Received Date:
10 October 2020 Revised Date:
20 November 2020 Available Online:
10 February 2021
Abstract:
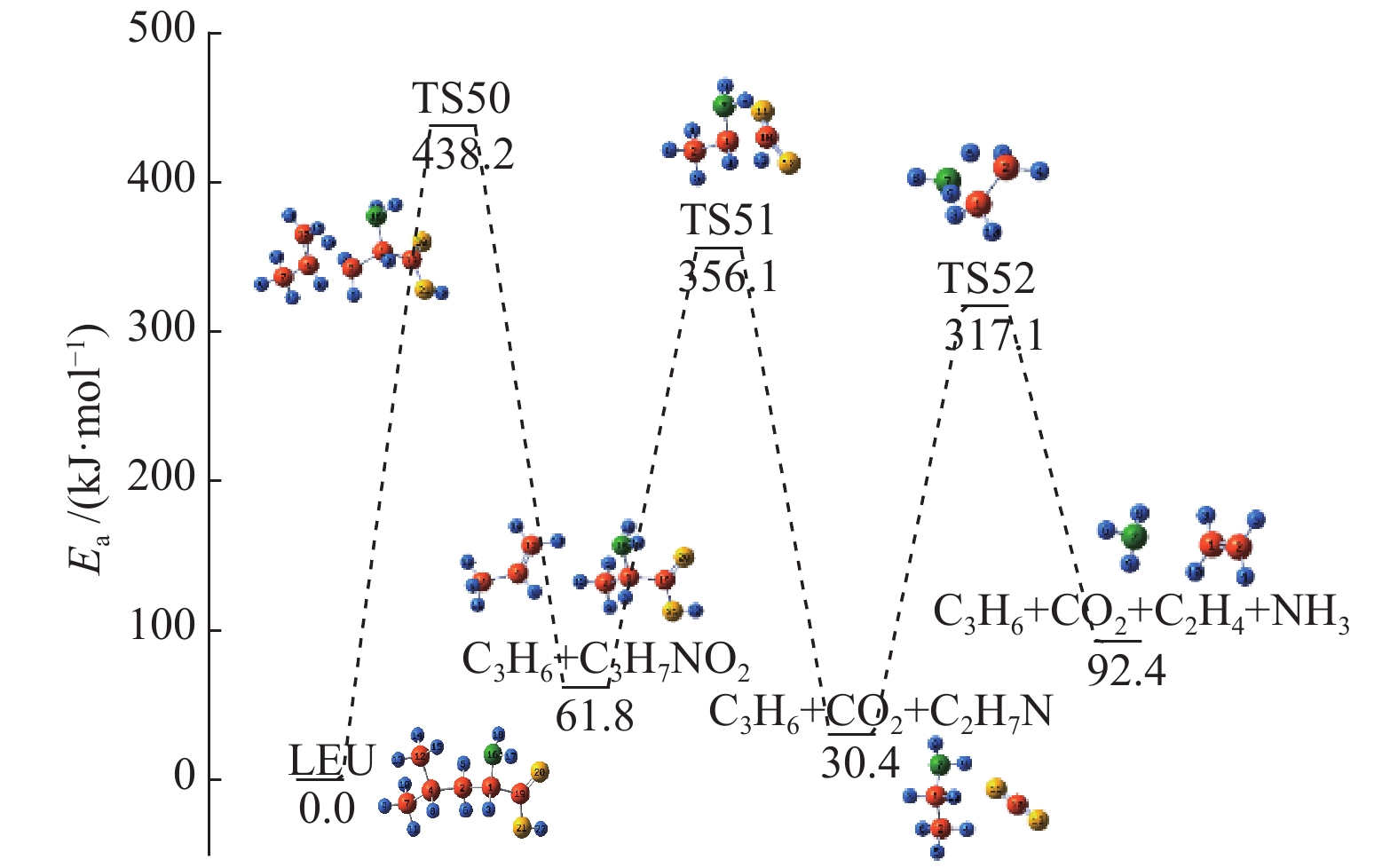
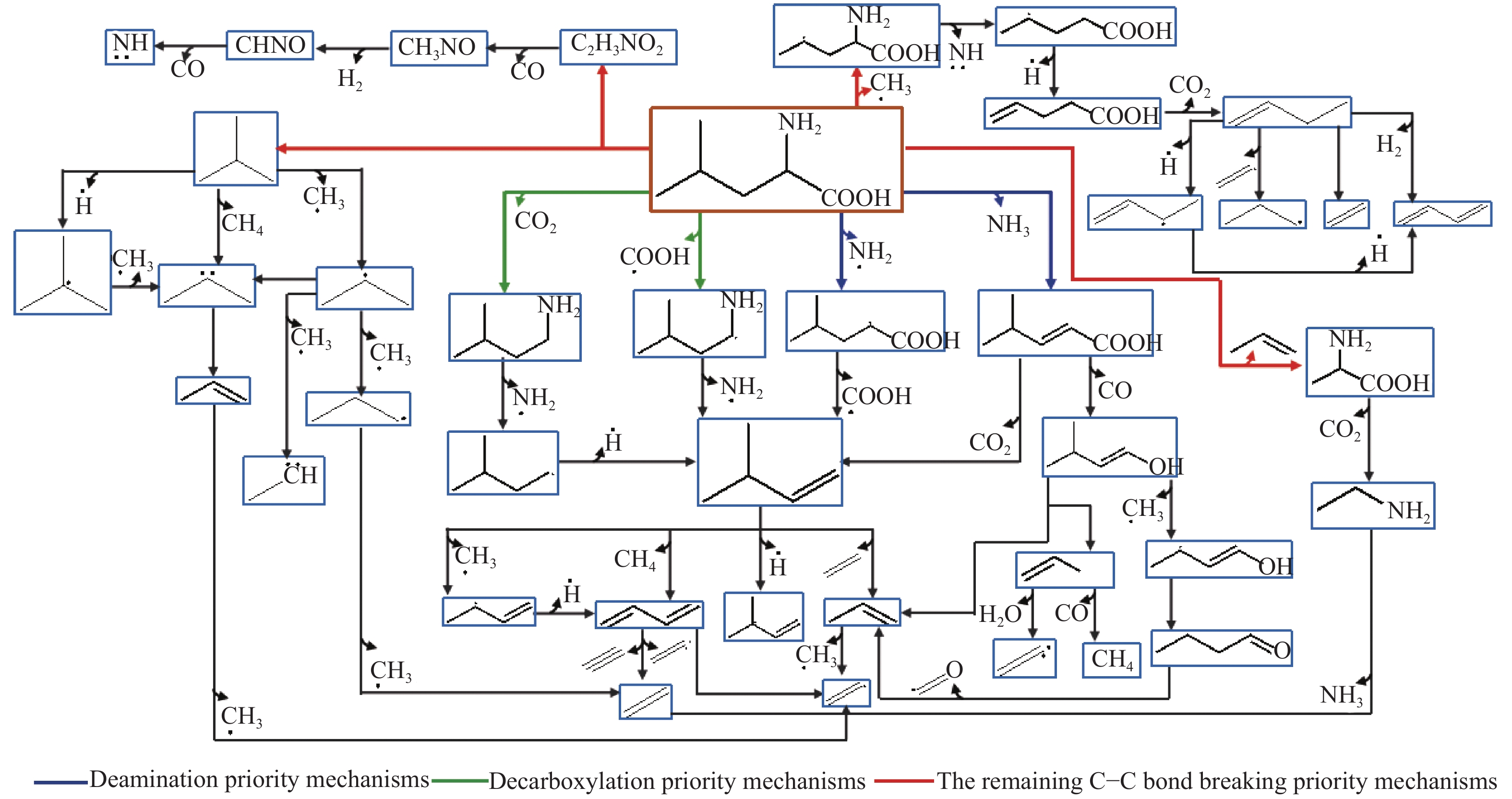
At present, non-thermal plasma technology has received extensive attention in the treatment of solid waste. Based on the density functional theory (DFT), the conversion path of leucine (LEU) as the model compound of protein in sludge during the non-thermal plasma treatment was simulated at the B3LYP/6-31G(d,p) level; 7 main conversion paths were considered, including the deamination priority mechanisms, decarboxylation priority mechanisms, and the remaining C−C bond breaking priority mechanisms. The results show that leucine is easy to lose the amino group and carboxyl group, generating C5H10 which is further decomposed into small molecular hydrocarbons. The CO2 product comes from the carboxyl group; although the reaction barrier to form CO is relatively high, CO2 is easily ionized into CO in the plasma, leading to the increase of CO concentration. The combination of small free radicals and the decomposition of other small molecules generate CH4 and H2. The energy required for all paths is within the maximum value of the high-energy electron energy in the non-thermal plasma.
DU C M, WU J. , Ma D Y, LIU Y, QIU P P, QIU R L, LIAO S S, GAO D. Gasification of corn cob using non-thermal arc plasma[J]. Int J Hydrog Energy,2015,40(37):12634−12649. doi: 10.1016/j.ijhydene.2015.07.111
[6]
孙世翼. 放电等离子体强化处理污泥减量及重金属去除[D]. 兰州: 西北师范大学, 2018.SUN Shi-yi. Discharge plasma enhanced treatment sludge reduction and heavy metal removal[D]. Lanzhou: Northwest Normal University, 2018.
