zr4+离子是占位取代Y3+.
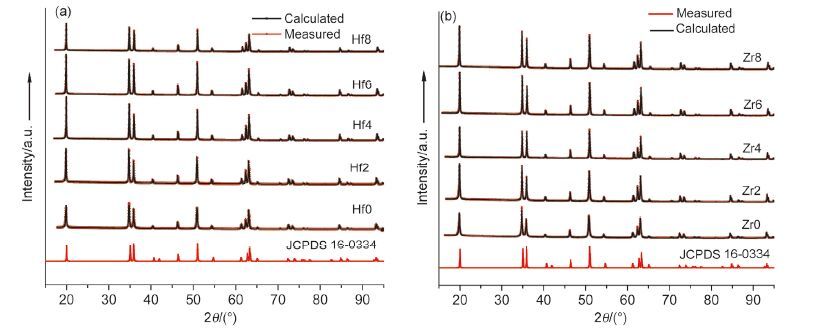 图 2
(a,b)分别是不同量Hf4+和Zr4+掺杂样品的实测XRD图谱(红线)和Rietveld全谱拟合计算图谱(黑线)及国际标准XRD衍射图谱JCPDS 16-0334
Figure 2.
(a,b) Measured (red line) and Rietveld calculated (black line) XRD patterns of different amount Hf4+and Zr4+ codoped samples,respectively; and international standard XRD pattern JCPDS 16-0334
图 2
(a,b)分别是不同量Hf4+和Zr4+掺杂样品的实测XRD图谱(红线)和Rietveld全谱拟合计算图谱(黑线)及国际标准XRD衍射图谱JCPDS 16-0334
Figure 2.
(a,b) Measured (red line) and Rietveld calculated (black line) XRD patterns of different amount Hf4+and Zr4+ codoped samples,respectively; and international standard XRD pattern JCPDS 16-0334
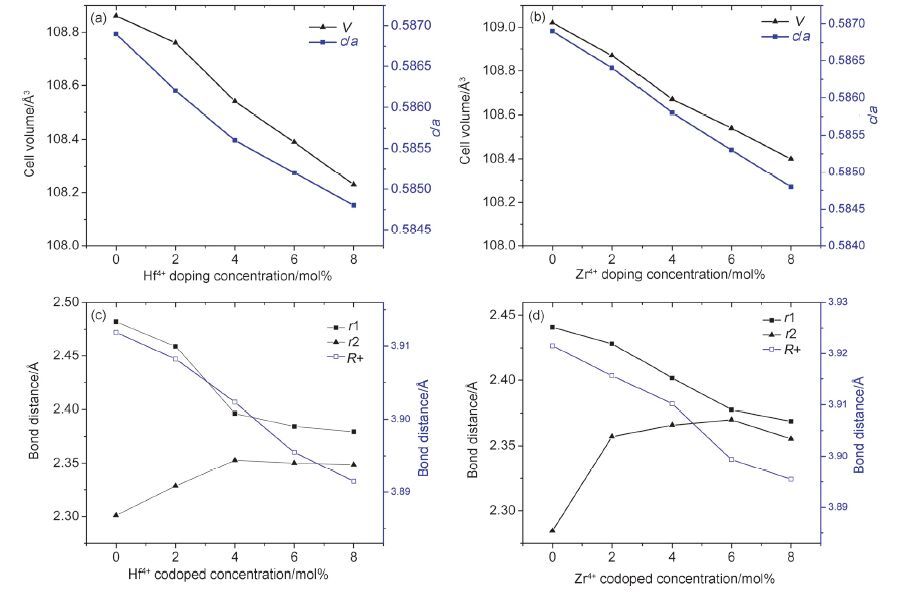 图 3
(a,b)分别是晶胞体积V和晶胞参数c/a随着Hf4+和Zr4+掺杂量增加的变化趋势图; (c,d)分别是F1-Y(r1)、F2-Y(r2)及Y-Y(R+)之间的键长随着Hf4+和Zr4+掺杂量增加的变化趋势图.
Figure 3.
(a,b) Changing trend of cell volume V and cell parameters c/a with the rising of Hf4+and Zr4+ codoped concentration,respectively. (c,d) changing trend of bond distance of F1-Y(r1),F2-Y(r2) and Y-Y(R+) with the rising of Hf4+ and Zr4+ codoped concentration,respectively
图 3
(a,b)分别是晶胞体积V和晶胞参数c/a随着Hf4+和Zr4+掺杂量增加的变化趋势图; (c,d)分别是F1-Y(r1)、F2-Y(r2)及Y-Y(R+)之间的键长随着Hf4+和Zr4+掺杂量增加的变化趋势图.
