图 1.
Co@C-X催化剂制备过程示意图
Figure 1.
Preparation process of Co@C-X catalyst

木质纤维素通常指植物材料,如农业残留物、林业废料和能源作物等,其主要由纤维素(22%−42%)、半纤维素(12%−27%)、木质素(11%−30%)三部分组成[1-4]。木质素是一种由不同类型的甲氧基苯丙基单元组成的复杂的三维非晶聚合物,通常是生物精炼厂和造纸厂的废弃物[5, 6]。目前,只有5%的木质素用于低价值的商业应用,如作为混凝土添加剂(木质素磺酸盐)[7]。将木质素转化为高附加值的化学品既能提高其经济效益,也有利于解决木质纤维素利用过程中的废弃物处理问题。
通过木质素的热解和催化解聚可以获得愈创木酚等酚类化合物。愈创木酚同时具有酚羟基和甲氧基,被认为是代表性的木质素解聚产物[8-10]。催化加氢脱氧(HDO)可将愈创木酚进一步转化为苯[11]、环己烷[12]、2-甲氧基环己醇[13]和环己醇[14]等化学品。贵金属催化剂在木质素模型化合物及其衍生酚类的催化转化方面具有较高的活性[15-17],但昂贵的价格往往限制了其在工业上的广泛应用。过渡金属硫化物在酚类HDO工业过程中被大量使用,但会对环境产生污染[18]。因此,有必要开发可替代的非贵金属催化剂。近年来,由金属离子和多官能团有机配体构成的金属有机骨架(MOFs),具有比表面积大、孔隙可调节等特点,引起了研究者的广泛关注[19, 20]。以MOFs为自牺牲模板,通过一步热解法制备碳基纳米催化剂,不需要氢气还原,与浸渍法相比制备工艺简单,已在多个催化领域得到了应用[21],但在木质素衍生酚类化合物的催化加氢转化方面的应用还鲜有报道。
本研究通过Co-MOF一步热解法制备了对木质素衍生酚类C−O键断裂具有高活性和高选择性的加氢转化催化剂Co@C,并推测了Co@C催化愈创木酚加氢转化的反应路径。考察了Co-MOF热解温度、反应温度、初始氢压和反应时间对Co@C催化愈创木酚加氢转化的影响,并考察了Co@C对木质素其他衍生酚类化合物的催化活性。
Co(NO3)2·6H2O、1,4-苯二甲酸(H2BDC)和愈创木酚购自上海麦克林生化有限公司。苯酚、3-甲基苯酚、4-甲基苯酚、4-甲基愈创木酚、4-乙基愈创木酚和4-丙基愈创木酚购自上海阿拉丁生化科技有限公司。N’N-二甲基甲酰胺(DMF)和正己烷购自国药集团化学试剂有限公司。实验所用试剂均为分析纯。
Co-MOF的合成参考了Wang等[22]的方法,流程示意图见图1。首先,将8 mmol Co(NO3)2·6H2O和4 mmol H2BDC溶于50 mL DMF中。为保证充分溶解,在40 ℃恒温水浴中搅拌30 min,再滴加1 mL盐酸充分搅拌。将得到的紫色溶液转移到200 mL特氟龙内胆的水热釜中,在180 ℃的烘箱中反应12 h。待水热釜冷却后,过滤出沉淀,并用DMF洗涤三次得到所需样品。最后,将样品在70 ℃真空干燥箱中烘干得到Co-MOF。将Co-MOF在管式炉中N2气氛下热解,升温速率3 ℃/min,分别在500、600和700 ℃下保持3 h,得到的催化剂标记为Co@C-X,X表示热解温度。