[7]
CHEN S S, DONG B, DAI X H, WANG H Y, LI N, YANG D H. Effects of thermal hydrolysis on the metabolism of amino acids in sewage sludge in anaerobic digestion[J]. Waste Manage,2019,88:309−318. doi: 10.1016/j.wasman.2019.03.060
[8]
郑燕, 李明, 朱锡锋. 城市污水污泥催化快速热解制备芳香烃和烯烃[J]. 化工学报,2016,67(11):4802−4807.ZHENG Yan, LI Ming, ZHU Xi-feng. Aromatics and olefines were prepared by fast catalytic pyrolysis of municipal sewage sludge[J]. CIESC Jo,2016,67(11):4802−4807.
[9]
PENG C, ZHAI Y B, HORNUNG A, WANG B, LI S H, WANG T F, LI C T, ZHU Y. In-depth comparison of morphology, microstructure, and pathway of char derived from sewage sludge and relevant model compounds[J]. Waste Management,2020,102:432−440. doi: 10.1016/j.wasman.2019.11.007
[10]
AZADI P, AFIF E, FOROUHI H, DAI T S, AZADI F. , FARNOOD R. Catalytic reforming of activated sludge model compounds in supercritical water using nickel and ruthenium catalysts[J]. Appl Catal B: Environ.,2013,134-135:265−273. doi: 10.1016/j.apcatb.2013.01.022
[11]
WANG C Y, FAN Y J, HORNUNG U, ZHU W, DANMEN N. Char and tar formation during hydrothermal treatment of sewage sludge in subcritical and supercritical water: Effect of organic matter composition and experiments with model compounds[J]. J Clean Prod,2020,242:1−9.
[12]
WEI F, CAO J P, ZHAO X Y, REN J, GU B, WEI X Y. Formation of aromatics and removal of nitrogen in catalytic fast pyrolysis of sewage sludge: A study of sewage sludge and model amino acids[J]. Fuel,2018,218:148−154. doi: 10.1016/j.fuel.2018.01.025
[13]
SUBRAHMANYAM P V R, SASTRY C A, RAO A V S P, PILLAI S C. Amino acids in sewage sludges[J]. J Water Pollut Control Fed,1960,32(4):344−350.
[14]
张军. 微波热解污水污泥过程中氮转化途径及调控策略[D]. 哈尔滨: 哈尔滨工业大学, 2013.ZHANG Jun. Nitrogen conversion pathways and control strategies in the process of microwave pyrolysis of sewage sludge[D]. Harbin: Harbin Institute of Technology, 2013.
[15]
LI J, WANG Z Y, YANG X, HU L, LIU Y W, WANG C X. Decomposing or subliming? An investigation of thermal behavior of l-leucine[J]. Thermochim Acta,2006,447(2):147−153. doi: 10.1016/j.tca.2006.05.004
[16]
ZHAO S H, BI X L, SUN R Y, NIU M M, PAN X J. Density functional theory and experimental study of cellulose initial degradation stage under inert and oxidative atmosphere[J]. J Mol Struct,2020,1204:1−10.
[17]
YANG X X, FU Z W, HAN D D, ZHAO Y Y, LI R, WU Y L. Unveiling the pyrolysis mechanisms of cellulose: Experimental and theoretical studies[J]. Renewable Energy,2020,147:1120−1130. doi: 10.1016/j.renene.2019.09.069
[18]
ZHANG Y Y, LIU C, XIE H. Mechanism studies on β-d-glucopyranose pyrolysis by density functional theory methods[J]. J Anal Appl Pyrolysis,2014,105:23−34. doi: 10.1016/j.jaap.2013.09.016
[19]
HUANG X Y, CHENG D G, CHEN F Q, ZHAN X L. A density functional theory study on the decomposition of aliphatic hydrocarbons and cycloalkanes during coal pyrolysis in hydrogen plasma[J]. J Energy Chem,2015,24(1):65−71. doi: 10.1016/S2095-4956(15)60285-6
[20]
黄金保, 武书彬, 雷鸣, 程皓, 梁嘉晋, 童红. 木质素二聚体模型化合物热解机理的量子化学研究[J]. 燃料化学学报,2015,43(11):1334−1343. doi: 10.3969/j.issn.0253-2409.2015.11.008HUANG Jin-bao, WU Shu-bin, LEI Ming, CHENG Hao, LIANG Jia-jin, TONG Hong. Quantum chemistry study on the pyrolysis mechanism of lignin dimer model compounds[J]. J Fuel Chem Technol,2015,43(11):1334−1343. doi: 10.3969/j.issn.0253-2409.2015.11.008
[21]
程小彩, 黄金保, 潘贵英, 童红, 蔡勋明. 聚苯乙烯热降解机理的理论研究[J]. 燃料化学学报,2019,47(7):884−896. doi: 10.3969/j.issn.0253-2409.2019.07.014CHENG Xiao-cai, HUANG Jin-bao, PAN Gui-ying, TONG Hong, CAI Xun-ming. Theoretical study on the thermal degradation mechanism of polystyrene[J]. J Fuel Chem Technol,2019,47(7):884−896. doi: 10.3969/j.issn.0253-2409.2019.07.014
[22]
HUANG X Y, GU J M, CHENG D G, CHEN F Q, ZHAN X L. Pathways of liquefied petroleum gas pyrolysis in hydrogen plasma: A density functional theory study[J]. J Energy Chem,2013,22(3):484−492. doi: 10.1016/S2095-4956(13)60063-7
[23]
CHEN L, CHENG D G, CHEN F Q, ZHAN X L. A density functional theory study on the conversion of polycyclic aromatic hydrocarbons in hydrogen plasma[J]. Int J Hydrog Energy,2020,45(1):309−321. doi: 10.1016/j.ijhydene.2019.10.208
[24]
黄金保, 刘朝, 任丽蓉, 童红, 李伟民, 伍丹. 木质素模化物紫丁香酚热解机理的量子化学研究[J]. 燃料化学学报,2013,41(6):657−666.HUANG Jin-bao, LIU Chao, REN Li-rong, TONG Hong, LI Wei-min, WU Dan. Studies on pyrolysis mechanism of syringol as lignin model compound by quantum chemistry[J]. J Fuel Chem Technol,2013,41(6):657−666.
[25]
HUANG X Y, CHENG D G, CHEN F Q, ZHAN X L. The decomposition of aromatic hydrocarbons during coal pyrolysis in hydrogen plasma: A density functional theory study[J]. Int J Hydrog Energy,2012,37(23):18040−18049. doi: 10.1016/j.ijhydene.2012.09.006
[26]
段毓, 程皓, 武书彬. 基于密度泛函理论研究木质素二聚体Cα-OH基团的修饰对其热解均裂历程的影响[J]. 燃料化学学报,2019,47(12):1440−1448. doi: 10.3969/j.issn.0253-2409.2019.12.004DUAN Yu, CHENG Hao, WU Shu-bin. Study on the effect of modification of Cα-OH group of lignin dimer on its pyrolysis homocracking process based on density functional theory[J]. J Fuel Chem Technol,2019,47(12):1440−1448. doi: 10.3969/j.issn.0253-2409.2019.12.004
[27]
MUDEDLA S K, KUMAR C V S, SURESH A, BASKAR P, DASH P S, SUBRAMANIAN V. Water catalyzed pyrolysis of oxygen functional groups of coal: A density functional theory investigation[J]. Fuel,2018,233:328−335. doi: 10.1016/j.fuel.2018.06.057
[28]
HUANG J B, LIU C, TONG H, LI W M, WU D. A density functional theory study on formation mechanism of CO, CO2 and CH4 in pyrolysis of lignin[J]. Comput Theor Chem,2014,1045:1−9. doi: 10.1016/j.comptc.2014.06.009
[29]
张晓星, 胡雄雄, 肖焓艳. 介质阻挡放电等离子体降解SF6的实验与仿真研究[J]. 中国电机工程学报,2017,37(8):2455−2465.ZHANG Xiao-xing, HU Xiong-xiong, XIAO Han-yan. Experimental and simulation study of SF6 degradation by dielectric barrier discharge plasma[J]. Proc CSEE,2017,37(8):2455−2465.
[30]
SIMMONDS P G, MEDLEY E E, RATCLIFF M A, Jr, SHULMAN G P. Thermal decomposition of aliphatic monoamino-monocar boxylic acids[J]. Anal Chem,1972,44(12):2060−2066. doi: 10.1021/ac60320a040
[31]
DEAN, A. M. Predictions of pressure and temperature effects upon radical addition and recombination reactions[J]. J. Phys. Chem. A,1985,89(21):4600−4608. doi: 10.1021/j100267a038
[32]
WESTBROOK C K, DRYER F L, SCHUG K P. Comprehensive mechanism for the pyrolysis and oxidation of ethylene[J]. Symp Combustion,1982,19(1):153−166. doi: 10.1016/S0082-0784(82)80187-2
[33]
TOWFIGHI J, NIAEI A, KARIMZADEH R, SAEDI G. Systematics and modelling representations of LPG thermal cracking for olefin production[J]. Korean J Chem Eng,2006,23(1):8−16. doi: 10.1007/BF02705685
[34]
王丽. 等离子体催化氨分解制氢的协同效应研究[D]. 大连: 大连理工大学, 2013.WANG Li. Study on the synergistic effect of plasma-catalyzed ammonia decomposition for hydrogen production[D]. Dalian: Dalian University of Technology, 2013.
[35]
刘广益. 污泥催化热解制取烃类化合物及转化途径研究[D]. 哈尔滨: 哈尔滨工业大学, 2016.LIU Guang-yi. Research on catalytic pyrolysis of sludge to produce hydrocarbon compounds and conversion route[D]. Harbin: Harbin Institute of Technology, 2016.