Figure 3.
(a,b) Changing trend of cell volume V and cell parameters c/a with the rising of Hf4+and Zr4+ codoped concentration,respectively. (c,d) changing trend of bond distance of F1-Y(r1),F2-Y(r2) and Y-Y(R+) with the rising of Hf4+ and Zr4+ codoped concentration,respectively
分别选取Hf0、Hf4、Hf8和Zr0、Zr4、Zr8样品,采用场发射扫描电镜对样品的晶粒形貌与大小进行分析,结果如图S2所示(见支持信息),晶粒都是六方柱状结构,长度约5 μm,宽度约1 μm,Hf4+和Zr4+共掺杂没有明显改变晶粒的大小和形貌. 利用200 kV的场发射透射电镜对Hf2样品进行形貌与结构表征,如图 4所示. 晶粒大小及形态与扫描电镜的表征结果一致,图 4b高分辨图显示有明显的晶格畸变条纹,说明2 mol% Hf4+离子掺杂有效调制六方相NaYF4的微观结构,晶格条纹间距为0.2987 nm,可归属于六方相NaYF4的(110)晶面.
 图 4
(a)样品Hf2的透射电镜照片,(b)是(a)中红色区域的高分辨电镜照片
Figure 4.
(a) TEM image of Hf2 sample,(b) HR-TEM image of red square area of (a)
图 4
(a)样品Hf2的透射电镜照片,(b)是(a)中红色区域的高分辨电镜照片
Figure 4.
(a) TEM image of Hf2 sample,(b) HR-TEM image of red square area of (a)
2.2 上转换发光性能
采用爱丁堡FS920荧光光谱仪,在980 nm光源的激发下,分别测试不同量Hf4+和Zr4+掺杂样品的上转换发射光谱,如图 5a,5b所示. 谱中各发射峰分别归属于Tm3+离子4f电子层中不同能级间的电子跃迁: 360 nm为电子从1D2能级到3H6的跃迁发射,450 nm为电子从1D2能级到3F4 能级的跃迁发射,475 nm为电子从1G4能级到3H6能级的跃迁发射,646 nm为电子从1G4 能级到3F4能级的跃迁发射,802 nm是电子从3H4能级到3H6能级的跃迁发射. 图 5a、S3a显示,Hf4+掺杂系列样品中Tm3+362 nm和450 nm发射带的荧光强度变化幅度比较大; 短波发射荧光强度占比随着Hf4+掺杂量的增加,先增加后减小,Hf4+掺杂量为4 mol%时达到最大值; 而图 5b、S3b显示Zr4+共掺杂系列样品的362 nm和450 nm发射带荧光强度变化幅度很小,且短波发射的荧光占比基本无变化; 476 nm和646 nm发射带荧光强度占比随着Zr4+掺杂量的增加反而逐渐减小,但长波802 nm的发光强度及荧光占比随着Zr4+掺杂量的增加则逐渐增强. 采用爱丁堡FS920荧光光谱仪及瞬态荧光模块,分别测试不同量Hf4+和Zr4+掺杂样品的475 nm和802 nm发射带的荧光衰减谱,如图 5c,e,g,i所示,并用单指数衰减函数
$I=a-{{b}_{1}}\exp (\frac{-t}{{{t}_{r}}})+{{b}_{2}}\exp (\frac{-t}{{{t}_{d}}})$ (a,b1,b2为常数,tr荧光增强时间常数,td是荧光衰减时间常数)拟合计算荧光增强时间常数和荧光衰减时间常数(寿命),计算结果如图 5d,f,h,j所示. Hf4+掺杂系列样品475 nm发射带荧光增强时间常数和荧光衰减时间常数都随着Hf4+掺杂量的增加,先减小后增大. 当Hf4+掺杂量为4 mol%时,荧光寿命最短. 而长波802 nm发射带荧光增强时间常数和衰减时间常数都随着Hf4+掺杂量的增加而延长. 对于Zr4+掺杂系列样品短波475 nm和长波802 nm发射带的荧光增强时间常数和荧光衰减时间常数都随着 Zr4+掺杂量的增加逐渐延长. 图 5
(a,b)分别是不同量Hf4+和Zr4+共掺杂样品的上转换发射光谱; (c,e,g,i)分别是不同量Hf4+和Zr4+掺杂样品的475 nm和802 nm发射带的荧光实测衰减谱和单指数衰减拟合计算谱; (d,f,h,j)分别是各样品475 nm和802 nm发射带的荧光增强时间常数和荧光寿命
Figure 5.