利用美国康塔Autosotrb-IQ2-MP-XR型物理吸附仪测定样品的比表面积和孔径分布,样品测定前先于300 ℃脱气5 h,之后于−196 ℃进行N2吸附-脱附,采用Brumaire-Emmett-Teller(BET)公式计算比表面积,采用Barret-Joyner-Halenda(BJH)模型计算平均孔径和孔容。利用德国布鲁克D8 ADVANCE型X射线衍射仪(XRD)对催化剂晶体形貌进行分析,使用陶瓷X射线管,管电压30 kV,管电流20 mA,5°−80°扫描。利用美国飞雅Quanta 250型扫描电子显微镜(SEM)对催化剂的表面形貌和元素分布进行表征。采用JEM-200 CX透射电子显微镜(TEM)对催化剂的微观形态、颗粒尺寸进行分析。利用美国Thermo Fisher ESCALAB 250 Xi型X射线光电子能谱仪(XPS)分析催化剂的元素组成及存在形式。催化剂中钴元素的实际含量采用激光烧蚀电感耦合等离子体质谱仪(LA-ICP-MS)进行测定。
将40 mg愈创木酚、20 mg催化剂和15 mL正己烷加入到100 mL的不锈钢高压反应釜中,密封反应釜,用1 MPa N2置换空气三次,调节釜内压力为设定的氢压,加热反应器至设定的温度。反应釜的搅拌速率控制在300 r/min。反应结束后,将反应釜冷却至室温,对反应混合物进行过滤分离出催化剂。利用美国安捷伦气相色谱质谱联用仪(GC/MS)分析产物组成。GC/MS的参数如下:流动相载气为高纯He(99.999%,流量1.0 mL/min),色谱柱型号为HP-5 MS型毛细管柱(30 m × 0.25 mm × 0.25 μm),电子电离源的电压和温度分别为70 eV和230 ℃,四级杆质量分析器温度为60 ℃,离子质量扫描30−500 Da,溶剂延迟设为4.5 min。液体混合物以分流比100:1进样,升温程序为:初始温度60 ℃,以10 ℃/min的升温速率升温至300 ℃,保留5 min。转化率和产物选择性通过以下公式计算:
|
$\begin{split} &{\text{转化率}}\left( \% \right) =\\ &\left( {1 - {\text{未反应底物的量}}/{\text{初始底物量}}} \right) \times 100\% \end{split}$ |
(1) |
|
$ {\text{选择性}}\left( \% \right) = \left( {\text{某一产物量}} \right)/\left( {\text{总产物量}} \right) \times 100\% $ |
(2) |
为考察催化剂的循环性能,过滤回收最佳反应条件下的Co@C-600催化剂,用乙醇洗涤三次,在真空干燥箱中70 ℃干燥12 h后循环使用。
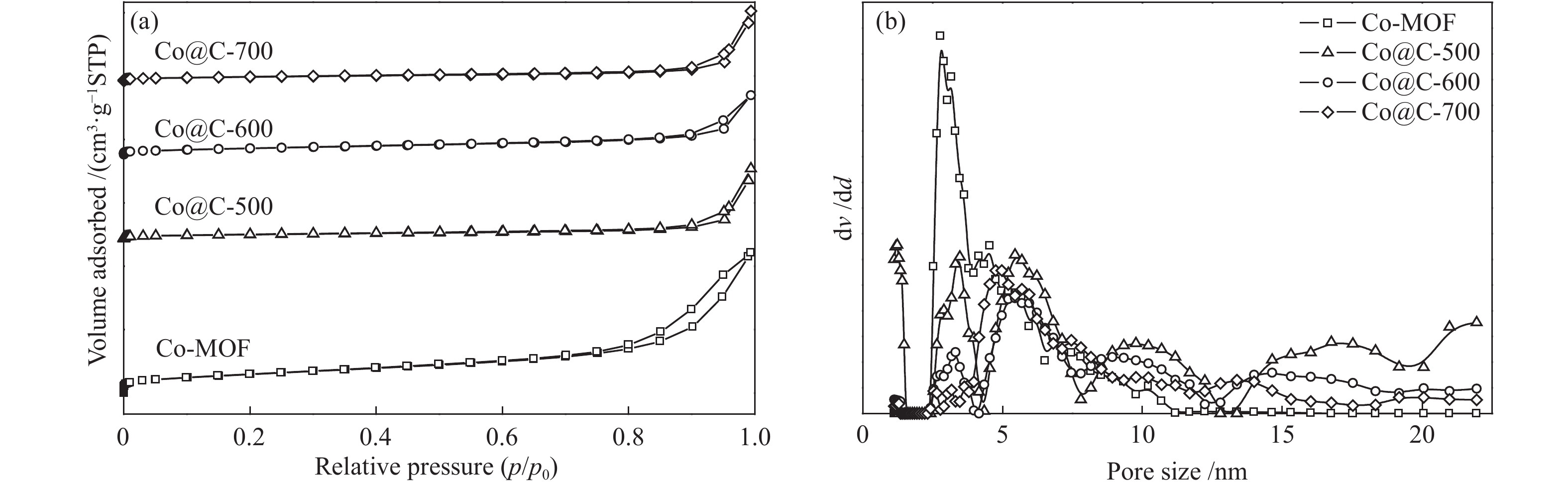
Co-MOF和Co@C-X的N2吸附-脱附等温线及相应的孔径分布见图2(a)和2(b)。根据IUPAC的分类,Co-MOF和Co@C-X的N2吸脱-附等温线均呈现典型的IV型曲线,Co@C-X具有H4型回滞环,说明其结构中主要以介孔为主。Co-MOF的孔径主要分布在2.5−11.2 nm,而Co@C-X的孔径分布较宽,这主要是由于热解过程中有机配体1,4-对苯二甲酸的分解导致孔径和孔体积变大(表1)。Co@C-X的比表面积随热解温度的升高而减小。热解温度为700 ℃时,比表面积下降至17.77 m2/g,这是由于温度过高导致孔结构坍塌。由于温度越高,有机配体分解的越彻底,Co@C催化剂中Co含量随热解温度升高而增加。