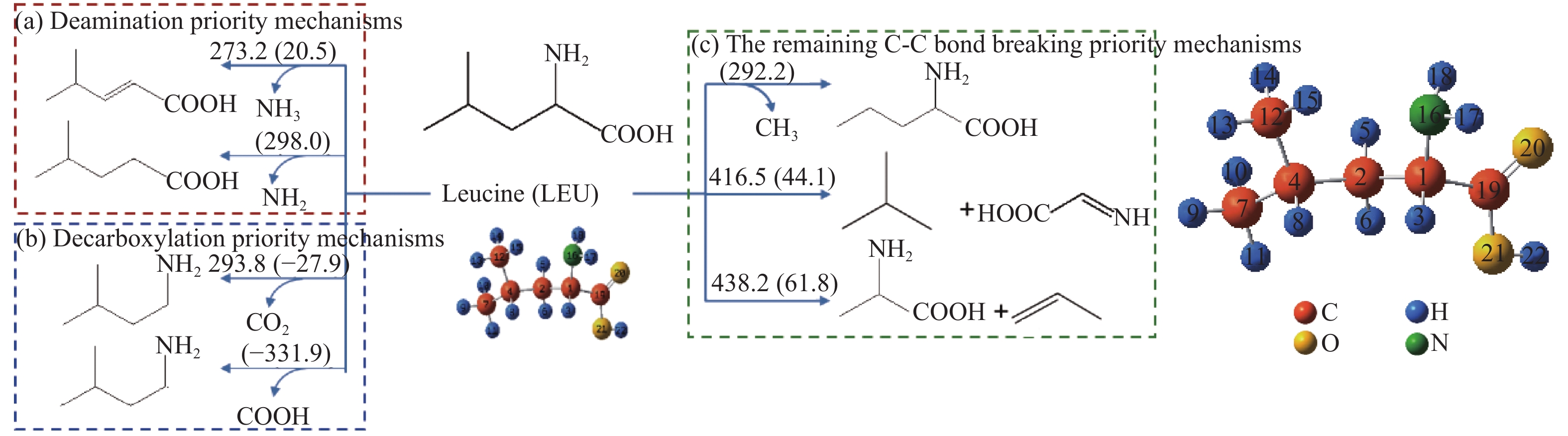
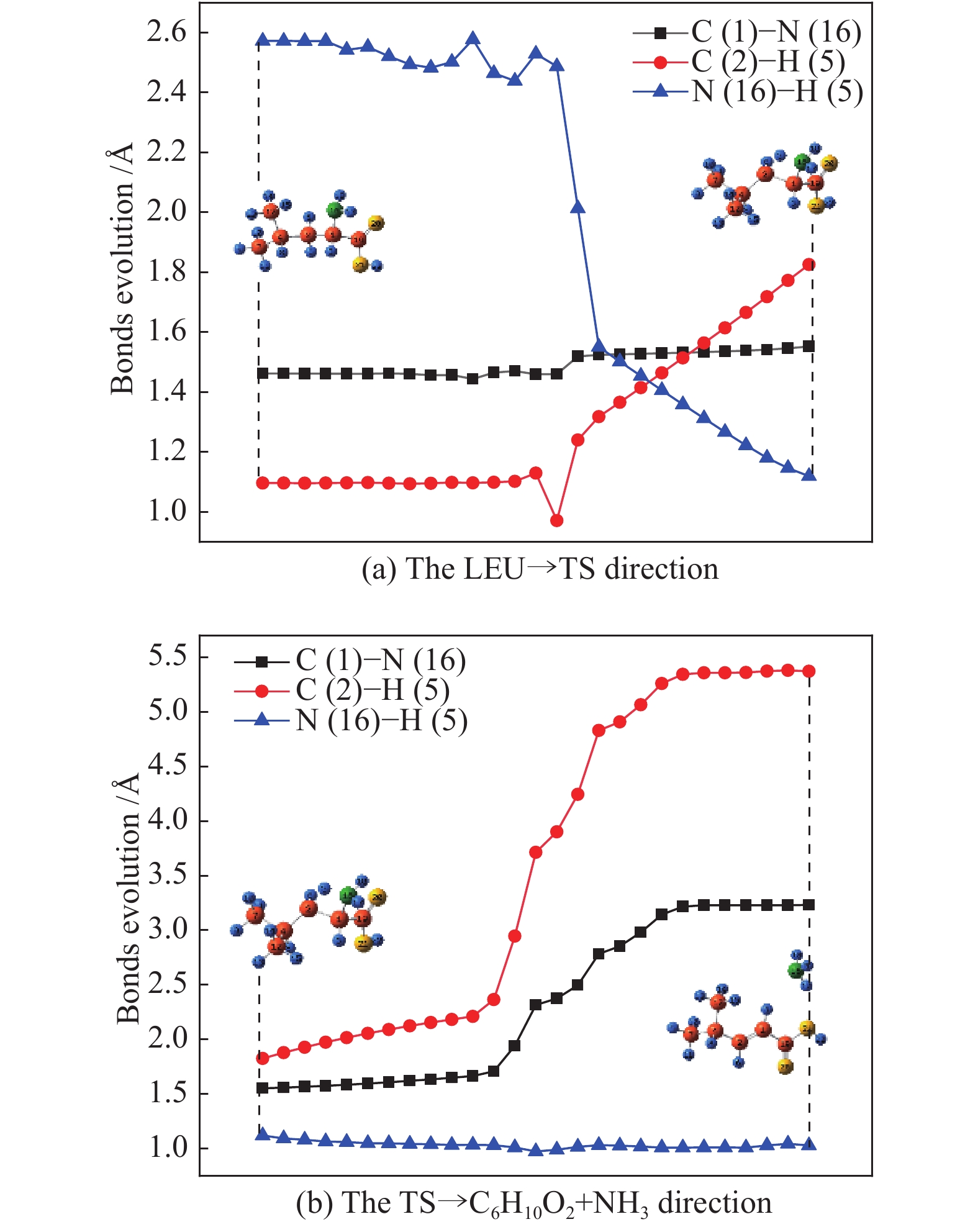
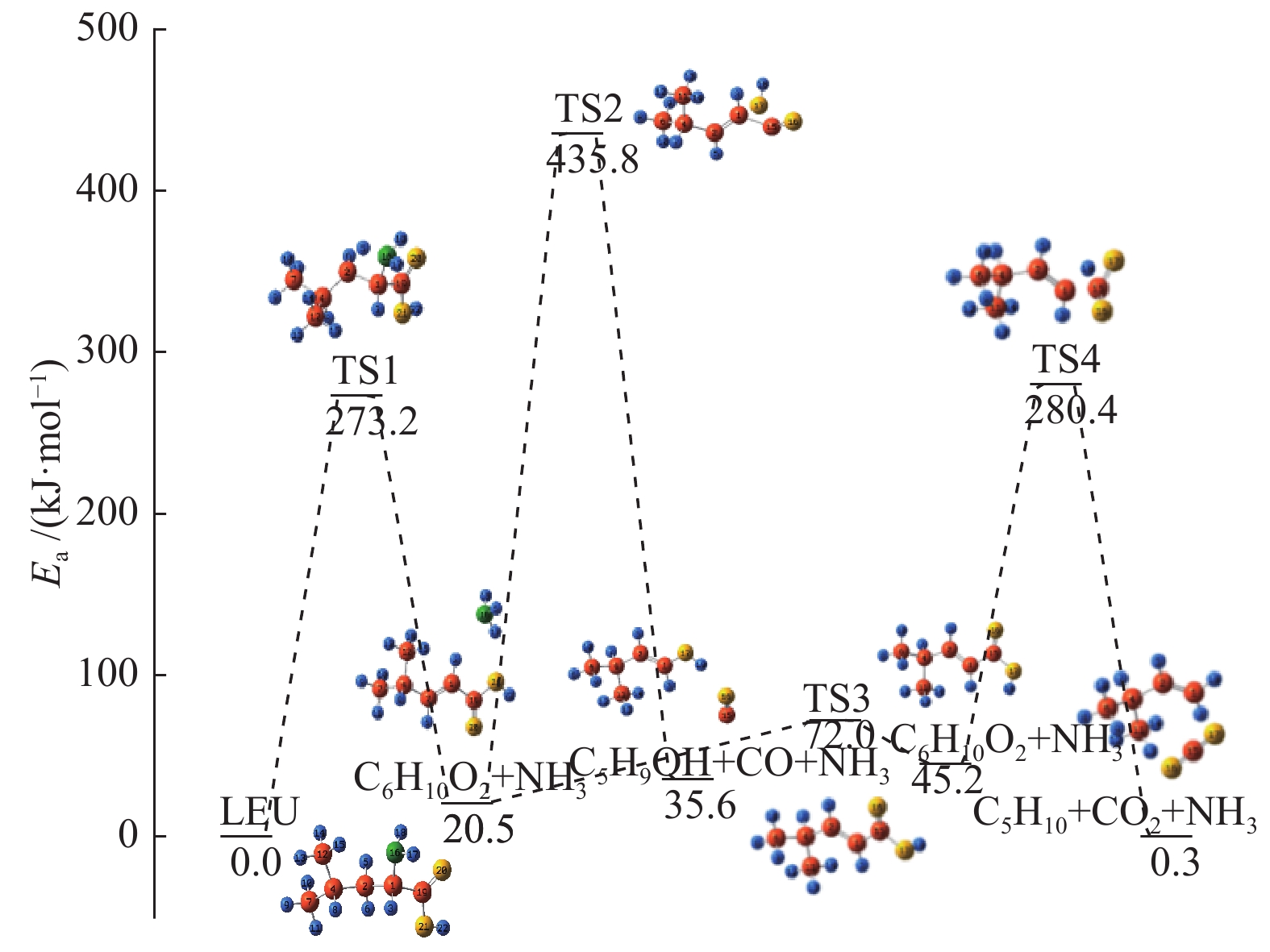
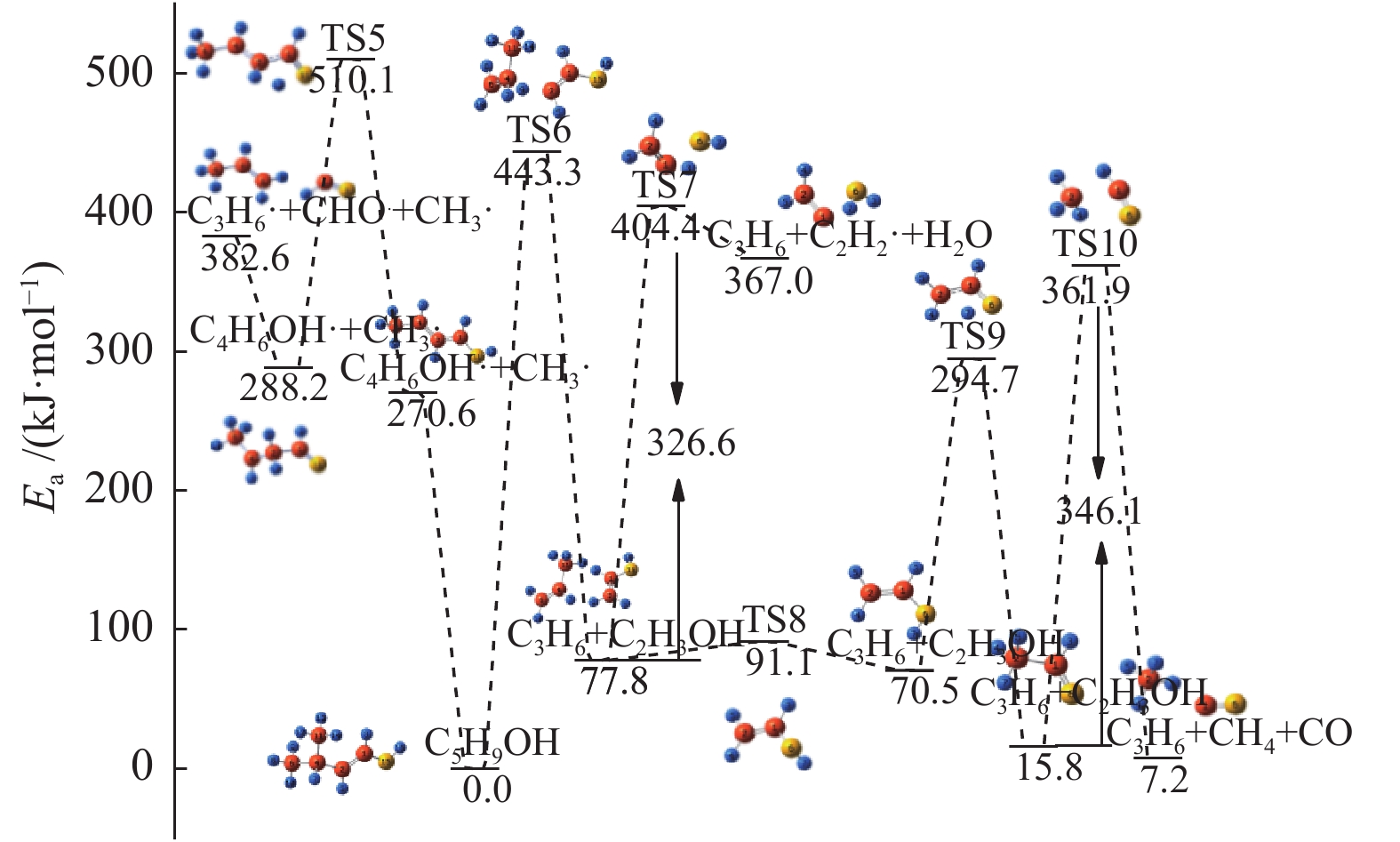