(a,b) upconversion spectra of different amount Hf4+and Zr4+ codoped samples,respectively; (c,e,g,i) measured and calculated decay curve of 475 nm and 802 nm emissions of different concentration Hf4+and Zr4+ codoped samples,respectively; (d,f,h,j) changing trend of rising and decay time constant of 475 nm and 802 nm emissions with the rising of Hf4+and Zr4+ codoped concentration,respectively
图 5
(a,b)分别是不同量Hf4+和Zr4+共掺杂样品的上转换发射光谱; (c,e,g,i)分别是不同量Hf4+和Zr4+掺杂样品的475 nm和802 nm发射带的荧光实测衰减谱和单指数衰减拟合计算谱; (d,f,h,j)分别是各样品475 nm和802 nm发射带的荧光增强时间常数和荧光寿命
Figure 5.
(a,b) upconversion spectra of different amount Hf4+and Zr4+ codoped samples,respectively; (c,e,g,i) measured and calculated decay curve of 475 nm and 802 nm emissions of different concentration Hf4+and Zr4+ codoped samples,respectively; (d,f,h,j) changing trend of rising and decay time constant of 475 nm and 802 nm emissions with the rising of Hf4+and Zr4+ codoped concentration,respectively
前文Hf4+和Zr4+掺杂样品的结构分析结果表明,三价阳离子Y3+与阴离子F-的配位键长随着Hf4+和Zr4+掺杂量的增加发生了明显变化,说明Hf4+或Zr4+共掺可有效调制六方相NaYF4的微观结构,结合样品的制备方法及Hf4+和Zr4+离子的核外电子结构特征,可将Hf4+或Zr4+共掺NaYF4: Yb3+/Tm3+引起的上转换发光性能变化做以下几方面的探讨:
首先,采用水热法制备的样品可使各种离子在溶液体系中得到均匀分散,但由于Yb3+、Tm3+、Y3+离子的半径非常接近且价态相同,这将不利于稀土离子Yb3+和Tm3+的均匀分散. 当Hf4+或Zr4+离子共掺时,由于Hf4+/Zr4+的半径比Yb3+、Tm3+、Y3+离子小,且价态也不同,由此,当Hf4+或Zr4+离子加入时,溶液体系将存在荷电分布梯度,荷电梯度将促进溶液中离子的运动与扩散,直至消除荷电梯度为止. 因此离子的运动分散过程将减少稀土离子团簇的形成,提高稀土离子的分散 性[35]. 因稀土离子的团簇会产生荧光猝灭,所以稀土离子团簇的减少就抑制了荧光团簇猝灭效应,从而在一定程度上提高稀土离子的上转换发光强度.
其次,根据Judd-Ofelt理论[36, 37],稀土发光强度的振子因子Ω2对稀土离子所处晶体场的对称性特别敏感,当晶体场对称性降低时,Ω2就会随之增大,从而提高稀土离子的发光强度. 在本研究中,Hf4+共掺杂系列样品的微观结构表征结果显示,随着Hf4+掺杂量的增加,六方NaYF4结构中键长r1和r2的差异越来越小,当掺杂量大于4 mol%时r1、r2的几乎无变化,说明Hf4+掺杂明显改变了晶体微观结构的对称性; 其发射谱显示各发射带的荧光强度随着Hf4+掺杂量的增加先增强,当Hf4+掺杂量大于4 mol%时其发射谱的变化较小,与结构的变化一致. 而对于Zr4+掺杂系列样品,随着Zr4+掺杂量的增加六方NaYF4结构中键长r1和r2逐渐接近,其长波荧光强度也逐渐增强,且荧光强度的变化趋势与晶格结构中键长的变化趋势一致. 以上结果说明,微观结构对称性与稀土Tm3+离子的上转换发光强度密切相关. 在以前等价离子掺杂六方NaYF4的研究中发现当键长r1和r2相差大时[38],晶场不对称高,上转换发光强度增强,而本研究结果却恰恰与之相反,键长r1和r2越接近其上转换发光强度越强. 这可能是由于高价离子Hf4+或Zr4+与稀土离子相邻时,它们的高价态使得它与F-离子的配位场比等价的强,它们与F-的配位键长越接近时,反而对稀土离子产生的是越强的晶场影响,这种晶场影响与等价掺杂引起的键长大小变化的作用一致. 配位阴离子F-的核外电子结合能变化规律可进一步证实该现象,如图 6所示. Hf4+、Zr4+掺杂系列样品中F-离子核外电子结合能随着Hf4+或Zr4+掺杂量增加的变化趋势是一致的,结合能随着Hf4+、Zr4+掺杂量的增加,先略有减小,后增大,然后再减小,而且样品Hf4、Hf6、Hf8中F-离子结合能基本没有什么变化,与Y-F配位键长r1、r2基本不变相一致; 而Zr4+掺杂系列样品中F-的核外电子结合能变化与Y-F配位健长r1、r2的变化一致. 同时F-核外电子结合能随着Hf4+或Zr4+掺杂量增加的变化趋势与等价Sc3+共掺杂六方NaYF4中F-离子核外电子结合能随晶格微观结构对称性的变化趋势一致[23]. 说明高价掺杂引起的键长r1和r2逐渐接近与等价掺杂引起的键长r1和r2差值变大造成的晶场影响是一致的. 所以Hf4+或Zr4+共掺明显提高802 nm发射带的上转换发光强度,也是由于802 nm发射的电子跃迁能级符合超敏跃迁定则,当晶场不对称性提高时,促进电子的超敏跃迁,提高电子在该激发态上的布居数,从而增强其荧光强度.