 下载:
导出CSV
下载:
导出CSV
| Sample | Surface
area/ (m2·g−1)a |
Pore
volume/ (cm3·g−1)b |
Pore diameter/
nmb |
Cobalt content
w/%c |
| Co-MOF | 56.30 | 0.03 | 3.41 | 37.3 |
| Co@C-500 | 35.51 | 0.03 | 3.83 | 50.6 |
| Co@C-600 | 31.93 | 0.04 | 3.81 | 56.2 |
| Co@C-700 | 17.77 | 0.04 | 3.82 | 64.8 |
| acalculated by BET method; bcalculated by BJH method; cmeasured with LA-ICP-MS | ||||
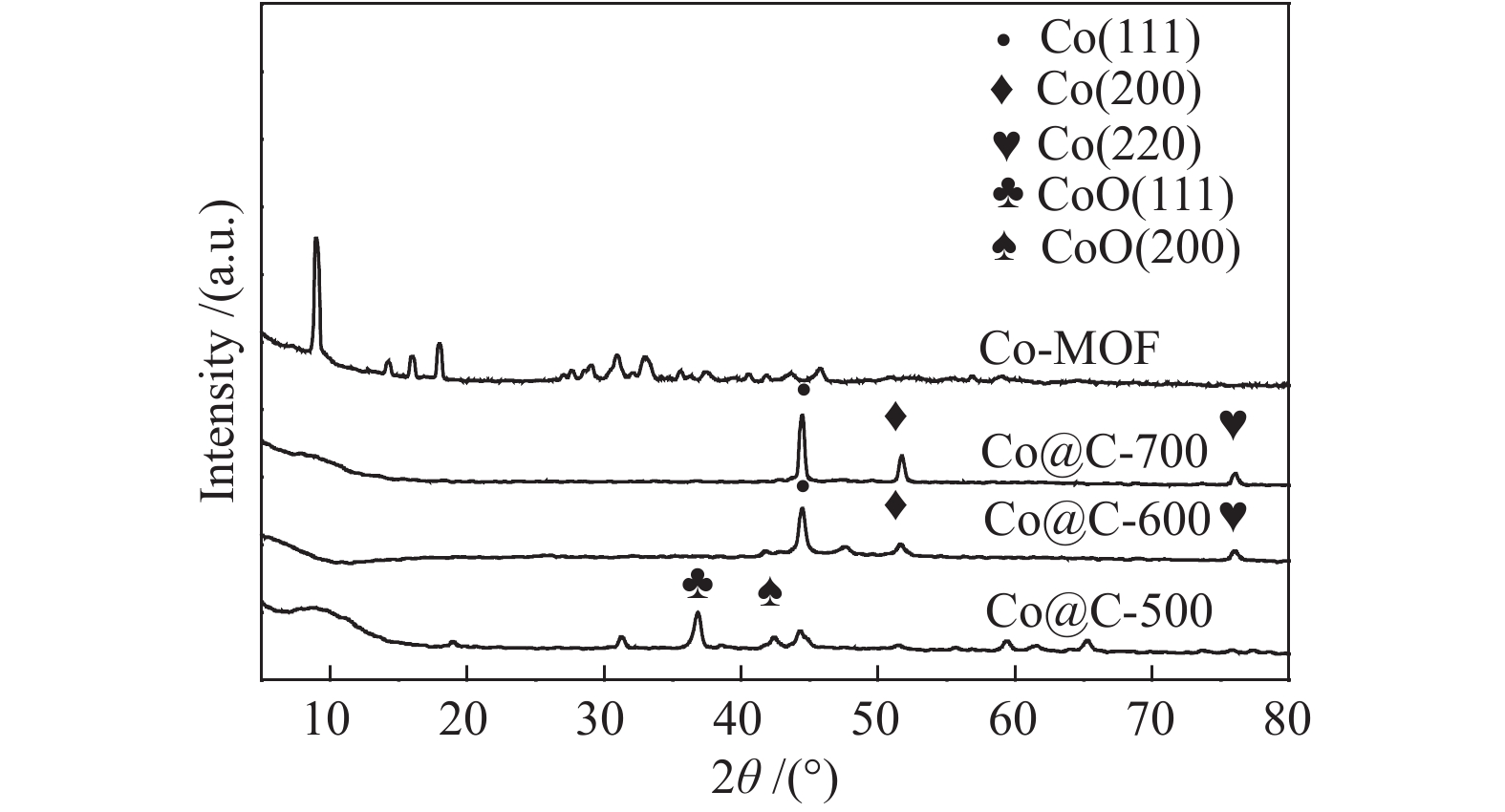
如图3所示,Co-MOF的XRD谱图与文献报道一致[23]。Co@C-700和Co@C-600在44.47°、51.59°和76.08°出现的衍射峰对应于Co的(111)、(200)和(220)晶面[24],这表明Co-MOF热解后直接得到了还原态的金属Co。与Co@C-600相比,Co@C-700的特征峰更尖锐,说明其结晶度更高,晶体粒径更大[25]。而Co@C-500的XRD谱图在36.81°和42.44°的两个衍射峰分别为CoO的(111)和(200)晶面[24],这说明CoO在500 ℃不能被完全还原。Cai等[24]指出MOFs经高温热解后,有机配体分解会导致碳基体的形成,碳基体可以使金属离子还原,形成沉积在碳基体上的金属纳米粒子,在形成金属纳米粒子的同时形成碳层以抑制其氧化和聚集。
利用XPS分析了Co@C-600表面的元素组成及价态。由图4(a)可知,Co@C-600表面同时存在C、O、Co三种元素,这与催化剂的原料元素组成一致。Co 2p可以分峰拟合为两个区域,780.5 eV(2p3/2)和796.5 eV(2p1/2)处的结合能归属于氧化态的Co2+,778.3 eV(2p3/2)和793.4 eV (2p1/2)处的结合能归属于金属态的Co0。氧化态Co2+的拟合峰面积大于金属态Co0,这可能是由于测试过程中催化剂与空气接触造成的。如图4(c)所示,C 1s谱图可分为三个峰,其结合能分别为289.5、286.2和284.8 eV,对应于催化剂表面的−COO、C−O和C−C//C=C官能团。
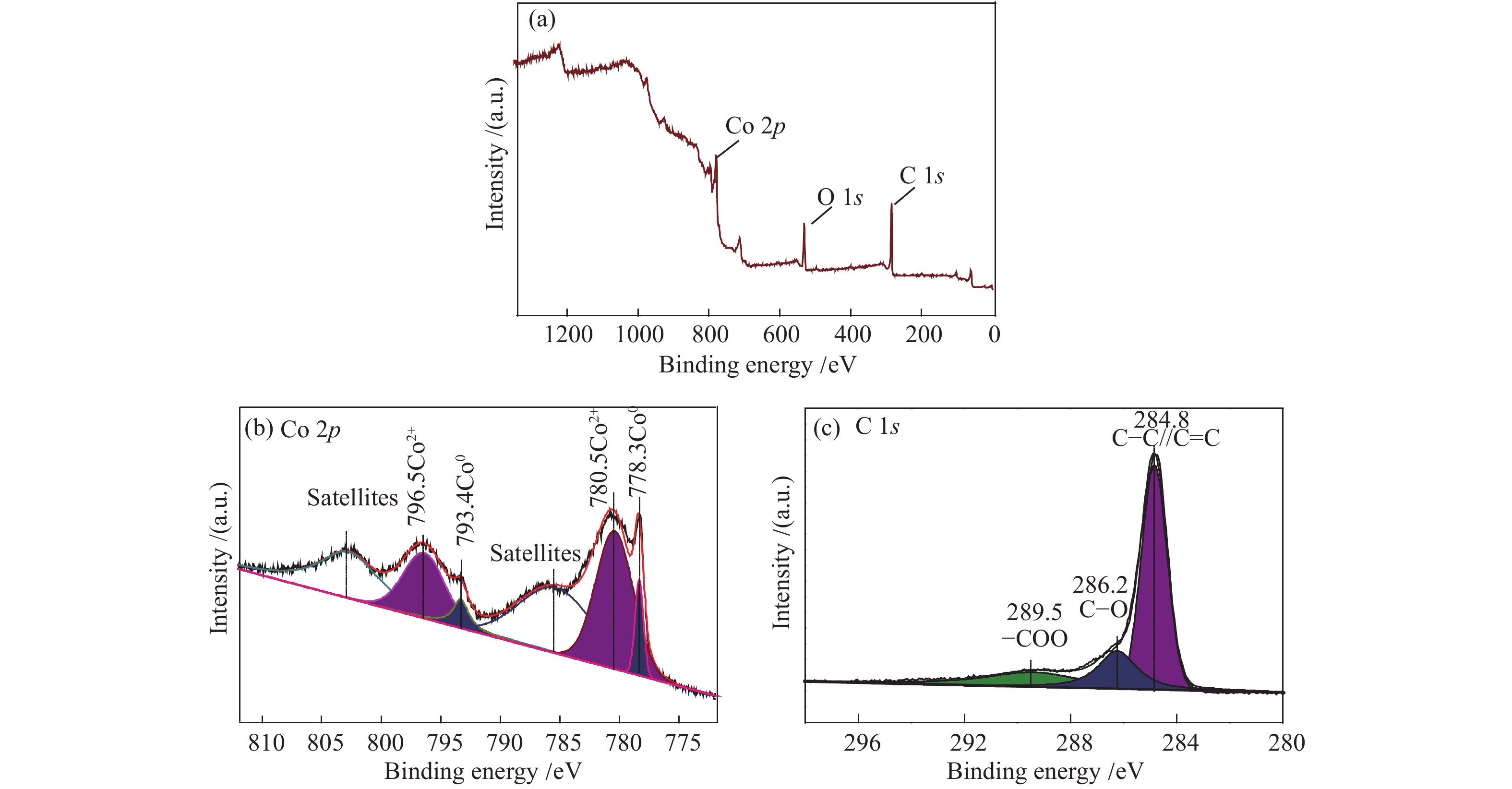
(a): survey; (b):Co 2p; (c):C 1s
图5为Co-MOF和Co@C-600的TEM和SEM照片,以及Co@C-600的SEM-EDS图。由图5(a)可以看出,Co-MOF为片层状结构,图5(c)更为清晰的展示了Co-MOF的表面形貌。600 ℃热解后,Co-MOF分解为不规则的球形颗粒(图5(b)和5(d))。Co@C-600的粒径主要分布在40−60 nm。从Co@C-600的元素分布图可以看到,Co、C、O元素分布均匀,其中,Co元素较为集中,这主要是由于热解后有机配体分解导致Co含量相对较高。