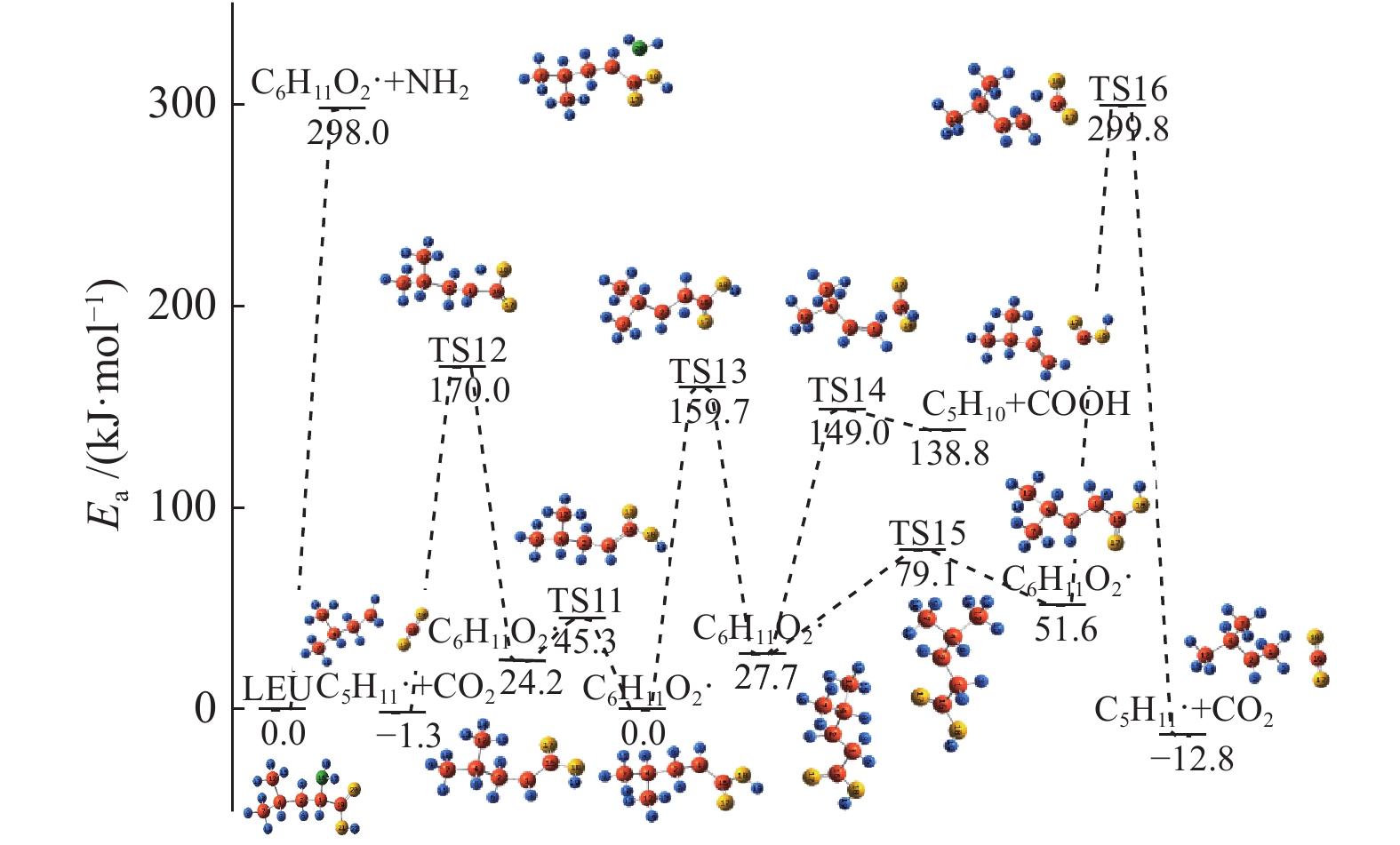
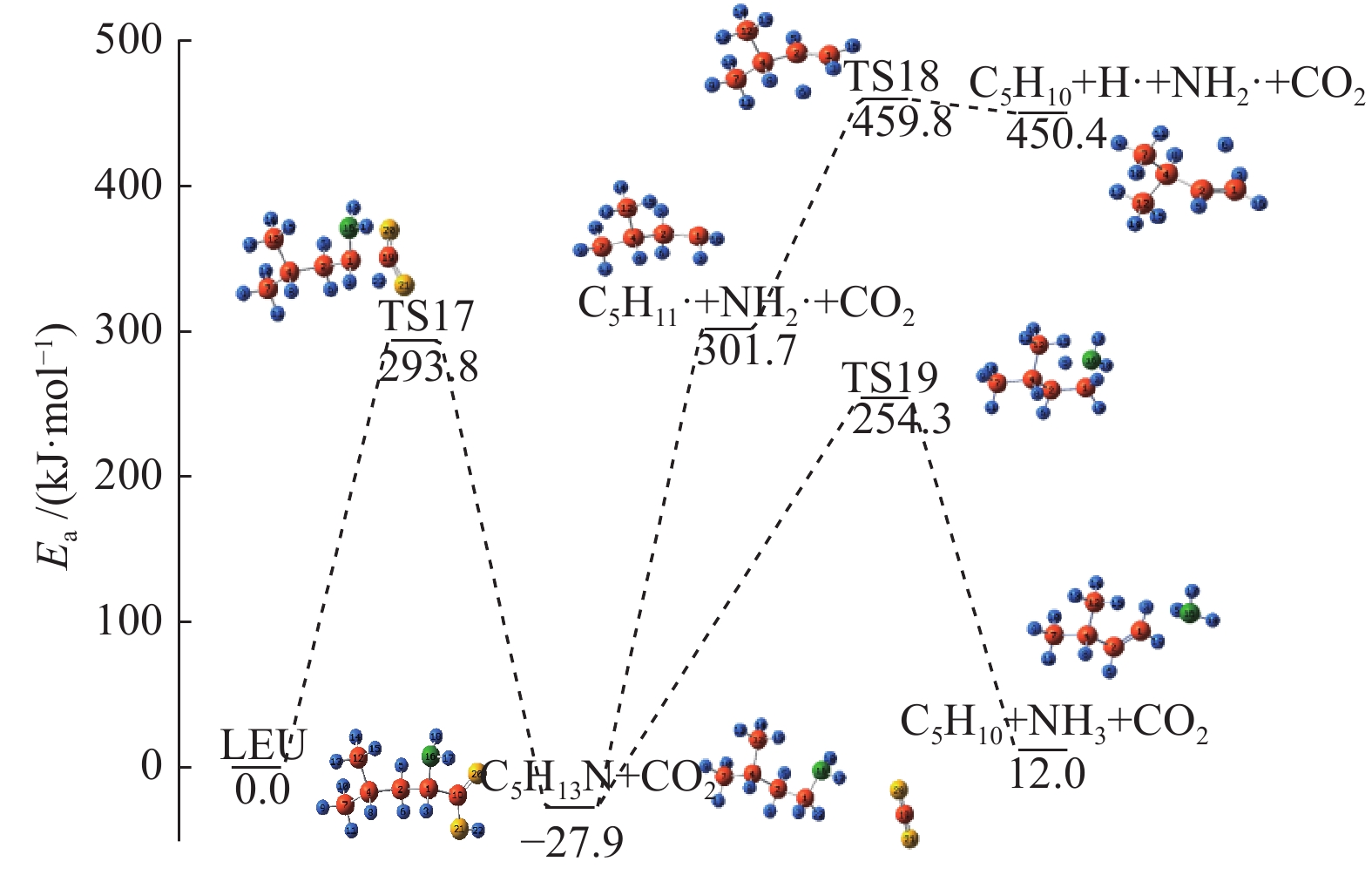
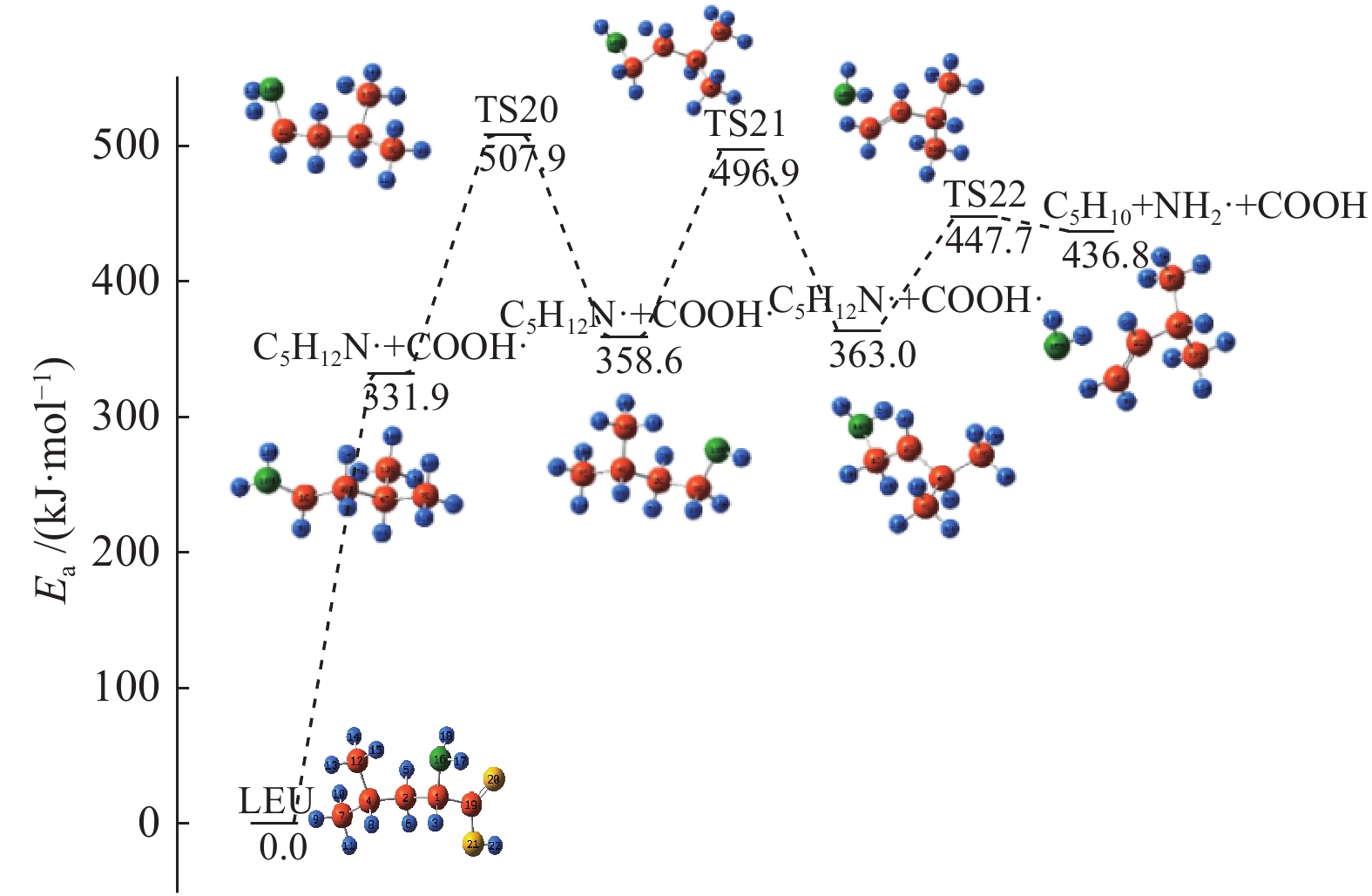