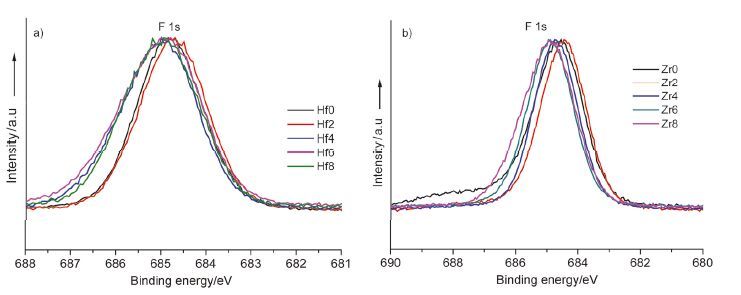 图 6
(a) Hf4+共掺杂样品中F-离子的XPS光谱,(b) Zr4+共掺杂样品中F-离子的XPS光谱
Figure 6.
(a) F- XPS spectra of different concentration Hf4+codoped samples; (b) F- XPS spectra of different concentration Zr4+ codoped samples
图 6
(a) Hf4+共掺杂样品中F-离子的XPS光谱,(b) Zr4+共掺杂样品中F-离子的XPS光谱
Figure 6.
(a) F- XPS spectra of different concentration Hf4+codoped samples; (b) F- XPS spectra of different concentration Zr4+ codoped samples
再次,稀土离子的荧光寿命取决于基质材料的声子能量高低和施主离子的作用,在Zr4+共掺杂系列样品中,因为Zr4+不具备稀土离子特性,无法参与稀土离子上转换发光的能量传递过程,施主离子只有敏化剂 Yb3+且各样品中Yb3+浓度一样,可以认为它对荧光寿命的影响是一致的,不会导致荧光寿命的明显变化. 那么影响不同Zr4+掺杂量样品荧光寿命变化的主要因素则是晶格缺陷和敏化离子与激活离子之间的间距,分析Zr4+共掺NaYF4:Yb3+/Tm3+样品的微观结构与荧光寿命的变化关系,发现荧光寿命与键长r1、r2大小的变化趋势一致,当键长r1、r2的大小越接近时,各发射带的荧光寿命越长,键长r1、r2的大小的差值越大时,荧光寿命越短; 键长的变化与三价阳离子间距随着Zr4+掺杂量的增加逐渐减小是相关的,三价离子间距缩小有利于敏化离子Yb3+与激活离子Tm3+之间的能量传递,提高敏化效率,延长荧光寿命. Zr4+掺杂样品各发射带的荧光增强时间常数与荧光衰减时间常数很接近,说明激发态能级上的电子布居是通过多次的能量转移及合作上转换来进行的,而不是通过快速的激发态吸收实现其激发态能级布居. 这使激活离子的荧光寿命与敏化剂的敏化效率之间的关系变得更为密切,因此随着三价阳离子间距的减小Zr4+掺杂系列样品475 nm和802 nm发射带的荧光寿命都随着Zr4+掺杂量的增加而延长.