为了研究Co-MOF热解温度对催化剂性能的影响,采用不同热解温度得到的催化剂在温度为180 ℃、初始H2压力为2 MPa、反应时间1 h的条件下催化愈创木酚加氢转化,结果如表2所示。以Co@C-500为催化剂时,愈创木酚的转化率仅为6.4%,而以Co@C-600为催化剂时,愈创木酚转化率达到92.5%,环己醇的选择性为90.8%,但Co@C-700几乎没有催化活性。热解温度较低时,有机配体分解较少,不足以完全还原CoO为金属Co,Cai等[24]对比了CoO和C/Co@C对苯乙炔加氢催化性能,CoO的转化率很低,表明金属Co比CoO具有更高的催化活性。此外,Zhu等[26]在400 ℃煅烧五倍子酸作为有机配体的Ni-MOF材料得到的催化剂XRD谱图中未出现明显的Ni衍射峰,催化活性低,而煅烧温度为500−800 ℃时Ni衍射峰明显,催化活性明显提高。这也说明金属是此类催化剂的主要活性成分。而热解温度过高,催化剂孔结构坍塌,比表面积减小,结晶度高,粒径大,分散性差,导致Co@C-700催化活性低[27]。因此,在后续实验中选择了Co@C-600为催化剂来探索反应条件的影响。
下载:
导出CSV
| Sample | Conversion/% | Selectivity/% | |