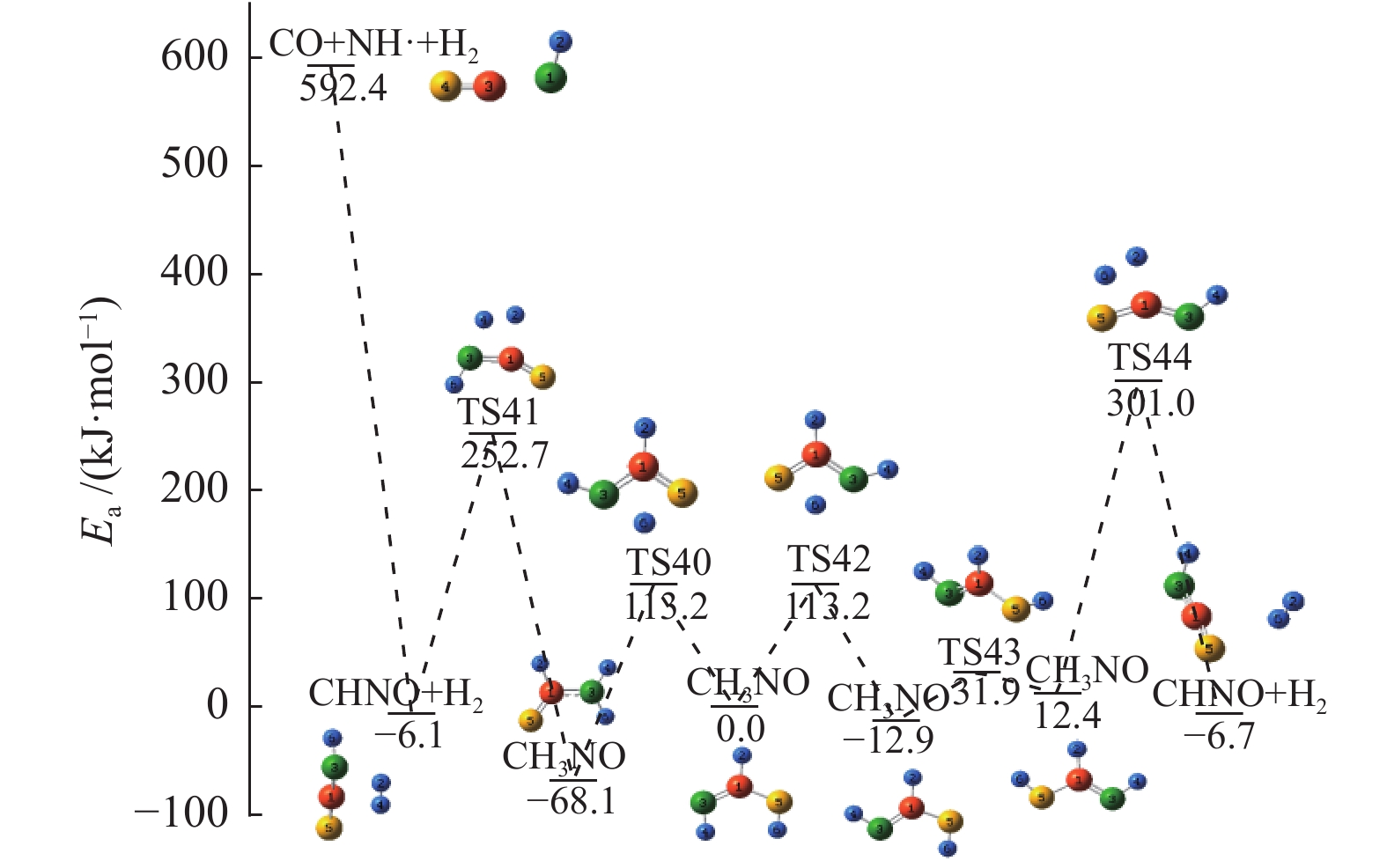
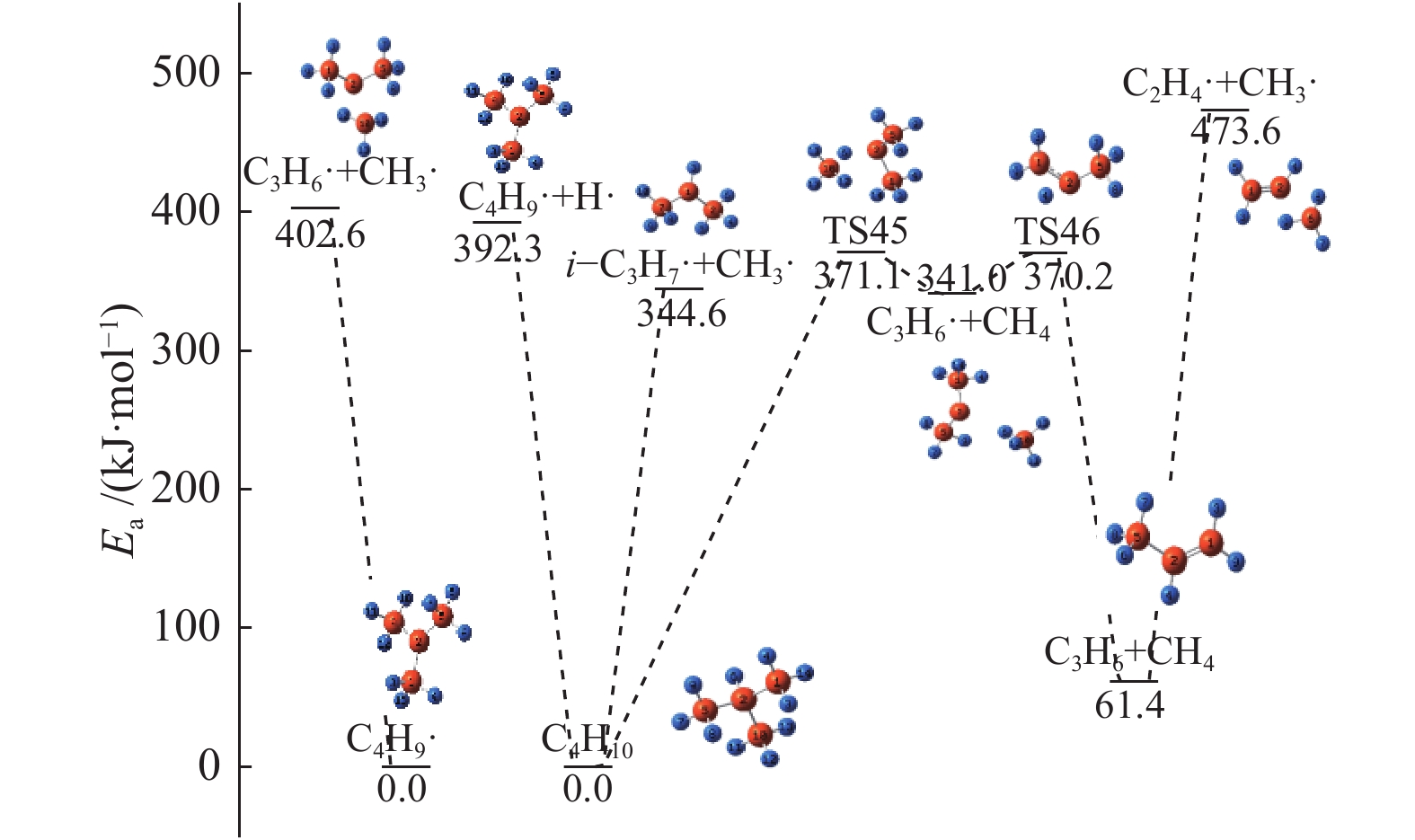
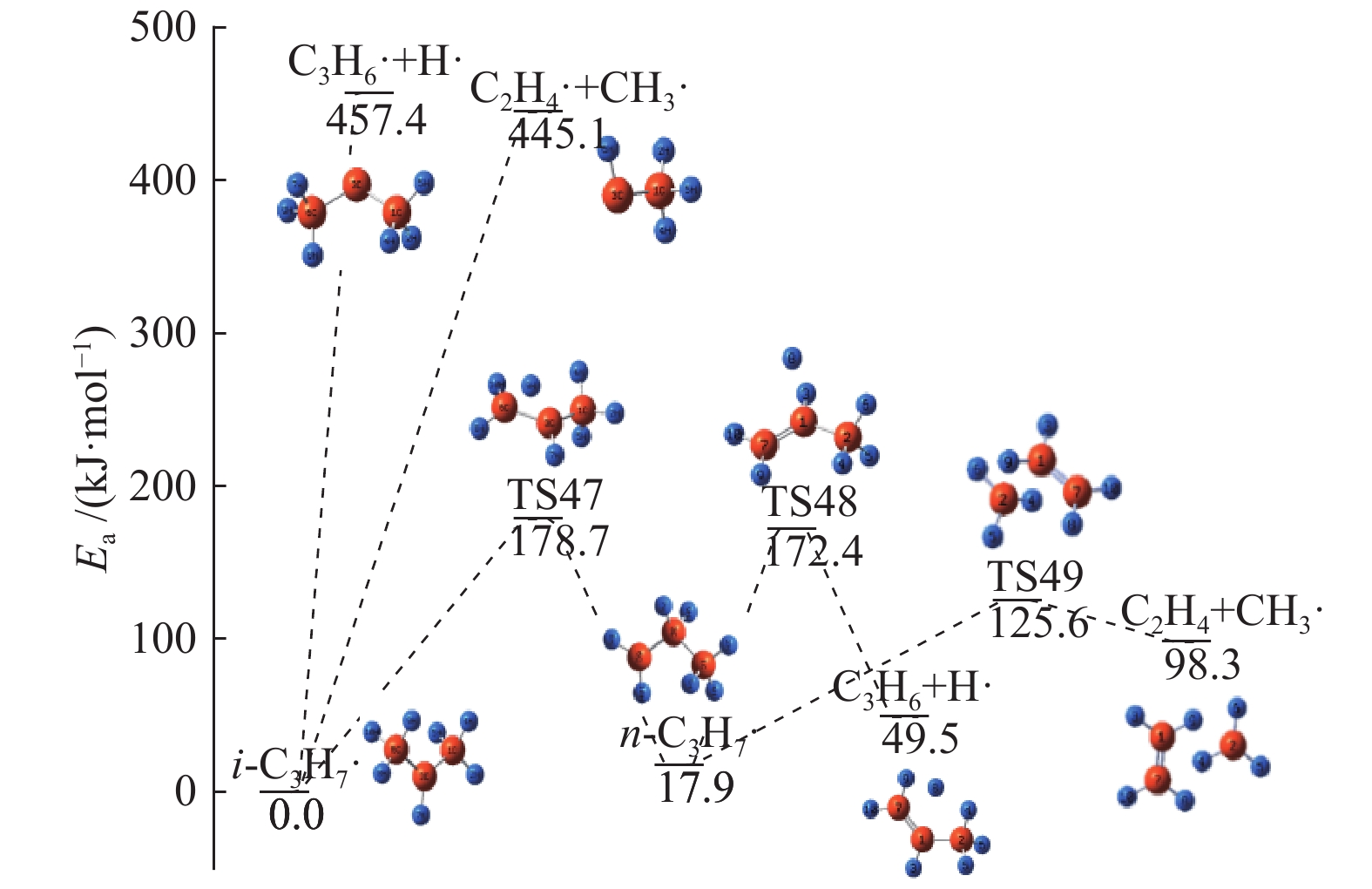
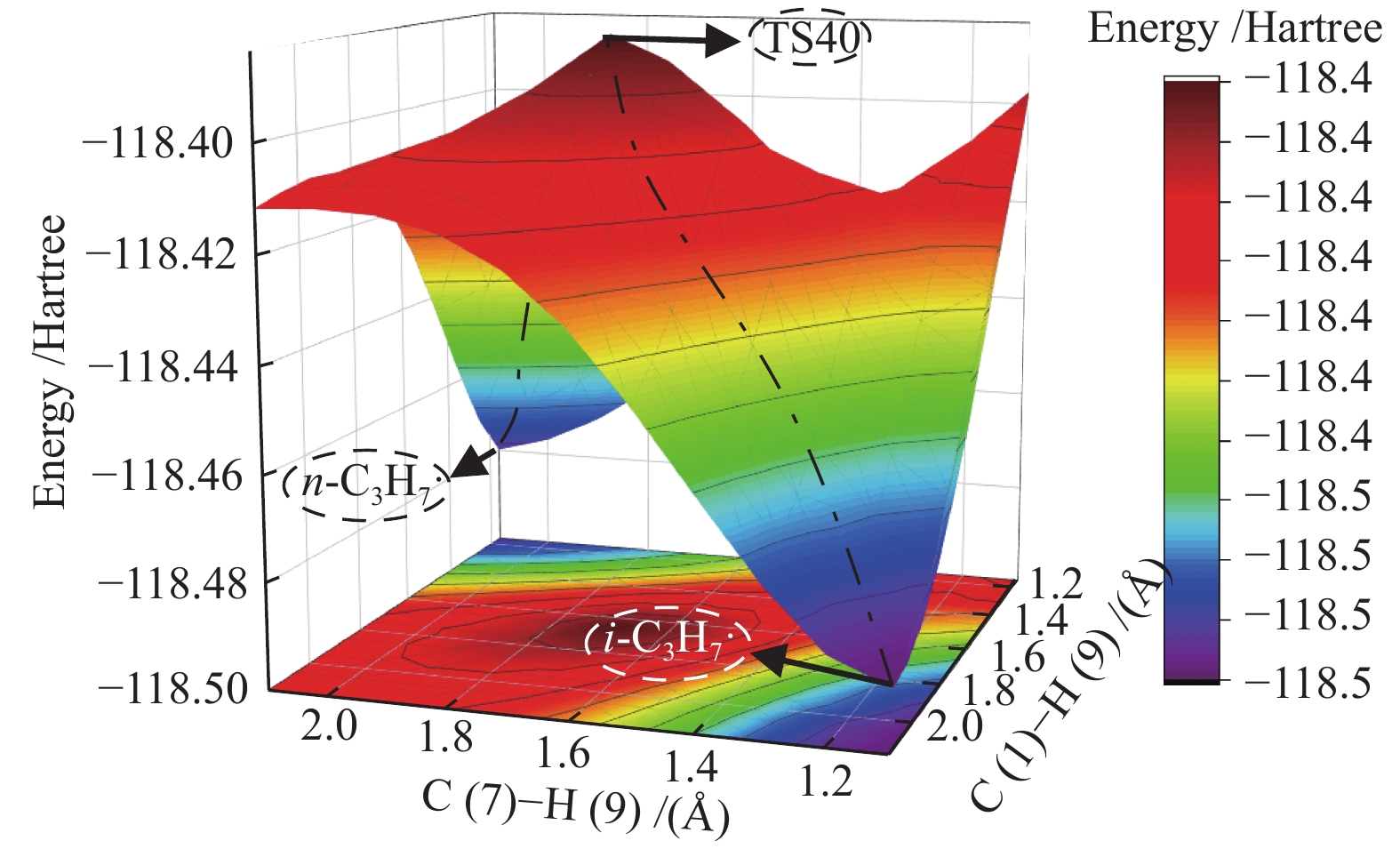