最后,比较Hf4+和Zr4+掺杂系列样品的上转换发射光谱,它们的谱带特征差异明显,Hf4+共掺杂样品随着Hf4+掺杂量的增加,短波500 nm以下发射带的荧光强度增强得很明显,而大于500 nm发射带的荧光强度增强幅度却很小,这与Zr4+掺杂样品的荧光强度随着掺杂量的增加的变化完全不同,如果只考虑晶场不对称性变化对荧光增强的贡献,Hf4+离子与Zr4+离子的半径非常接近,其对晶场对称性的影响差异不大,在荧光光谱特征上不会有如此大的差异. 那是否是因为Hf4+离子参与Tm3+离子的上转换的发光过程呢? 众所周知Hf4+与稀土离子都属于镧系,它们都具有4f层未充满电子轨道; 由图 1可知,Hf4+离子的多个能级与Tm3+离子的能级相匹配,这些相匹配的能级间将会发生能量的弛豫转移. 通过Tm3+离子基态吸收、敏化离子Yb3+能量转移、 Tm3+低能级激发态与Yb3+离子的合作上转换,将能量转移给邻近的Hf4+离子,使Hf4+离子基态电子吸收能量后跃迁布居到4P5/2、4P3/2 、4P1/2,而后通过能量调谐迁移的方式,将能量再转移给Tm3+离子的1D2与1G4激发态能级,这两个能级上的布居电子分别以1D2能级到3H6能级跃迁,发射出360 nm的荧光,以1D2能级到3F4 能级跃迁,发射出450 nm的荧光,以1G4能级到3H6能级跃迁发射出475 nm荧光. 同时部分布居在Tm3+离子 1D2与1G4激发态能级上的电子,以无辐射的形式将能量转移给Hf4+离子的4P5/2、4P3/2、4P1/2能级,将能量暂时存蓄,而后又通过能量调谐转移的方式,回传给Tm3+离子,发挥蓄能离子的作用. 所以当Hf4+掺杂量小于4 mol%时,由于Hf4+离子的浓度较低,发生团簇的少,与稀土离子相邻的Hf4+离子都可以成为能量转移离子和蓄能离子,它们之间的合作能量上转换和调谐能量转移上转换,提高了Tm3+离子高能激发态能级的电子布居数,从而明显提高了360 nm和450 nm发射带的荧光强度; 但由于Hf4+离子与Tm3+之间的这种能量传递,使得3H4激发态跃迁回基态的能量转移给Hf4+,导致符合超敏跃迁定则的802 nm发射带的荧光强度不如Zr4+掺杂系列样品的显著. 同时由于Hf4+离子与Tm3+之间的这种能量传递,导致475 nm发射带的荧光寿命随着掺杂量的增加先减小,但当Hf4+掺杂量超过4 mol%时,由于Hf4+离子的浓度增大,出现了团簇,从而减少其与稀土离子相邻的离子数,即减少Hf4+与Tm3+之间的能量传递机率,475 nm荧光强度减弱,荧光寿命也因此而开始延长. Tm3+离子中能级3H6到3H4的跃迁符合超敏跃迁定则,所以802 nm发射带受晶场不对称性的影响大,Zr4+掺杂系列样品随着Zr4+掺杂时的增加,荧光强度逐渐增强. 激发态能级3H4上的电子主要是通过激发态吸收布居,敏化剂作用下的合作上转换布居和部分高激发态能级弛豫布居. 其寿命取决于能级本征寿命、激活离子与敏化离子间的能量传递及高能激发态的弛豫. 随着Hf4+掺杂量的增加,三价离子间距的减小,提高激活离子与敏化离子之间的能量传递,延长激发态能级的电子布居时间; 高能态的本征寿命在延长,所以弛豫布居时间也相应延长,所以其荧光增强时间常数和荧光寿命都逐渐延长. 另一可能因素是当Hf4+或Zr4+掺杂时,键长r1与r2的差异逐渐变小,原子的温度因子减小,振动减小、体系的声子能量变小,所以475 nm的荧光寿命也逐渐延长.
3 结论
Hf4+、Zr4+共掺NaYF4:Yb3+/Tm3+可有效调控基质材料的微观结构,不同量Zr4+共掺杂,可控调制稀土离子在六方相NaYF4晶体结构中的晶场对称性,促进符合超敏跃迁定则能级间的电子跃迁,提高相应激发态能级的电子布居,增强上转换发光强度; 同时由于Zr4+掺杂量的增加,减小三价阳离子间距,提高敏化剂与激活离子之间的能量传递机率,延长荧光寿命. Hf4+共掺NaYF4:Yb3+/Tm3+时,参与Tm3+上转换发光的能量传递过程,有效增强Tm3+离子短波发射带的荧光强度,但由于Tm3+离子的高能级激发态不符合超敏跃迁选择定则,以及Tm3+与Hf4+的能级并不完全匹配,所以影响Hf4+离子与Tm3+之间的能量传递效率,使得Tm3+短波发射带的荧光强度提高的不显著,最大仅提高一倍. 但该研究可启发 人们通过选择与Hf4+离子能级匹配度更好且高能激发态符合超敏跃迁定则的激活离子来制备高性能的上转换发光材料.