|

(trans-, cis-) |
||
| Co@C-500 | 6.4 | 77.6 | 22.4 |
| Co@C-600 | 92.5 | 90.8 | 9.2 |
| Co@C-700 | trace | − | − |
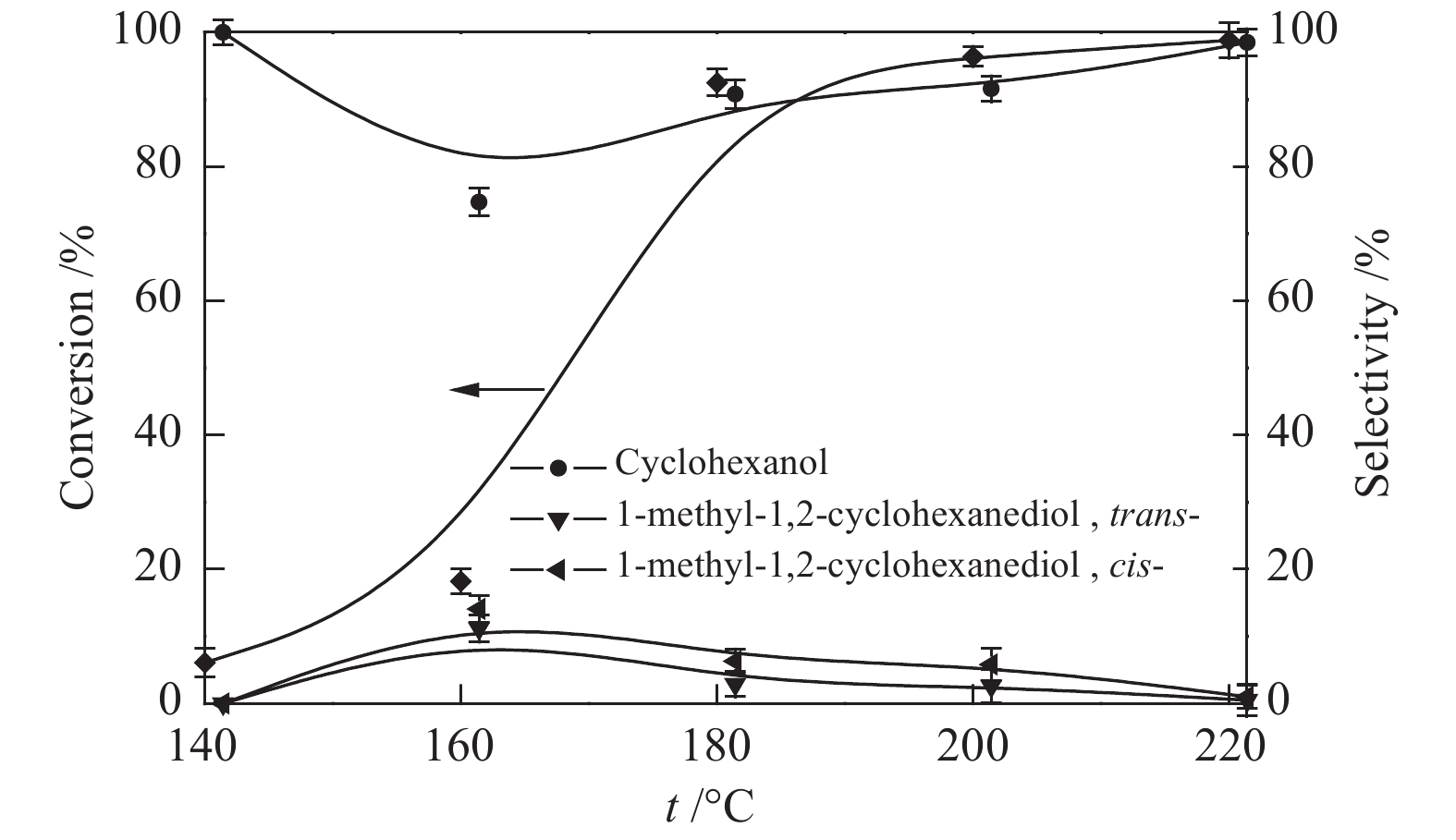
图6为反应温度对愈创木酚转化率和环己醇选择性的影响。当反应温度从140 ℃升至160 ℃时,愈创木酚的转化率由6.1%提高到18.2%,环己醇的选择性由100%降低到74.7%。升温有利于愈创木酚的转化,同时也促进了副产物1-甲基-1,2-环己二醇(顺式-,反式-)的生成。当反应温度升至180 ℃时,愈创木酚的转化率提高到90.8%。继续升温至220 ℃,愈创木酚转化率和环己醇选择性随温度升高而缓慢增加,1-甲基-1,2-环己二醇(顺式-,反式-)的选择性呈下降趋势。高温有利于提高转化率和环己醇的选择性,抑制副产物的生成。综合考虑,选择180 ℃作为后续研究的反应温度。
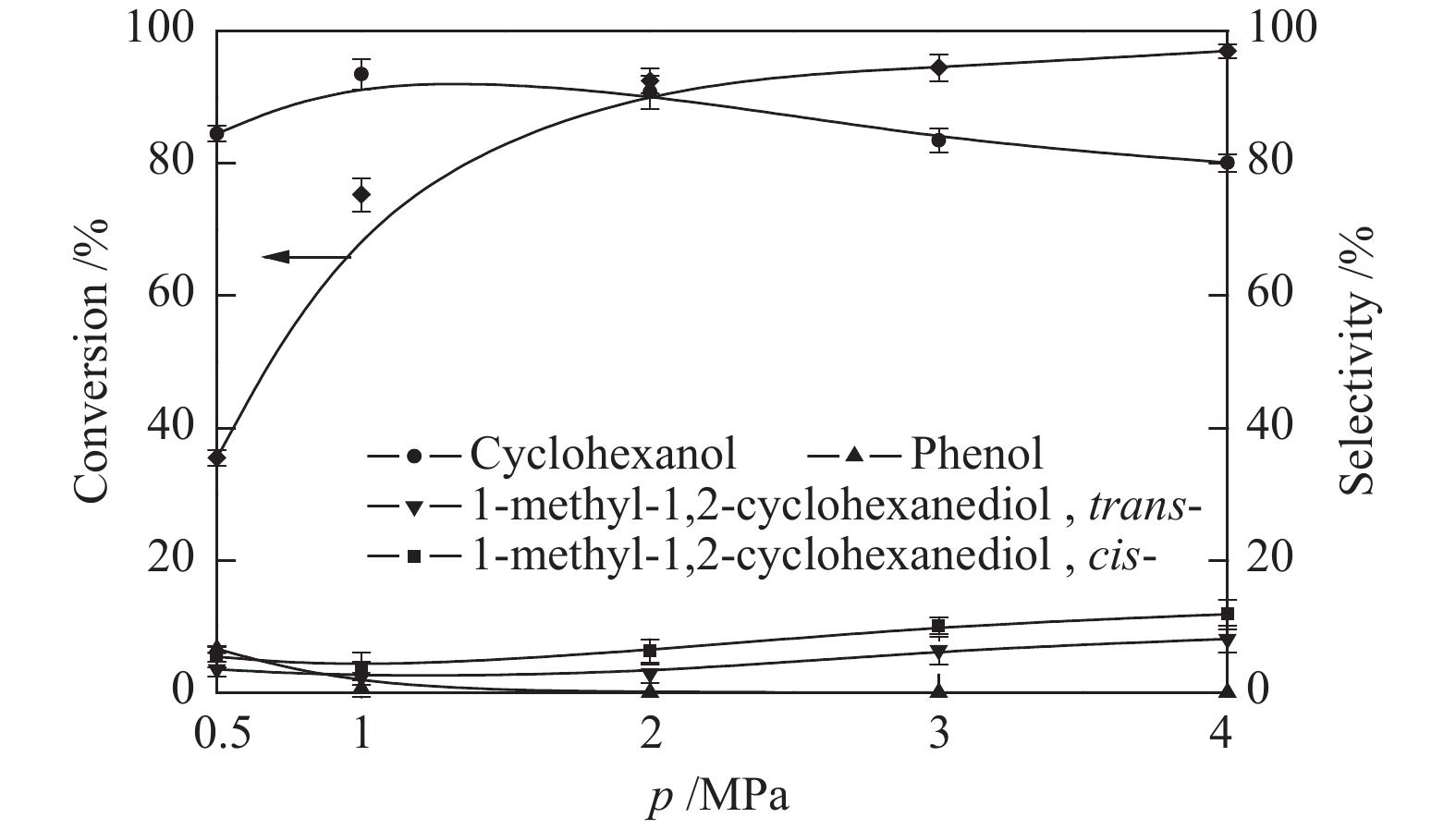
在180 ℃条件下考察了初始氢气压力(0.5−4.0 MPa)对愈创木酚转化的影响。图7为愈创木酚转化率和选择性随氢气压力的变化。当氢压较低时(0.5、1 MPa),愈创木酚的转化率较低,并且在产物中有苯酚,这说明低氢压有利于愈创木酚脱甲氧基生成苯酚,而不利于苯环加氢。当氢气压力增加到2 MPa时,产物中的苯酚消失。增加氢气压力使得溶液中溶解的氢量增加,更容易到达催化剂的活性位点。随着氢压继续升高,1-甲基-1,2-环己二醇(顺式-,反式-)的选择性升高,环己醇的选择性降低,愈创木酚的转化率没有明显增加。Schutyser等[28]指出,低氢压有利于提高环己醇的选择性。综合考虑愈创木酚的转化率和环己醇的选择性,选择2 MPa作为后续实验的初始氢气压力。
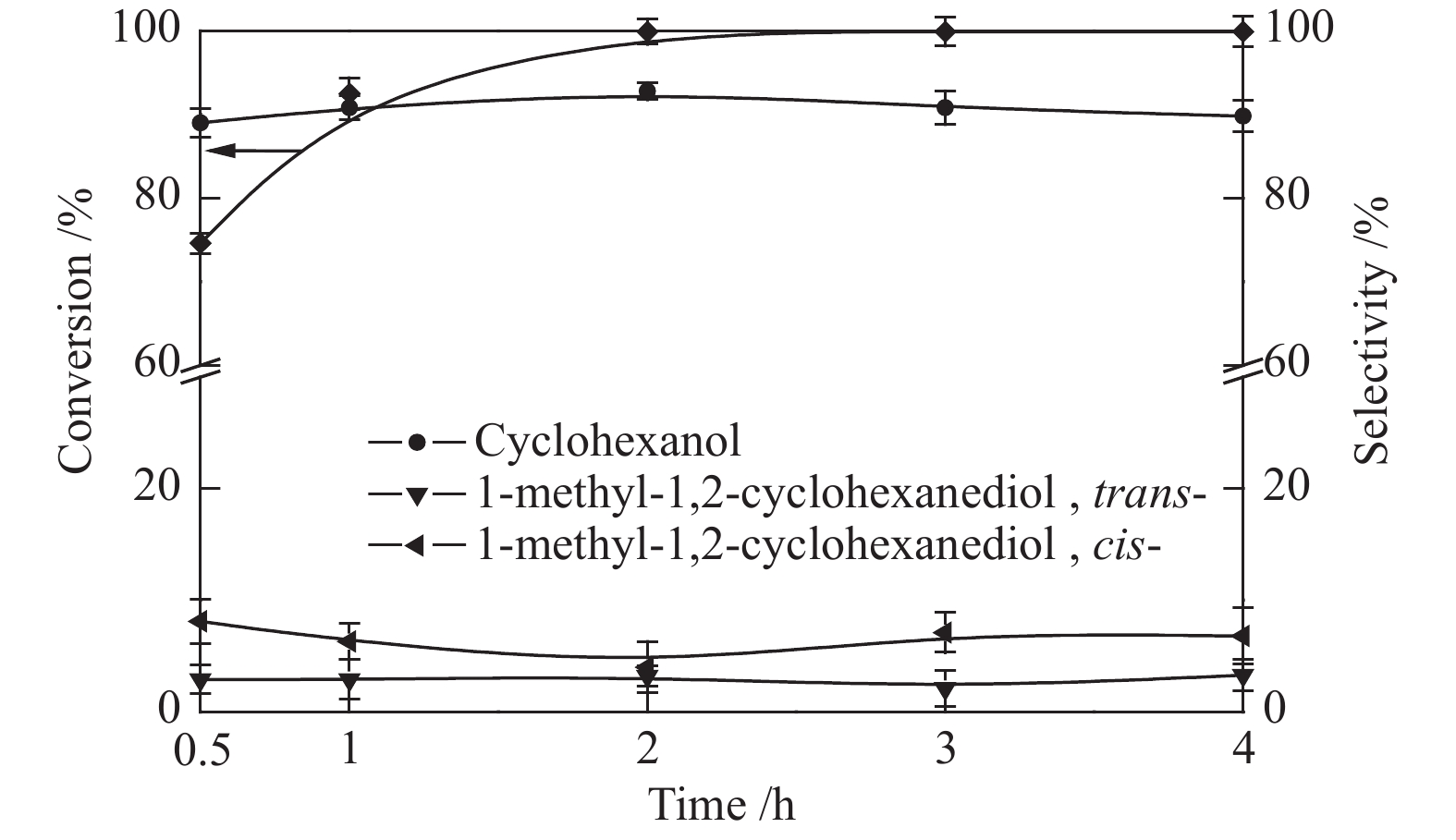