图 1
亮氨酸的初步分解路径及其分子构型
Figure 1
Leucine preliminary decomposition pathway and its molecular configuration
(a): deamination priority mechanisms; (b): decarboxylation priority mechanisms; (c): the remaining C−C bond breaking priority mechanisms

 下载:
下载:
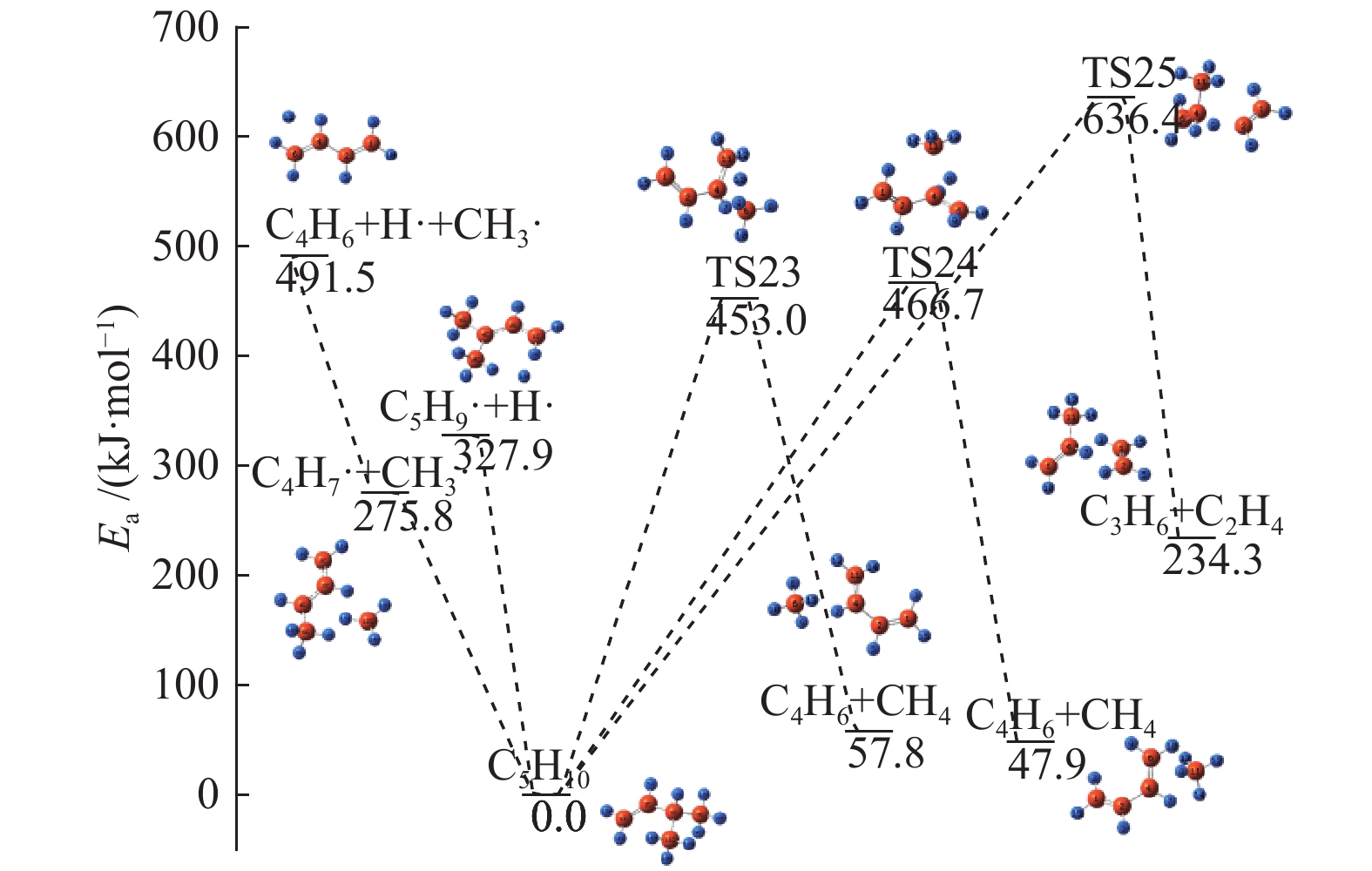