4 实验部分
4.1 样品的制备
将分析纯稀土氧化物RE2O3 (RE=Tm,Yb)、Y2O3和HfO2、ZrO2分别用浓硝酸和氢氟酸在加热的条件下溶解,并除去过量的酸,然后用去离子水配置成0.2 mol/L的备用溶液. 稀土离子Yb3+和Tm3+在NaYF4中相对于三价总离子数浓度固定为5 mol%和2 mol%,Hf4+和Zr4+相对于三价离子总量的掺杂浓度分别为0、2、4、6、8 mol%制备五个样品,分别命名为Hf0、Hf2、Hf4、Hf6、Hf8和Zr0、Zr2、Zr4、Zr6、Zr8. 典型的样品制备过程是将设计化学计量配比的0.2 mol/L的三价、四价离子备用液在磁力搅拌下逐滴加入20 mL含有0.04 mmol EDTA二钠盐的溶液中,并持续搅拌30 min. 然后在持续磁力搅拌下逐滴加入25 mL含有0.2 mol/L NaF和0.2 mol/L NH4HF2的乙醇溶液,滴完后用1 mol/L的HF或NaOH将混合溶液的pH值调至3.0左右,再持续搅拌30 min后,将乳浊液转移至聚四氟乙稀高压釜内,把高压釜放入烘箱,在180 ℃下反应12 h; 待高压釜自然冷却后,将沉淀物取出,用乙醇和去离子水分别洗涤数次,将洗干净的样品在60 ℃下烘干8 h,制得Hf4+、Zr4+与稀土Tm3+、Yb3+共掺六方NaYF4样品.
4.2 仪器与表征
X射线多晶衍射图谱采用帕纳科X’pert Pro MPD 仪器进行测试,测试条件为Co靶波长0.178901 nm,管压40 kV,电流40 mA,加Fe滤波片,扫描速度1 (°)/min,扫描范围10°~95°. 采用ZEISS IGMA HD/VP 场发射扫描电子显微镜观察拍摄样品的晶粒大小和形貌; 采用场发射电镜FEI Tecnai G2 F20 S-TWIN仪器观察晶粒形貌和高分辨图像,其工作电压为200 kV. 采用爱丁堡 FSLP920 仪器对样品的上转换发射光谱和荧光时间衰减谱进行测试表征,泵浦光源为OPTEK OPO激光器发射的波长980 nm的激光,泵浦功率为5 W,重复频率为10 ns,使用积分球. 采用美国热电ESCALAB 250的X射线光电子能谱仪对掺杂样品中各元素的核外电子结合能进行测试,所有样品的测试都在室温下进行.
-
-
[1]
Cates, E. L.; Chinnapongse, S. L.; Kim, J. H. Environ. Sci. Technol. 2012, 46(22), 12316. 10.1021/es303612p
-
[2]
Li, C. X.; Lin, J. J. Mater. Chem.2010, 20(33), 6831. 10.1039/c0jm00031k
-
[3]
Feng, W.; Han, C.; Li, F. Adv. Mater. 2013, 25(37), 5287.
-
[4]
Buenzli, J.-C. G.; Eliseeva, S. V. J. Rare Earths 2010, 28(6), 824. 10.1016/S1002-0721(09)60208-8
-
[5]
Zhou, L.; Li, Z.; Liu, Z.; Yin, M.; Ren, J.; Qu, X. Nanoscale 2014, 6(3), 1445.
-
[6]
Lin, M.; Zhao, Y.; Wang, S. Q.; Liu, M.; Duan, Z. F.; Chen, Y. M.; Li, F.; Xu, F.; Lu, T. J. Biotechnol. Adv. 2012, 30(6), 1551. 10.1016/j.biotechadv.2012.04.009
-
[7]
Wang, F.; Banerjee, D.; Liu, Y.; Chen, X.; Liu, X. Analyst 2010, 135(8), 1839.
-
[8]
Chen, Z.; Wu, X.; Hu, S.; Hu, P.; Yan, H.; Tang, Z.; Liu, Y. J. Mater. Chem. C 2015, 3(1), 153. 10.1039/C4TC01766H
-
[9]
Min, Y.; Li, J.; Liu, F.; Padmanabhan, P.; Yeow, E. K. L.; Xing, B. Nanomaterials 2014, 4(1), 129. 10.3390/nano4010129
-
[10]
Ni, D.; Bu, W.; Zhang, S.; Zheng, X.; Li, M.; Xing, H.; Xiao, Q.; Liu, Y.; Hua, Y.; Zhou, L.; Peng, W.; Zhao, K.; Shi, J. Adv. Funct. Mater. 2014, 24(42), 6613. 10.1002/adfm.v24.42
-
[11]
Yang, D.; Kang, X.; Ma, P. a.; Dai, Y.; Hou, Z.; Cheng, Z.; Li, C.; Lin, J. Biomaterials 2013, 34(5), 1601.