反应时间对愈创木酚催化转化的影响见图8。在之前筛选的反应温度和初始氢气压力(180 ℃,2 MPa)下,愈创木酚在30 min内的转化率达74.6%,且未检测到苯酚等中间产物,说明苯酚能在短时间内快速加氢生成环己醇。反应时间为2 h时,愈创木酚完全转化,环己醇的选择性达到最大值(92.8%)。继续延长反应时间,环己醇和1-甲基-1,2-环己二醇(顺式-,反式-)的选择性变化不明显。
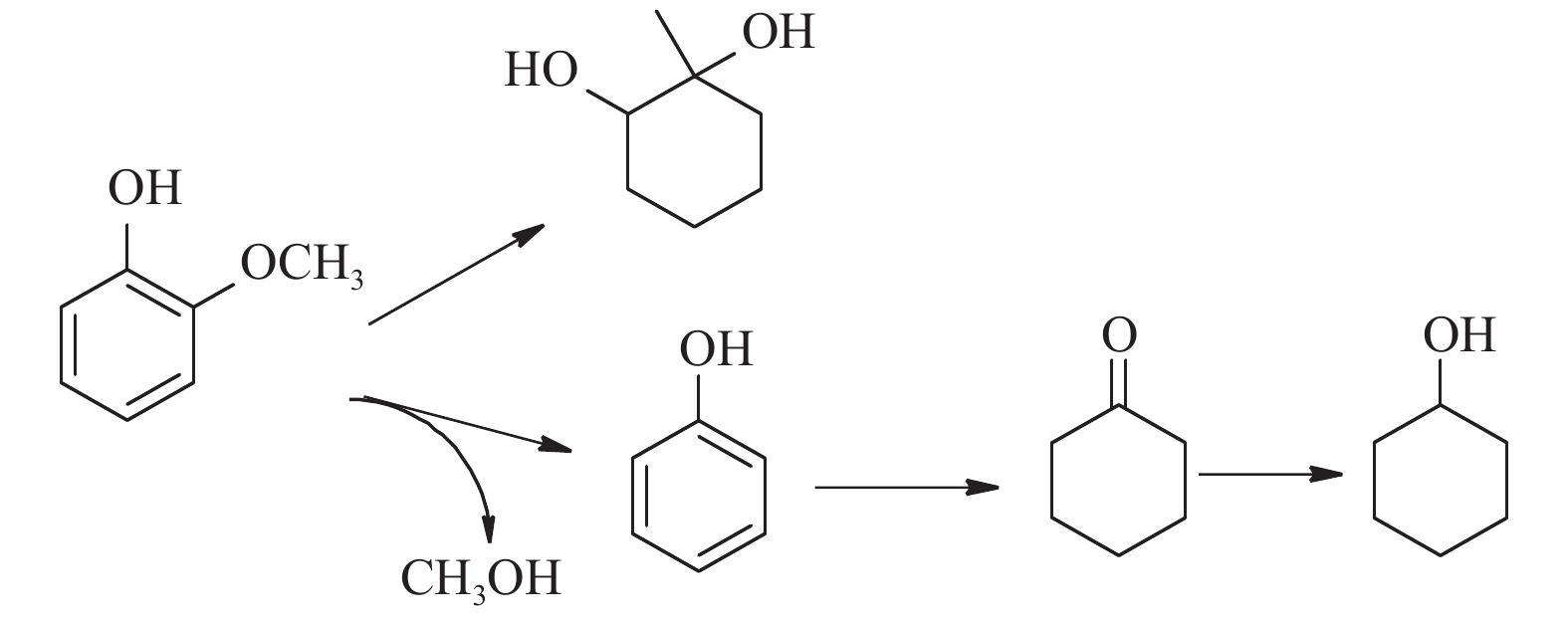
Nakagawa等[29]报道了Ru/C + MgO催化愈创木酚转化为环己醇的两条可能的反应路径,一条为愈创木酚脱甲氧基生成苯酚,继续加氢形成环己醇;另一条为愈创木酚直接加氢生成2-甲氧基环己醇,但2-甲氧基环己醇很难继续脱甲氧基生成环己醇。Long等[30]并未在产物中检测到2-甲氧基环己醇,主要是由于酚羟基作为Brønsted酸,易与Lewis碱MgO结合,从而削弱了Ar-OCH3的碳氧键,使得愈创木酚更易脱甲氧基生成苯酚。本研究中愈创木酚转化的产物种类较少,主要是环己醇和副产物1-甲基-1,2-环己二醇(顺式-,反式-)(图9)。副产物的生成是由于愈创木酚加氢脱氧过程中苯环的加氢以及甲基的转移[31]。低氢气压力下检测出苯酚,这也说明环己醇是由愈创木酚脱甲氧基生成的苯酚加氢而成。产物中未检测到环己酮,这是由于苯酚加氢的速率过快。
在愈创木酚转化的最佳条件下(180 ℃, H2 2.0 MPa, 2 h),考察了催化剂的通用性(表3)。当底物为苯酚时,环己醇的选择性为100%。当底物为间甲氧基苯酚和对甲氧基苯酚时,转化率仍维持在较高水平,但环己醇的选择性存在差异。间甲氧基苯酚为底物时环己醇的选择性为75.1%,而对甲氧基苯酚为底物时环己醇的选择性仅为2.6%,推测可能是由于甲氧基为给电子基团,其在对位取代时,推电子共轭效应远大于吸电子诱导效应,使得酚羟基酸性减小,不易附着在催化剂表面,Ar-OCH3的碳氧键没有被削弱,导致主要生成的是苯环加氢产物[32]。当底物为4-甲基愈创木酚、4-乙基愈创木酚、4-丙基愈创木酚时,Co@C-600催化剂仍能催化这些底物的甲氧基完全脱除并生成对应的醇,这表明空间位阻对催化剂的活性影响较小。
下载:
导出CSV
| Substrate | Conversion/% | Selectivity/% | ||