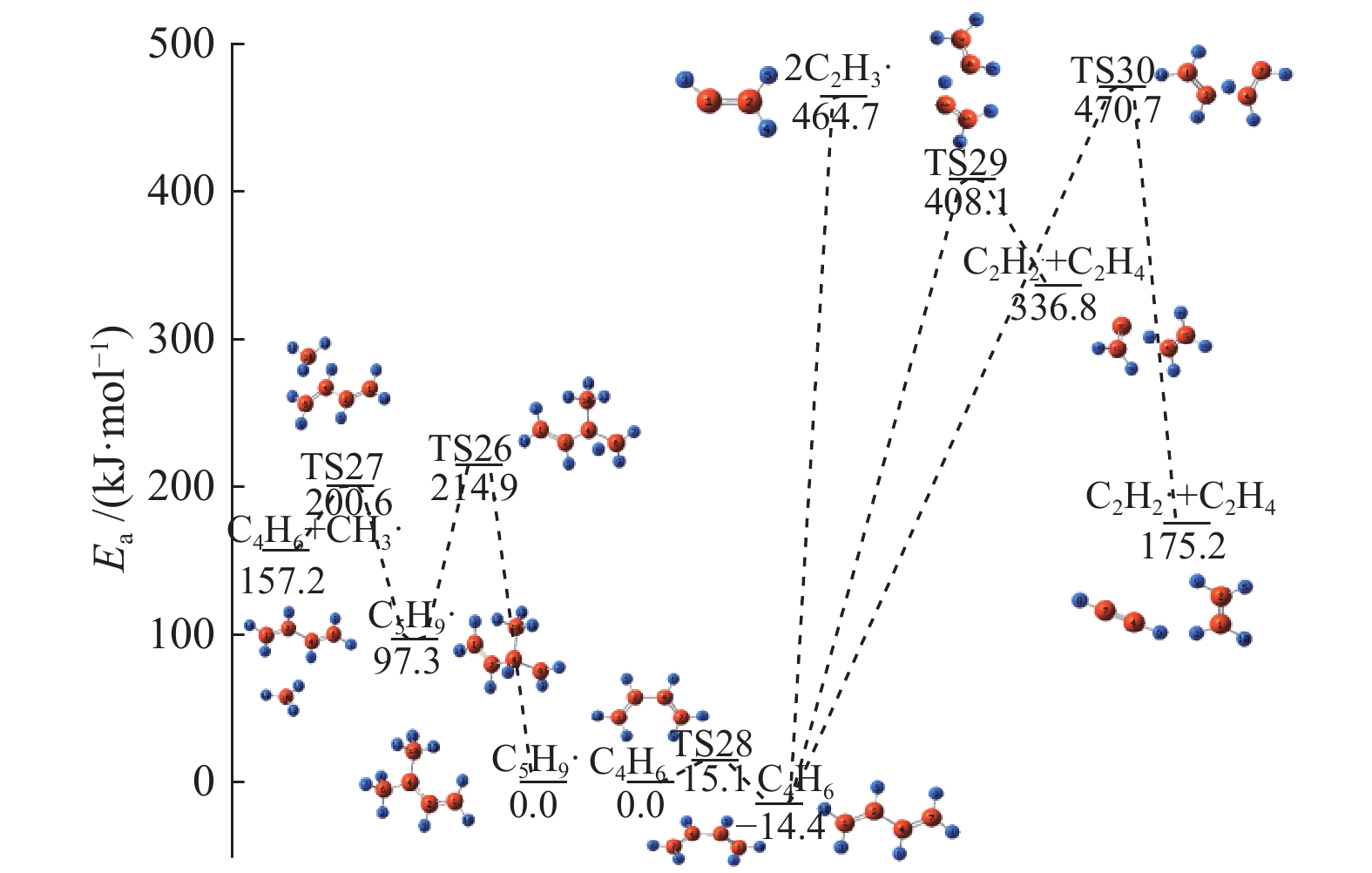
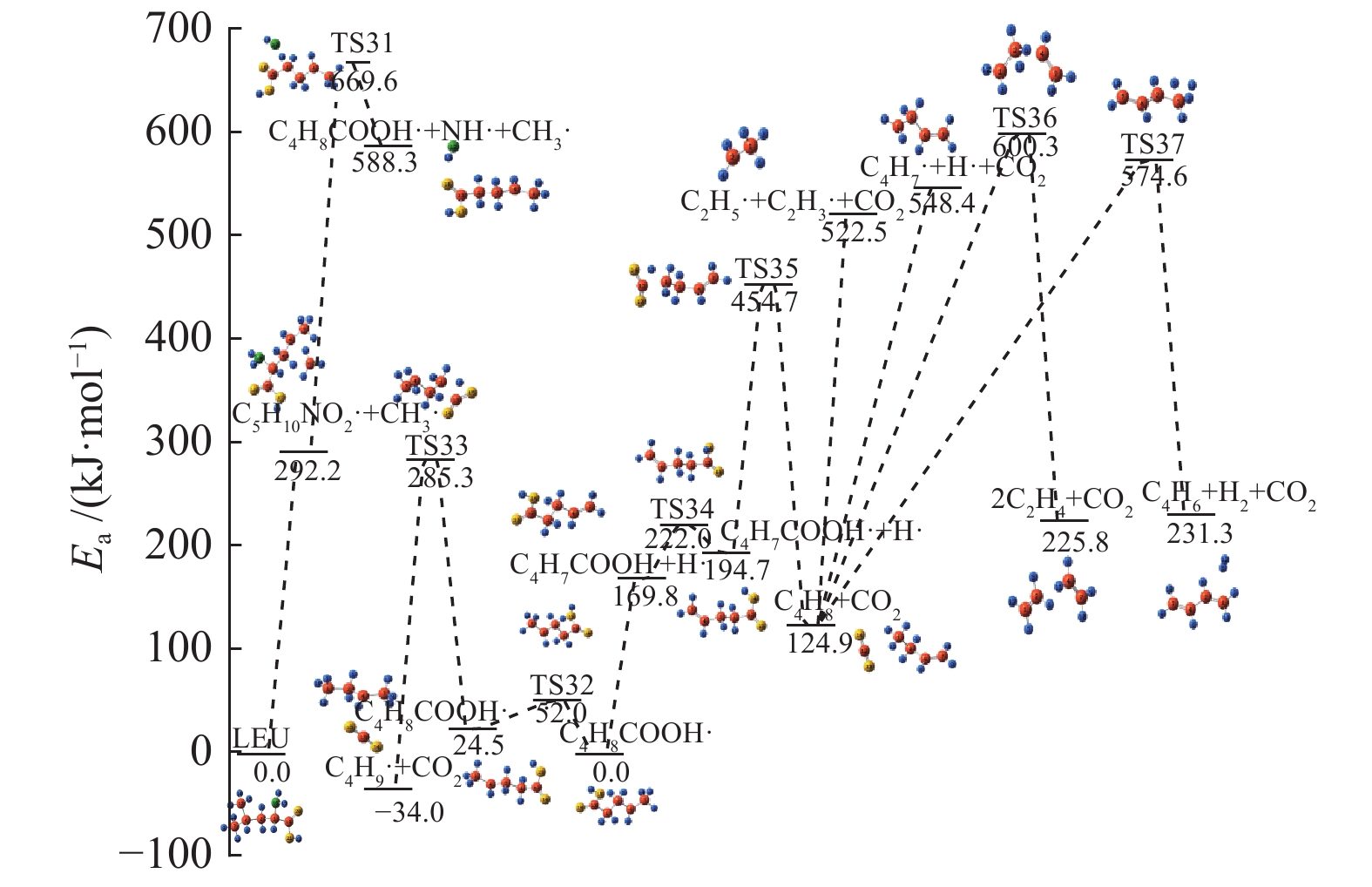
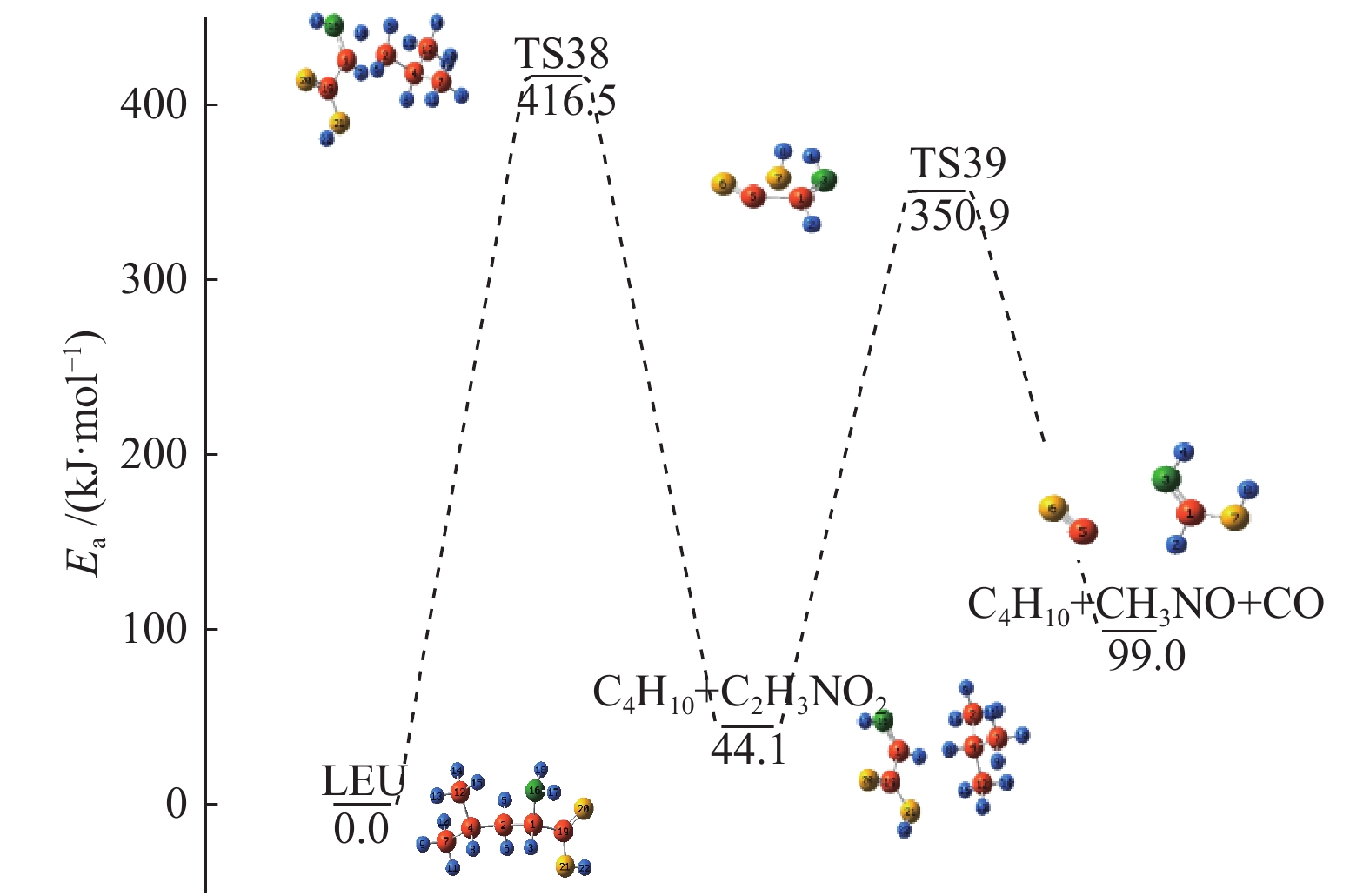

 下载:
下载:



