-
[12]
Tian, G.; Gu, Z. J.; Liu, X. X.; Zhou, L. J.; Yin, W. Y.; Yan, L.; Jin, S.; Ren, W. L.; Xing, G. M.; Li, S. J.; Zhao, Y. L. J. Phys. Chem. C 2011, 115(48), 23790. 10.1021/jp209055t
-
[13]
Park, Y.; Kim, H. M.; Kim, J. H.; Moon, K. C.; Yoo, B.; Lee, K. T.; Lee, N.; Choi, Y.; Park, W.; Ling, D.; Na, K.; Moon, W. K.; Choi, S. H.; Park, H. S.; Yoon, S. Y.; Suh, Y. D.; Lee, S. H.; Hyeon, T. Adv. Mater. 2012, 24(42), 5755.
-
[14]
Vetrone, F.; Naccache, R.; Zamarron, A.; de la Fuente, A. J.; Sanz-Rodriguez, F.; Maestro, L. M.; Rodriguez, E. M.; Jaque, D.; Sole, J. G.; Capobianco, J. A. ACS Nano 2010, 4(6), 3254. 10.1021/nn100244a
-
[15]
Shaoshuai, Z.; Kaimo, D.; Xiantao, W.; Guicheng, J.; Changkui, D.; Yonghu, C.; Min, Y. Opt. Commun. 2013, 291, 138.
-
[16]
Wang, W.; Ding, M.; Lu, C.; Ni, Y.; Xu, Z. Appl. Catal. B-Environ. 2014, 144, 379. 10.1016/j.apcatb.2013.07.035
-
[17]
Fix, T.; Ferblantier, G.; Rinnert, H.; Slaoui, A. Sol. Energy Mater. Sol. Cells 2015, 132, 191. 10.1016/j.solmat.2014.09.009
-
[18]
Huang, X.; Han, S.; Huang, W.; Liu, X. Chem. Soc. Rev. 2013, 42(1), 173. 10.1039/C2CS35288E
-
[19]
Khan, A. F.; Yadav, R.; Mukhopadhya, P. K.; Singh, S.; Dwivedi, C.; Dutta, V.; Chawla, S. J. Nanopart. Res. 2011, 13(12), 6837. 10.1007/s11051-011-0591-9
-
[20]
Gao, D.; Zhang, X.; Zheng, H.; Shi, P.; Li, L.; Ling, Y. Dalton Trans. 2013, 42(5), 1834.
-
[21]
Zhang, H. B.; Bu, Y. Y.; Yang, X. L.; Xiao, S. G.; Ding, J. W. Mater. Sci. Eng. B-Adv. 2011, 176(3), 256. 10.1016/j.mseb.2010.12.006
-
[22]
Wang, F.; Deng, R.; Wang, J.; Wang, Q.; Han, Y.; Zhu, H.; Chen, X.; Liu, X. Nat. Mater. 2011, 10(12), 968.
-
[23]
Zhao, J. Z.; Ji, S. M.; Guo, H. M. RSC Adv. 2011, 1(6), 937. 10.1039/c1ra00469g
-
[24]
Huang, Q.; Yu, H.; Zhang, X.; Yu, J. Acta Chim. Sinica 2013, 71(7), 1071. (黄清明, 俞瀚, 张新奇, 俞建长, 化学学报, 2013, 71(7), 1071.) http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract342074.shtml
-
[25]
Wang, F.; Deng, R. R.; Wang, J.; Wang, Q. X.; Han, Y.; Zhu, H. M.; Chen, X. Y.; Liu, X. G. Nat. Mater. 2011, 10(12), 968. 10.1038/nmat3149
-
[26]
Vetrone, F.; Naccache, R.; Mahalingam, V.; Morgan, C. G.; Capobianco, J. A. Adv. Funct. Mater. 2009, 19(18), 2924. 10.1002/adfm.v19:18
-
[27]
Su, Q. Q.; Han, S. Y.; Xie, X. J.; Zhu, H. M.; Chen, H. Y.; Chen, C. K.; Liu, R. S.; Chen, X. Y.; Wang, F.; Liu, X. G. J. Am. Chem. Soc. 2012, 134(51), 20849. 10.1021/ja3111048
-
[28]
Luo, Q.; Chen, Y.; Li, Z.; Zhu, F.; Chen, X.; Sun, Z.; Wei, Y.; Guo, H.; Wang, Z. B.; Huang, S. Nanotechnology 2014, 25(18), 185401. 10.1088/0957-4484/25/18/185401
-
[29]
Cheng, Q.; Sui, J. H.; Cai, W. Nanoscale 2012, 4(3), 779. 10.1039/C1NR11365H
-
[30]