|
99.9 |

100 |
||

|
99.9 |

75.1 |

2.8 |

22.1 |

|
99.9 |

2.6 |

97.4 |
|

|
99.9 |

100.0 |
||

|
99.9 |

100.0 |
||

|
99.9 |

100.0 |
||
为了研究催化剂的稳定性,回收了反应温度180 ℃,初始H2压力2.0 MPa,反应时间2 h条件下的Co@C-600催化剂,循环使用性见图10(a)。第二次循环时,愈创木酚仍能完全转化,而环己醇的选择性略微降低。第三次循环时,愈创木酚的转化率降低至34.5%,环己醇的选择性为68.4%。图10(b)为第三次循环实验后催化剂的TEM图。与新鲜的催化剂相比(图5(b)),第三次循环实验后的催化剂发生了团聚,分散度下降,这可能是催化性能下降的主要原因。此外,循环过程中金属颗粒从载体脱落也可能是催化剂失活的一个原因[13]。
通过Co-MOF一步热解法制备了对愈创木酚加氢转化具有高活性和高选择性的Co@C催化剂。在180 ℃,初始H2压力2.0 MPa,反应时间2 h的条件下,愈创木酚的转化率和环己醇的选择性分别为99.9%和92.8%。愈创木酚催化加氢转化的主要反应路径为先脱甲氧基生成苯酚,苯酚再加氢生成环己醇,而副产物1-甲基-1,2-环己二醇(顺式-,反式-)的生成主要是由于苯环的加氢和甲氧基的转移。Co@C-600对苯酚、对甲氧基苯酚和4-甲基愈创木酚等木质素衍生其他酚类单体也具有良好的催化活性。
SHERWOOD J. The significance of biomass in a circular economy[J]. Bioresour Technol,2020,300:122755. doi: 10.1016/j.biortech.2020.122755
余强, 庄新姝, 袁振宏, 亓伟, 王琼, 谭雪松, 许敬亮, 张宇, 徐慧娟, 马隆龙. 木质纤维素类生物质制取燃料及化学品的研究进展[J]. 化工进展,2012,31(4):784−791.YU Qiang, ZHUANG Xin-shu, YUAN Zhen-hong, QI Wei, WANG Qiong, TAN Xue-song, XU Jin-liang, ZHANG Yu, XU Hui-juan, MA Long-long. Research progress on fuel and chemicals production from lignocellulose biomass[J]. Chem Ind Eng Prog,2012,31(4):784−791.
朱晨杰, 张会岩, 肖睿, 陈勇, 柳东, 杜风光, 应汉杰, 欧阳平凯. 木质纤维素高值化利用的研究进展[J]. 中国科学: 化学,2015,45(5):454−478. doi: 10.1360/N032014-00280ZHU Chen-jie, ZHANG Hui-yan, XIAO Rui, CHEN Yong, LIU Dong, DU Feng-guang, YING Han-jie, OUYANG Ping-kai. Research progress of high value utilization of lignocellulose[J]. Sci Sin Chim,2015,45(5):454−478. doi: 10.1360/N032014-00280
袁正求, 龙金星, 张兴华, 夏莹, 王铁军, 马隆龙. 木质纤维素催化转化制备能源平台化合物[J]. 化学进展,2016,28(1):103−110.YUAN Zhen-qiu, LONG Jin-xing, ZHANG Xing-hua, XIA Ying, WANG Tie-jun, MA Long-long. Catalytic conversion of lignocellulose into energy platform chemicals[J]. Prog Chem,2016,28(1):103−110.
UPTON B M, KASKO A M. Strategies for the conversion of lignin to high-value polymeric materials: review and perspective[J]. Chem Rev,2015,116(4):2275−2306.
PEREZ J, MUNOZ-DORADO J, DE LA RUBIA T, MARTINEZ J. Biodegradation and biological treatments of cellulose, hemicellulose and lignin: An overview[J]. Int Microbiol,2002,5(2):53−63. doi: 10.1007/s10123-002-0062-3
袁亮. 木质素磺酸盐在混凝土外加剂方面的应用[J]. 特钢技术,2012,18(2):58−61. doi: 10.3969/j.issn.1674-0971.2012.02.018YUAN Liang. Application of lignosulfonate in concrete admixture[J]. Spec Steel Techonl,2012,18(2):58−61. doi: 10.3969/j.issn.1674-0971.2012.02.018
TYMCHYSHYN M, YUAN Z, ZHANG Y, XU C C. Catalytic hydrodeoxygenation of guaiacol for organosolv lignin depolymerization-catalyst screening and experimental validation[J]. Fuel,2019,254:115664. doi: 10.1016/j.fuel.2019.115664
VERMA S, NADAGOUDA MN, VARMA RS. Visible light-mediated and water-assisted selective hydrodeoxygenation of lignin-derived guaiacol to cyclohexanol[J]. Green Chem,2019,21(6):1253−1257. doi: 10.1039/C8GC03951H
于玉肖, 徐莹, 王铁军, 马隆龙, 张琦, 张兴华, 张雪. 木质素降解模型化合物愈创木酚及苯酚原位加氢制备环己醇[J]. 燃料化学学报,2013,41(4):443−448. doi: 10.3969/j.issn.0253-2409.2013.04.009YU Yu-xiao, XU Ying, WANG Tie-jun, MA Long-long, ZHANG Qi, ZHANG Xing-hua, ZHANG Xue. In-situ hydrogenation of lignin depolymerization model compounds to cyclohexanol[J]. J Fuel Chem Technol,2013,41(4):443−448. doi: 10.3969/j.issn.0253-2409.2013.04.009
LUO Z C, ZHENG Z X, WANG Y C, SUN G, JIANG H, ZHAO C. Hydrothermally stable Ru/HZSM-5-catalyzed selective hydrogenolysis of lignin-derived substituted phenols to bio-arenes in water[J]. Green Chem,2016,18(21):5845−5858. doi: 10.1039/C6GC01971D
WANG X, ZHU S, WANG S, HE Y, LIU Y, WANG J G, FAN W B, LV Y K. Low temperature hydrodeoxygenation of guaiacol into cyclohexane over Ni/SiO2 catalyst combined with Hβ zeolite[J]. RSC Adv,2019,9(7):3868−3876. doi: 10.1039/C8RA09972C
WANG X, ZHU S, WANG S, WANG J G, FAN W B, LV Y K. Ni nanoparticles entrapped in nickel phyllosilicate for selective hydrogenation of guaiacol to 2-methoxycyclohexanol[J]. Appl Catal A: Gen,2018,568:231−241. doi: 10.1016/j.apcata.2018.10.009
LU J Q, LIU X, YU G Q, LV J K, RONG Z M, WANG M, WANG Y. Selective Hydrodeoxygenation of guaiacol to cyclohexanol catalyzed by nanoporous nickel[J]. Catal Lett,2019,150(3):837−848.
GUO M, PENG J, YANG Q H, LI C. Highly active and selective RuPd bmetallic NPs for the cleavage of the diphenyl ether C–O bond[J]. ACS Catal,2018,8(12):11174−11183. doi: 10.1021/acscatal.8b03253
HUA M L, SONG J L, XIE C, WU H R, HU Y, HUANG X, HAN B X. Ru/hydroxyapatite as a dual-functional catalyst for efficient transfer hydrogenolytic cleavage of aromatic ether bonds without additional bases[J]. Green Chem,2019,21(18):5073−5079. doi: 10.1039/C9GC02336D
严龙, 庞欢, 黄耀兵, 傅尧. Pd催化木质素醚类二聚体分子内氢转移断裂C-O键研究[J]. 化学学报,2014,72(9):1005−1011. doi: 10.6023/A14050397YAN Long, PANG Huan, HUANG Yao-bing, FU Yao. Supported Pd catalysts for the C-O cleavage of the lignin derived model dimers through intramolecular hydrogenolysis reaction[J]. Acta Chim Sin,2014,72(9):1005−1011. doi: 10.6023/A14050397
邱泽刚, 尹婵娟, 李志勤, 冯跃阔. 酚类加氢脱氧催化剂研究进展[J]. 化工进展,2019,38(8):3658−3669.QIU Ze-gang, YIN Chan-juan, LI Zhi-qin, FENG Kuo-yue. Recent advances in hydrodeoxygenation catalysts for phenols[J]. Chem Ind Eng Prog,2019,38(8):3658−3669.
YUAN S, FENG L, WANG K C, PANG J D, BOSCH M, LOLLAR C, SUN Y J, QIN J S, YANG X Y, ZHANG P, WANG Q, ZOU L F, ZHANG Y M, ZHANG L L, FANG Y, LI J L, ZHOU H C. Stable metal-organic frameworks: design, synthesis, and applications[J]. Adv Mater,2018,30(37):1704303. doi: 10.1002/adma.201704303
黄刚, 陈玉贞, 江海龙. 金属有机骨架材料在催化中的应用[J]. 化学学报,2016,74(2):113−129. doi: 10.6023/A15080547HUANG Gang, CHEN Yu-zhen, JIANG Hai-long. Metal-organic frameworks for catalysis[J]. Acta Chim Sin,2016,74(2):113−129. doi: 10.6023/A15080547
SHEN K, CHEN X D, CHEN J Y, LI Y W. Development of MOF-derived carbon-based nanomaterials for efficient catalysis[J]. ACS Catal,2016,6(9):5887−5903. doi: 10.1021/acscatal.6b01222
WANG J, ZHONG Q, XIONG Y H, CHENG D Y, ZENG Y Q, BU Y F. Fabrication of 3D Co-doped Ni-based MOF hierarchical micro-flowers as a high-performance electrode material for supercapacitors[J]. Appl Surf Sci,2019,483:1158−1165. doi: 10.1016/j.apsusc.2019.03.340
REZAEE S, SHAHROKHIAN S. Facile synthesis of petal-like NiCo/NiO-CoO/nanoporous carbon composite based on mixed-metallic MOFs and their application for electrocatalytic oxidation of methanol[J]. Appl Catal B: Environ,2019,244:802−813. doi: 10.1016/j.apcatb.2018.12.013
CAI J Y, CHEN Y, SONG H T, HOU L X, LI Z H. MOF derived C/Co@C with a “one-way-valve”-like graphitic carbon layer for selective semi-hydrogenation of aromatic alkynes[J]. Carbon,2020,160:64−70. doi: 10.1016/j.carbon.2020.01.006
LIU X H, XU L J, XU G Y, JIA W D, MA Y F, ZHANG Y. Selective hydrodeoxygenation of lignin-derived phenols to cyclohexanols or cyclohexanes over magnetic CoNx@NC catalysts under mild conditions[J]. ACS Catal,2016,6(11):7611−7620. doi: 10.1021/acscatal.6b01785
ZHU J, CHEN F Q, ZHANG Z G, LI M, YANG Q W, YANG Y W, BAO Z B, REN Q L. M-gallate (M = Ni, Co) metal-organic framework-derived Ni/C and bimetallic Ni-Co/C catalysts for lignin conversion into monophenols[J]. ACS Sustainable Chem Eng,2019,7(15).
DONG L, YIN L L, XIA Q E, LIU X H, GONG X Q, WANG Y Q. Size-dependent catalytic performance of ruthenium nanoparticles in the hydrogenolysis of a β-O-4 lignin model compound[J]. Catal Sci Technol,2018,8(3):735−745. doi: 10.1039/C7CY02014G
SCHUTYSER W, VAN DEN BOSSCHE G, RAAFFELS A, VAN DEN BOSCH S, KOELEWIJN S F, RENDERS T, SELS B F. Selective conversion of lignin-derivable 4-alkylguaiacols to 4-alkylcyclohexanols over noble and non-noble-metal catalysts[J]. ACS Sustainable Chem Eng,2016,4(10):5336−5346. doi: 10.1021/acssuschemeng.6b01580
NAKAGAWA Y, ISHIKAWA M, TAMURA M, TOMISHIGE K. Selective production of cyclohexanol and methanol from guaiacol over Ru catalyst combined with MgO[J]. Green Chem,2014,16(4):2197−2203. doi: 10.1039/C3GC42322K
LONG J X, SHU S Y, WU Q Y, YUAN Z Q, WANG T J, XU Y, ZHANG X H, ZHANG Q, MA L L. Selective cyclohexanol production from the renewable lignin derived phenolic chemicals catalyzed by Ni/MgO[J]. Energy Convers Manage,2015,105:570−577. doi: 10.1016/j.enconman.2015.08.016
ZHOU M H, WANG Y, WANG Y B, XIAO G M. Catalytic conversion of guaiacol to alcohols for bio-oil upgrading[J]. J Energy Chem,2015,24(4):425−431. doi: 10.1016/j.jechem.2015.06.012
郭荷民. 利用电子效应解释取代基的位置及种类对芳香化合物化学性质的影响[J]. 大学化学,2008,(5):54−57.GUO He-min. The effects of substituent position and type on the chemical properties of aromatic compounds were explained by electron effect[J]. Univ Chem,2008,(5):54−57.

图 2 (a) N2吸附-脱附等温线和(b) Co-MOF和Co@C-X催化剂的孔径分布
Figure 2 N2 BET characterization of Co-MOF and Co@C-X: (a) N2 adsorption-desorption isotherms and (b) pore size distribution of Co-MOF

图 4 Co@C-600的XPS谱图
Figure 4 XPS spectra of Co@C-600 catalyst
(a): survey; (b):Co 2p; (c):C 1s

图 5 Co-MOF (a)、Co@C-600 (b)的TEM照片,Co-MOF (c)、Co@C-600 (d)的SEM照片,Co@C-600 (e)、Co (f)、C (g)和O (h)元素的SEM-EDS照片
Figure 5 TEM images of Co-MOF (a) and Co@C-600 (b); SEM images of Co-MOF (c) and Co@C-600 (d); SEM-EDS mapping images of Co@C-600 catalyst (e) with Co (f), C (g) and O (h) elements

图 6 温度对愈创木酚催化加氢脱氧的影响
Figure 6 Effect of temperature on the guaiacol hydrodeoxygenation

图 7 初始氢压对愈创木酚催化加氢脱氧的影响
Figure 7 Effect of initial H2 pressure on the guaiacol hydrodeoxygenation
表 1 Co-MOF及Co@C-X的理化性质
Table 1. Physicochemical properties of Co-MOF and Co@C-X
| Sample | Surface
area/ (m2·g−1)a |
Pore
volume/ (cm3·g−1)b |
Pore diameter/
nmb |
Cobalt content
w/%c |
| Co-MOF | 56.30 | 0.03 | 3.41 | 37.3 |
| Co@C-500 | 35.51 | 0.03 | 3.83 | 50.6 |
| Co@C-600 | 31.93 | 0.04 | 3.81 | 56.2 |
| Co@C-700 | 17.77 | 0.04 | 3.82 | 64.8 |
| acalculated by BET method; bcalculated by BJH method; cmeasured with LA-ICP-MS | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 愈创木酚在不同催化剂上的转化及产物分布
Table 2. Guaiacol conversion and product distribution over different catalysts
| Sample | Conversion/% | Selectivity/% | |
|
|
(trans-, cis-) |
||
| Co@C-500 | 6.4 | 77.6 | 22.4 |
| Co@C-600 | 92.5 | 90.8 | 9.2 |
| Co@C-700 | trace | − | − |
下载: 导出CSV
表 3 Co@C-600对木质素衍生酚类单体的催化
Table 3. Catalysis of Co@C-600 to lignin-derived phenolic monomers
| Substrate | Conversion/% | Selectivity/% | ||
|
|
99.9 |
100 |
||
|
|
99.9 |
75.1 |
2.8 |
22.1 |
|
|
99.9 |
2.6 |
97.4 |
|
|
|
99.9 |
100.0 |
||
|
|
99.9 |
100.0 |
||
|
|
99.9 |
100.0 |
||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们