Yu, H.; Huang, Q.; Cao, W.; Zhang, X.; Yu, J. Acta Chim. Sinica 2013, 71(12), 1639. (俞瀚, 黄清明, 曹文兵, 张新奇, 俞建长, 化学学报, 2013, 71(12), 1639.) http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract343607.shtml
-
[31]
Chen, D. Q.; Yu, Y. L.; Huang, F.; Huang, P.; Yang, A. P.; Wang, Y. S. J. Am. Chem. Soc. 2010, 132(29), 9976. 10.1021/ja1036429
-
[32]
Tian, G.; Gu, Z. J.; Zhou, L. J.; Yin, W. Y.; Liu, X. X.; Yan, L.; Jin, S.; Ren, W. L.; Xing, G. M.; Li, S. J.; Zhao, Y. L. Adv. Mater. 2012, 24(9), 1226. 10.1002/adma.v24.9
-
[33]
Wang, F.; Han, Y.; Lim, C. S.; Lu, Y. H.; Wang, J.; Xu, J.; Chen, H. Y.; Zhang, C.; Hong, M. H.; Liu, X. G. Nature 2010, 463(7284), 1061. 10.1038/nature08777
-
[34]
Richardson, F. S. Inorg. Chem.1980, 19(9), 2806. 10.1021/ic50211a063
-
[35]
Huang, Q.; Yu, H.; Ma, E.; Zhang, X.; Cao, W.; Yang, C.; Yu, J. Inorg. Chem. 2015, 54(6), 2643.
-
[36]
Judd, B. R. Phys. Rev. 1962, 127, 750. 10.1103/PhysRev.127.750
-
[37]
Ofelt, G. S. J. Chem. Phys. 1962, 37, 511. 10.1063/1.1701366
-
[38]
Huang, Q.; Yu, J.; Ma, E.; Lin, K. J. Phys. Chem. C 2010, 114(10), 4719. 10.1021/jp908645h
-
[1]
-

图 1 离子Yb3+、Tm3+和Hf4+之间的可能能量转移机制图
Figure 1 Proposed energy transfer mechanism among Yb3+,Hf4+and Tm3+

图 2 (a,b)分别是不同量Hf4+和Zr4+掺杂样品的实测XRD图谱(红线)和Rietveld全谱拟合计算图谱(黑线)及国际标准XRD衍射图谱JCPDS 16-0334
Figure 2 (a,b) Measured (red line) and Rietveld calculated (black line) XRD patterns of different amount Hf4+and Zr4+ codoped samples,respectively; and international standard XRD pattern JCPDS 16-0334

图 3 (a,b)分别是晶胞体积V和晶胞参数c/a随着Hf4+和Zr4+掺杂量增加的变化趋势图; (c,d)分别是F1-Y(r1)、F2-Y(r2)及Y-Y(R+)之间的键长随着Hf4+和Zr4+掺杂量增加的变化趋势图.
Figure 3 (a,b) Changing trend of cell volume V and cell parameters c/a with the rising of Hf4+and Zr4+ codoped concentration,respectively. (c,d) changing trend of bond distance of F1-Y(r1),F2-Y(r2) and Y-Y(R+) with the rising of Hf4+ and Zr4+ codoped concentration,respectively

图 4 (a)样品Hf2的透射电镜照片,(b)是(a)中红色区域的高分辨电镜照片
Figure 4 (a) TEM image of Hf2 sample,(b) HR-TEM image of red square area of (a)

图 5 (a,b)分别是不同量Hf4+和Zr4+共掺杂样品的上转换发射光谱; (c,e,g,i)分别是不同量Hf4+和Zr4+掺杂样品的475 nm和802 nm发射带的荧光实测衰减谱和单指数衰减拟合计算谱; (d,f,h,j)分别是各样品475 nm和802 nm发射带的荧光增强时间常数和荧光寿命
Figure 5 (a,b) upconversion spectra of different amount Hf4+and Zr4+ codoped samples,respectively; (c,e,g,i) measured and calculated decay curve of 475 nm and 802 nm emissions of different concentration Hf4+and Zr4+ codoped samples,respectively; (d,f,h,j) changing trend of rising and decay time constant of 475 nm and 802 nm emissions with the rising of Hf4+and Zr4+ codoped concentration,respectively
-


 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 2672
- HTML全文浏览量: 479
 下载:
下载:
