
引用本文:
魏雪莹, 吴玮, 乃永宁, 姜梦圆, 田诗伟, 毛国梁. 膦胺铬和双膦铬催化乙烯选择性齐聚研究进展[J]. 应用化学,
2021, 38(2): 136-156.
doi:
10.19894/j.issn.1000-0518.200250

Citation: Xue-Ying WEI, Wei WU, Yong-Ning NAI, Meng-Yuan JIANG, Shi-Wei TIAN, Guo-Liang MAO. Research Progress on the Selective Oligomerization of Ethylene Catalyzed by Phosphoramine Chromium and Diphosphinoamine Chromium[J]. Chinese Journal of Applied Chemistry, 2021, 38(2): 136-156. doi: 10.19894/j.issn.1000-0518.200250
Citation: Xue-Ying WEI, Wei WU, Yong-Ning NAI, Meng-Yuan JIANG, Shi-Wei TIAN, Guo-Liang MAO. Research Progress on the Selective Oligomerization of Ethylene Catalyzed by Phosphoramine Chromium and Diphosphinoamine Chromium[J]. Chinese Journal of Applied Chemistry, 2021, 38(2): 136-156. doi: 10.19894/j.issn.1000-0518.200250
膦胺铬和双膦铬催化乙烯选择性齐聚研究进展
摘要:
乙烯选择性齐聚制备线性α-烯烃是在学术界和工业界均受到高度重视,具有目标产物选择性高、产品易分离和经济效益显著等特点。铬系配合物催化剂在催化乙烯选择性齐聚反应中综合性能较好,有良好的发展和应用前景。近年来的研究表明,配体结构对催化剂性能有重要影响;反应体系中通入H2可以改善齐聚产物的分布并抑制低聚物的生成;开发不以甲基铝氧烷为助催化剂的催化体系可降低催化剂成本;而乙烯选择齐聚机理及金属中心氧化态对催化剂的设计有重要的指导意义。本文主要从以上几个方面介绍了膦胺铬和双膦铬配合物在催化乙烯选择性齐聚中的研究进展,分析了目前乙烯选择性四聚工业化急需解决的问题,展望了该领域未来的发展方向。
English
Research Progress on the Selective Oligomerization of Ethylene Catalyzed by Phosphoramine Chromium and Diphosphinoamine Chromium
Abstract:
The selective oligomerization of ethylene to prepare linear α-olefins has been highly valued in both academia and industry because of its high target product selectivity, easy product separation, and significant economic benefits. Chromium-based catalysts have good overall performance in catalyzing selective oligomerization of ethylene, and are a class of catalysts with broad application prospects. Recent studies have shown that the ligand structure has an important effect on the catalyst performance; the introduction of hydrogen in the reaction system can improve the distribution of oligomerization products and inhibit the formation of polymers; development of the catalyst without methylaluminoxane as a cocatalyst can reduce the costs of the catalyst; the ethylene selective oligomerization mechanism and the oxidation state of the central metal have important guiding significance to the design of the catalyst. Recent progress in the research of phosphoamine chromium and diphosphinoamine chromium complexes in the selective oligomerization of ethylene is reviewed from the above aspects, with the problems to be solved in the industrializaion of ethylene selective tetramerization and the further development in ethylene selective oligomerization.
-
线性α-烯烃是一类重要化工原料,可用于生产聚乙烯、合成润滑油、增塑剂、表面活性剂等[1-3]。近年来,乙烯共聚物生产商消耗了约一半的线性α-烯烃,主要包括1-丁烯、1-己烯和1-辛烯,后两种作为共聚单体生产的聚乙烯由于性能优良而变得越来越具有吸引力[4]。传统的非选择性齐聚工艺生产得到宽分布的烯烃混合物,必须经过复杂的分离过程方可使用[5]。而乙烯选择性齐聚工艺主要得到特定碳数的线性α-烯烃,得到的产品线性化程度高、质量好、更降低了分离成本。催化乙烯高选择性生产α-烯烃,催化剂是其关键技术之一[6-9]。乙烯二聚制1-丁烯和乙烯三聚生产1-己烯已经实现工业化,其中乙烯选择性二聚催化体系以Ti(OBu)4/三乙基铝(TEA)作为催化剂对1-丁烯的选择性达到93%[10]。在乙烯三聚生产1-己烯方面,Chevron-Phillips公司率先实现突破,该工艺采用吡咯/铬盐/烷基铝组成催化体系成功应用于工业化生产并以93%的选择性生成1-己烯[11-12],这一突破性进展极大地推动了科研人员对乙烯三聚催化体系的研究,此后1-己烯选择性高达99%的乙烯三聚催化体系不断被开发[13-14]。乙烯四聚生产1-辛烯也有工业化的报道,Sasol公司采用铬系双膦胺(PNP)催化体系,形成了首个乙烯四聚生产1-辛烯的工业化装置[15]。
近年来,多种金属的配合物被用于催化乙烯选择性齐聚,如钛、锆、铬等前过渡金属以及铁、钴、镍等后过渡金属[16-20],其中铬系金属性能优异受到了更多的关注和更充分的研究。这类乙烯齐聚催化剂研究主要集中在配体的修饰、反应条件的改进、提高催化性能以及探索反应机理[21-24]。本文介绍了近几年膦胺铬和双膦铬催化体系催化乙烯选择性齐聚的研究进展,从配体结构、H2的作用和助催化剂等方面对催化剂催化性能展开讨论,总结了乙烯选择性齐聚催化机理的研究进展。
1. 配体结构对铬系乙烯选择性齐聚催化剂性能的影响
2004年,Bollmann等[25]通过改变乙烯三聚催化剂的PNP配体的结构,将P原子上芳基所连的甲氧基去除,首次发现了铬系乙烯四聚催化剂。研究结果表明,配体的结构对催化剂的催化性能影响至关重要,通过改变配体的取代基、空间位阻、咬合角和骨架的类型都能影响催化活性和选择性[26-28]。以膦配体为代表的铬系催化剂研究极为活跃,由于膦配体自身结构易于调控,可引入O、N和S等供电原子形成多齿配体。研究人员相继制备了大量新型碳-膦配体、氮-膦配体、氧-膦配体[29-31],这类配体与金属中心配合形成的催化剂催化乙烯选择性齐聚性能优异,极大地推动了乙烯选择性齐聚的应用研究。
1.1 PNP配体对乙烯齐聚催化性能的影响
PNP配体因骨架中N、P原子均含孤对电子,总体呈6e结构,配位能力强,研究空间巨大。Bollmann等[25]合成9种PNP结构的配体,如图 1所示,研究了配体结构对乙烯四聚反应的影响,结果表明N原子上为异丙基取代基的PNP配体对1-辛烯选择性最高,可达2.72×105 g/(mol·h)。Jiang等[32]设计合成了一系列N-芳基双膦胺配体,如图 2所示,增加苯环上间位取代基的空间位阻显著地提高了催化体系的活性,最高可达4.98×106 g/(mol·h)。Visser等[33]研究发现,PNP配体中N原子上取代基是影响催化体系选择性的重要因素之一,通过增大N-烷基取代基的空间位阻有利于1-辛烯的生成,1-辛烯选择性从62.8%升至69.4%,如图 3所示。综上所述,Cr-PNP催化体系催化乙烯选择性齐聚性能优异,并且容易改变配体中N和P原子上的取代基。配体结构可在相当大的范围内进行调整。
图 1
图 2
图 3

刚性配体相比柔性配体构型变化小,与金属中心配位后形成的配合物调控简单、结晶能力强。Churakov等[34]制备了能与Cr(CO)4配位的刚性PNP配体,如图 4所示,但所得的催化剂用于乙烯齐聚反应中是无活性的。当与Cr(acac)3反应时,配体26和29形成的催化剂,在乙烯齐聚反应中显示出126900和312500 h-1的TOF(每摩尔催化剂在1 h内转化的单体物质的量)。研究发现配体26形成的配合物在正庚烷中催化乙烯齐聚得到乙烯三聚和四聚混合物。配体28催化乙烯四聚反应时,对1-辛烯的选择性达到了84%。利用磷原子上的取代基与桥接基团直接结合形成刚性骨架,并且发现当苯基上邻位甲氧基数量增加,TOF也随之增加。
图 4
具有树枝状结构的催化剂是一种分子结构精确、单分散性良好、催化活性高和热稳定性强的催化剂。基于这些特性,Wang等[35]合成了一系列树枝状PNP铬配合物用于乙烯齐聚反应,如图 5所示。烷基链长度直接影响催化剂催化活性。反应温度为25 ℃,乙烯压力为0.9 MPa,甲基铝氧烷(MAO)活化下,随着桥联碳链的变短催化活性也随着升高,最高活性达到2.15×105 g/(mol·h)。C6和C8齐聚物的总选择性达到36.67%,这是因为β-氢化物消除速率慢于链增长速率,产物的分布与二者的竞速关系有直接联系。
图 5
配体骨架中软供体原子的存在在决定催化循环选择性方面起着微妙的作用,大大拓展了其在乙烯选择性齐聚中的应用。Labially等[36]用二甲基甲硅烷基取代SNS配体骨架中与氮相邻的亚甲基,深刻影响着Cr/Si-SNS催化体系的催化性能。随后,Jiang等[37]设计合成了一系列Cr/Si-PNP催化体系,如图 6所示。在乙烯压力为4 MPa和温度为15 ℃,助催化剂为甲基铝氧烷(DMAO)/Et3Al活化下,配体31c的催化体系对1-辛烯选择性高达83%,催化活性为4.35×104 g/(mol·h),而当用2, 6-二异丙基苯及环戊基替代异丙基来增加主链N原子取代基的空间体积,催化活性和对1-辛烯的选择性降低,1-己烯及聚合物的选择性提高。配体32b与Cr配位形成的催化体系选择性从四聚反应急剧转变为三聚反应(1-C8, 8%;1-C6, 91%),且催化活性很低,晶体结构研究表明,较大的P1-Cr-P2咬合角和较小的CPh-P-Cr角可以使这些催化剂中的C8选择性向C6移动。另一方面,配体骨架长度的改变也可改变配位模式,从而将四聚体系转换为三聚催化体系。
图 6
当配体取代基含有给电子体原子时,易与金属中心发生电子给体-受体作用。Liu等[38]设计合成了N-四氢糠基PNP配体,分别与CrCl3(THF)3和Cr(CO)6合成铬配合物,当n(Al)/n(Cr)=600、温度为35 ℃、乙烯压力为4.5 MPa时, 配合物33的催化活性为1.59×104 g/(mol·h), 1-辛烯和1-己烯的选择性分别为63.6%和10%,聚合物的质量分数为16.4%。通过进一步的研究发现,氮原子上所连接的环状烷氧取代基可能减弱了氧原子对金属活性中心的给电子作用, 因而CrC8中间体的稳定性提高,有利于C8组分的形成,协同的3, 9-H迁移导致选择性生成1-C8 为主要产物。
图 7

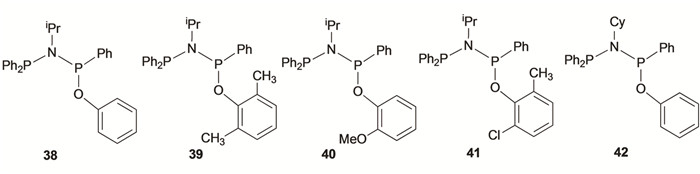
目前,基于PNP配体双膦骨架两端对称的研究较多,非对称骨架的报道较少,因此有必要对这类非对称配体的研究进行概述。Rosenthal等[39]合成了非对称PNP配体(图 8,36),通过配体36对一侧P原子取代基的改变,其负载的铬催化剂在MAO活化下生成1-己烯和1-辛烯齐聚物。之后,Wass等[40]制备了非对称PNP配体(图 8,37),配体37通过与CrCl3(THF)3和MAO结合进行选择性乙烯齐聚反应,1-C6的选择性为62.4%。Zhang等[41]报道了类似结构的不对称PNP配体,如图 9所示,在改性甲基铝氧烷(MMAO-3A)活化下催化乙烯选择性三聚四聚反应,配体38-42形成了在7.85×104~3.17×105 g/(mol·h)范围的催化活性。通过进一步修改氮原子和氧原子上的取代基,解释了改变配体结构对催化性能的影响。以氯苯为溶剂,Cr(acac)3为铬源,配体38表现出7.85×104 g/(mol·h)的高活性和88.8%的1-己烯的选择性。对于氮原子连有环己烷的配体42的铬配合物形成的催化体系,在氯苯为溶剂下催化活性达到3.17×105 g/(mol·h)的高活性,对1-己烯和1-辛烯的总选择性达到85.1%。配体38和42的铬催化体系表现出优异活性与氧参与了金属中心的配位有关。
图 8
图 9
Zhang等[42]设计合成了一系列对称/非对称N,N-二磷配体为载体的苯氧膦基Cr(Ⅲ)催化剂,对N、P原子上连接的取代基分别进行了改变,如图 10所示。通过将配体43中的异丙基取代基换为环己基来增大体积,催化活性由1.02×105 g/(mol·h)提高至2.32×105 g/(mol·h),配体43对1-辛烯和1-己烯的选择性也显著提高,并且在使用配体43时观察到较少的聚合物。与非对称的配体43和44催化体系相比,使用对称配体45和配体46的铬系催化体系,活性显著降低,分别产生较少的1-己烯和1-辛烯,以及较多的环状C6产物。对称配体45和46负载的催化剂活性较低,可能是由于它们中两个苯氧膦结构的刚性结构对催化性能产生影响。
图 10

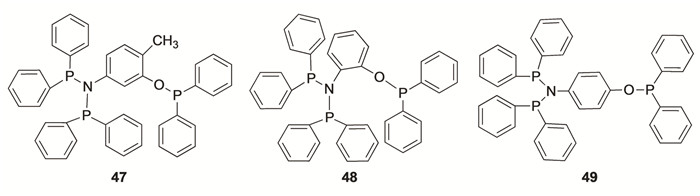
本课题组合成了一系列双膦胺及膦氧结构的多位点配体[43]。5-氨基邻甲酚的衍生物(47)/Cr(acac)3/MAO催化体系,产物主要以低碳烯烃为主,反应温度为50 ℃、反应压力为2.5 MPa、n(Al)/n(Cr)=700时, 催化剂活性可达5.91×106 g/(mol·h), 且1-辛烯选择性高达72.94%, 对1-己烯和1-辛烯总的选择性为82.11%。而2-氨基苯酚(48)/Cr(acac)3/MAO和4-氨基苯酚(49)/Cr(acac)3/MAO催化体系的催化活性和对1-辛烯的选择性相对较低, 这是因为邻位的氨基酚类配体的P—O结构和PNP较近,阻碍了乙烯分子与金属中心的配位,从而影响催化体系催化性能。
1.2 P-C-C-P配体对乙烯选择性齐聚催化性能影响
自PNP/Cr/MAO乙烯四聚催化体系被研发后,Sasol公司研究人员尝试设计合成双膦碳桥配体(PCCP),组成PCCP/Cr/MAO体系催化乙烯四聚反应,得到选择性为60%的1-辛烯。SK Energe的研究人员设计合成了一系列含环己基骨架的PCCP配体[44],与金属中心形成配合物后1-辛烯的选择性约为60%~70%,如图 12所示。研究表明,这类双膦配体与铬配合后催化乙烯选择性齐聚能拥有优异的1-C6和1-C8总选择性。目前,对PCCP配体的研究主要集中在对桥连键的长度、取代基、碳—碳键饱和度的修饰。
图 11

图 12
Cheredilin等[45]设计合成了1, 2-双(二苯基膦基)苯铬配合物,如图 13所示,在芳环的不同位置引入了取代基,以评估它们对齐聚反应的活性和选择性的影响。选择配合物CrCl3(THF)3为铬源,MAO为助催化剂进行乙烯齐聚反应,研究发现54a-54c配合物组成的催化体系催化活性较高(2.53×105~2.55×105 g/(mol·h)),配合物55参与的乙烯齐聚反应以1-C6为主,即在双膦配体磷原子上的一个苯基取代基中引入邻位官能团,可使乙烯齐聚制1-己烯的过程具有90%及以上的选择性,这是因为杂原子与金属原子的额外配位和形成刚性的金属复合结构,决定了乙烯分子在中心金属原子处的配位方式,从而实现了高选择性。
图 13

为了探究桥连骨架上不同取代基对催化乙稀选择性齐聚效果的影响,Klosin等[46]设计合成了一系列双膦配体,以CrCl3(THF)3为活性金属中心,MMAO-3A为助催化剂,反应温度60 ℃,反应压力为1.5 MPa时进行乙烯齐聚反应。含有二甲基双膦(56a,57a)和二异丙基双膦(56c,57c)配体导致了低活性催化剂的出现,但含有二乙基双膦基团(56b,57b)的配体的活性较高,分别为1.14×105和3.49×105 g/(mol·h)。带有两个邻甲氧基取代基配体58,是一种优良的乙烯三聚反应催化剂,催化活性为5.29×105 g/(mol·h),对1-己烯的选择性97.2%。体积较大的甲基/叔丁基双膦配体59对1-己烯选择性达到78%。配体60由两个五元膦环组成,对1-辛烯和-己烯选择性分为达到50.0%和31.8%。聚乙烯的质量分数3.5%。
图 14
出于形成适当咬合角的目的,并深入了解配体咬合角与催化活性之间的相关性,Zhang等[47]报道了一系列双膦噻吩配体,如图 15所示。在温度为45 ℃,3.5 MPa乙烯压力下,MMAO-3A为助催化剂,通过在配体62a的C2位置用三甲基硅烷基取代来增加噻吩主链的空间体积,显著增强了催化活性和1-己烯选择性。在C2处含有更大的SiEt3或SiPh3基团,进一步增加噻吩骨架取代基的空间体积导致配体62b和配体62c的活性下降,分别为2.07×105和3.33×105 g/(mol·h)。在C2和C3位置含有两个PPh2基团的配体63比在C3和C4位置具有两个PPh2基团的配体62活性高很多(2.64×105 g/(mol·h) vs 1.62×105 g/(mol·h))。研究人员认为两个磷原子的距离产生的配位咬合角与催化活性高度相关,对配体61和配体63的Cr配合物的配位模式来进行结构研究。双膦配体主链中两个相邻桥联碳原子的键长决定了两个磷原子间的距离,从而决定了配体咬合角的大小。在之前的报道中也提到了配位结构的研究,揭示了两个相邻桥联碳原子的键长与催化活性之间的直接相关性[48],从而很好地理解骨架取代基对催化剂性能的微妙影响。带有叔丁基取代基双膦配体的铬配合物66在3 MPa压力和40 ℃温度下催化活性为2.19×106 g/(mol·h),1-己烯和1-辛烯的总选择性为76.6%。与配合物65和66相比,配合物64具有两个相邻桥联碳原子和最大咬合角的最长键长,这可能是造成活性差的原因,也是使用配体60的铬催化剂时产生的更多聚合物的原因。因为两个相邻的桥联碳原子的键长较短,形成的较小的配位咬合角导致较高的催化活性。
图 15
在乙烯选择性齐聚催化过程中反应温度是影响催化剂寿命和催化活性的主要原因,温度较高时催化剂很容易失活,对催化反应不利,但是一些研究学者认为在高于100 ℃的高温下可溶解反应所产生的聚乙烯。Lee等[49]研究了一系列具有邻氟取代基的非对称碳桥联双膦配体,以MMAO-3A为助催化剂进行乙烯齐聚反应,克服了高温反应不利的局限性,并能保持良好的催化活性。研究人员根据数量和位置引入了氟原子,与双键所连取代基同一侧含有邻位F原子的配体组成的催化剂的催化活性要大于邻位没有被F原子取代的催化剂的催化活性(67a<67b,68a<68d,69a<69d),配体68b组成的催化体系,在反应温度为100 ℃,压力为3 MPa的条件下催化乙烯选择性齐聚,催化活性达到3.25×106 g/(mol·h),1-己烯和1-辛烯的选择性分别为74%和19%。配体67d,双键上碳原子连接体积最小的正丁基并且R1和R2均被F取代的催化体系,实验结果表明产生了大量的聚合物,温度由60 ℃升至100 ℃聚合物从9.2%增加至32%。R1连有F原子取代基的配体67c、68c、69c、70b的催化体系表现为中等活性。据推测,这是由于两侧取代的氟原子的空间效应所致。邻氟原子在芳基膦金属中心上的作用在类似研究中曾有报道,如图 17结构71,在FI催化剂中邻氟苯基配体对金属中心的稳定作用,在非共价C—F····H—C相互作用下,FI催化剂与邻位氟化亚体系配位稳定,乙烯和α-烯烃等单体可以连续插入,因此结构72能得到相似结论。另一种可能的原因是烷基铝具有高电子亲和力能促进氟化物原子配位。
图 16
图 17
双核金属催化剂将两个活性中心划分在同一配体骨架中,由于两个活性金属中心的距离较近,配合物会具有某种特性。考虑到金属核之间可能存在相互影响,Zhang等[50]构建了双核结构的桥联多膦配体,将双核桥联多膦配体与铬在MMAO-3A活化下,用于乙烯三聚四聚反应,考察不同条件下催化反应性能。当反应温度为40 ℃,乙烯压力3.5 MPa,催化活性达到3.14×106 g/(mol·h),1-C6和1-C8总选择性为77.6%。当以Cr(acac)3为铬源时,聚合活性和选择性都高于以CrCl3(THF)3为铬源。当采用单核配体体系时催化活性下降,与单核催化体系相比双核催化体系活性中心周围空间位阻较大。
图 18
2. H2对乙烯选择性齐聚催化剂催化性能的影响
在聚烯烃工业生产中,经常将H2作为链转移剂来控制聚烯烃的相对分子质量,链增长活性中心向氢发生链转移反应影响聚合物相对分子质量。Yu等[51]认为在乙烯齐聚成环过程中H2的加入使金属七元环中间体的选择路径增加,加速了β-H转移速率,目标产物生成更有利,从而能提高了催化活性。
Jiang等[52]以Ph2PN(i-Pr)PPh2/CrCl3(THF)3为催化剂进行了乙烯四聚反应,探讨了H2对催化活性和选择性的影响,并推测了乙烯四聚反应中H2的作用机理。当H2分压为0.5 MPa时,催化剂的催化活性为2.44×107 g/(mol·h)。随着H2分压的升高,乙烯的分压会降低,1-己烯和1-辛烯的总体选择性略有降低。研究发现H2的加入能促进乙烯选择性齐聚催化活性中心的形成。推测的H2的活化作用机理有:H2影响活性中心的数量;少量H2能使处于休眠状态的活性中心重新活化,提高催化剂催化活性。
由于铬前驱体在形成活性金属中心的过程中需要经过一系列的基团转化,此过程可能会产生聚合物[53]。另外,助催化剂MAO中所含的烷基铝也会导致产生聚合物。尽管聚合反应结束后产物会通过碱洗去除助催化剂并析出聚乙烯[54]。但黏附在反应器内壁、搅拌桨及管壁上的高分子聚合物会影响反应装置的稳定运行甚至导致管路堵塞,因此需要消耗人力物力及时清除。事实上,高相对分子质量副产物的存在也是影响乙烯四聚工艺实现工业化的一个主要障碍。减少甚至避免聚合物的生成成为一项有重要意义的研究工作。近年来,有报道称在催化反应体系中加入H2可减少聚合物的生成。2016年,Jiang等[55]研究了H2对PNP/Cr(Ⅲ)/MAO催化体系乙烯四聚反应性能的影响,当H2压力为0.03 MPa,催化活性从2.41×107 g/(mol·h)提升至4.71×107 g/(mol·h),H2压力增加到0.50 MPa时,聚乙烯含量从0.72%降低至0.02%。而在停止通入H2后,可观察到催化活性及聚合物的收率又回到最初程度。这一研究成果对解决高相对分子质量聚合物带来问题提供了一个简便可行的解决思路。
Bahri等[56]研究了H2与催化中心之间在乙烯三聚反应方面的相互作用,齐聚反应器除使用催化组分和乙烯单体外,还加入了0.25和0.5 MPa H2,考察了H2用量对Chevron-Phillips三聚体系即铬(Ⅲ)三(2-乙基己酸)/2, 5-二甲基吡咯/三乙基铝/四氯乙烷催化活性和选择性的影响。结果表明,加入H2对聚合物的形成和催化剂活性有显著影响,在添加0.5 MPa H2后聚合物减少60%,活性提高了38%,但对1-己烯选择性的影响不明显,这一结果与Jiang等[55]的发现相符。
Liu等[57]采用了实验与理论结合的方法研究了H2对乙烯选择性齐聚催化剂选择性和活性的影响,构建了Cr/Me2PN(Me)PMe2催化剂模型。密度泛函理论(DFT)计算表明H2与铬金属中心有缔合、解离作用,这可能是提高活性和降低1-辛烯选择性的关键因素。H2在催化乙烯齐聚反应中的作用机制尚不清楚,需要进一步的研究。
3. 乙烯选择性齐聚催化体系中的助催化剂
在乙烯选择齐聚反应中催化体系中除了包含主催化剂,还需要助催化剂。目前,常用的助催化剂有MAO、结构修饰过的MAO(MMAO)、烷基铝等。甲基铝氧烷性能优异,是目前使用最广泛的助催化剂,但MAO价格昂贵,合成高效且价格低廉的可取代甲基铝氧烷的新型助催化剂也是乙烯选择性齐聚领域急需解决的问题之一[58]。经过该领域研究学者的不断探索,非配位阴离子有机硼化合物作为助催化剂的研究竞相涌现,这类非配位阴离子与催化剂前体等物质的量反应形成阳离子活性中心[59],并能稳定阳离子活性中心。2017年,Agapie等[60]首次证明了质子化后乙烯四聚催化剂的产生,当在Cr/PNP配合物中用[H(Et2O)2]-[BAr′4]处理会导致芳基或甲基配体质子分解,随后添加乙烯形成高活性齐聚产物,不需要额外的烷基铝试剂。这些助催化剂相比MAO更容易制备,价格低廉,更避免了助催化剂复杂结构难表征的问题,为催化机理研究提供了便利。
2006年,Mcguinness等[61]制备的PNP/Cr配合物在分别被MAO和AlR3/B(C6F5)3活化后均具有不同程度的乙烯三聚催化作用,如图 19所示。实验结果显示,三烷基铝的用量对活性有很大影响。当AlMe3只有5化学计量时,活性较差(TOF=955 h-1),而10倍化学计量会产生较高的活性(TOF=21610 h-1), 显著地高于使用MAO时的活性(TOF=14465 h-1)。值得注意的是,该活化方法可产生非常高的总选择性(99.6%1-己烯)。利用较为便宜的三烷基铝作为乙烯选择性齐聚的活化剂节约了催化成本。
图 19
Lee等[62]制备了与常见的[B(C6F5)4]-阴离子配对的CrII配合物,结构为[(PNP)CrCl2L2]+[B(C6F5)4]-,分别使用三甲基铝、三乙基铝、三异丁基铝作为活化剂,当温度为75 ℃,n(Al)/n(Cr)=300时,催化体系[75a-CrCl2]+[B(C6F5)4]-/Me3Al在氯苯中表现出1.55×105 g/(mol·h)的高活性,对1-己烯和1-辛烯的选择性分别为42%和44%。具有长烷基链的[75b-CrCl2(CH3CN)2]+[B(C6F5)4]-催化体系使用甲基环己烷为溶剂,Et3Al或iBu3Al活化时,活性分别为7.70×104和8.40×104 g/(mol·h),环己烷为溶剂时活性分别为2.24×105和2.21×105 g/(mol·h),这与Sasol/MAO催化体系活性相当。
图 20

2019年, Lee等[63]继续报道了一系列无MAO的PNP催化系统,将CrCl3(THF)3、AlCl3、Ag+[B(C6F5)]-以1∶1∶2的物质的量比反应。随后添加长烷基链PNP配体,与iBu3Al混合,当反应温度为60 ℃,压力为4.5 MPa,n(Al)/n(Cr)=300时,催化活性1.11×106 g/(mol Cr·h)。在P原子连接的苯环对位上引入甲氧基和三氟甲基的配体76b和76f与Cr前体[CrAlCl3(acac)]2+[B(C6F5)4]-2反应,温度为60℃,用iBu3Al活化无活性。相同条件下,当苯环上带有吸电子F、Cl、Br的配体76c-76e进行反应时,催化活性在9.3×104~2.57×106 g/(mol·h)范围内变化,当引入甲基取代基活性降低至5.5×104 g/(mol·h)。通过在配体中的苯基对位引入诸如SiMe3(76i)和Si(nBu)3(76k)等体积大的三烷基硅基取代基时,活性显著提高,聚乙烯含量明显下降。配体76i组成的催化体系活性为2.96×106 g/(mol·h),增加了3倍,产生0.22%的少量聚乙烯。拥有更庞大的配体取代的76g和76k配体组成的催化体系进一步提高了活性,后者催化活性达到3.34×106 g/(mol·h)。
图 21

2019年,Agapie等[64]报道了将[H(OEt2)2][BAr′4](Ar′=3, 5-(CF3)2-C6H3)加入iPrN(PPh2)2(iPrPNP)和Cr(C6H4(CH2)2OCH3)3混合物中进行乙烯选择性齐聚反应,最后获得(PNP)CrR2+(R=o-C6H4(CH2)2OMe配合物, 其中PNP=iPrN(PPh2)2),该配合物可以在没有任何烷基铝试剂的情况下即可产生用于催化乙烯四聚的活性物质。以氯苯为溶剂,温度为25 ℃时,乙烯压力为0.7 MPa时,催化活性和对1-辛烯的选择性与相同条件下CrCl3(THF)3/MMAO催化体系相当。
图 22
4. 乙烯选择性齐聚机理的研究
乙烯选择性齐聚催化机理的研究对新型催化剂的研究发展具有重要的指导作用,鉴于铬基齐聚和聚合催化剂在工业上的重要地位,充分认识乙烯选择性齐聚反应机理是开发高催化活性、高选择性乙烯齐聚催化剂必备步骤之一[65]。自从Ti(OR')4/AlR3二聚体系被提出,人们认为反应路径是遵循Cossee-Arlman增长机理。Manyik提出金属环状乙烯三聚反应路径后,金属环化机理成为目前普遍接受的乙烯三聚反应路径[66-67]。在乙烯四聚反应的研究中,研究学者们提出了可以替代乙烯四聚单核金属环机理的双核金属环机理,这引起了乙烯四聚反应路径在两种机理间的争论。
4.1 Cossee-Arlman金属环机理
Cossee[66-69]提出了金属催化乙烯齐聚的一般机理,即通过配位、迁移、插入实现链增长。乙烯插入金属氢化物或金属-烷基键引发链增长,通过末端β-H消除或通过将β链转移至乙烯来诱导链终止,从而形成所需碳数的1-烯烃。当β-H转移速率远快于链增长速率时,长链1-烯烃就不能生成,因此实现了乙烯的二聚。随后,通过Alphabutol工艺的氘代实验和分子计算利用密度泛函理论进一步证实了该机理[70-71]。
图 23
4.2 乙烯三聚金属环机理
1977年,Manyik等[72]认为乙烯要插入到铬的金属中心则需要空的配位点,反应开始于两个乙烯分子的氧化偶联,形成金属环戊烷中间体,金属环戊烷与另一个乙烯分子配位,最后通过β-H转移和还原消除释放1-己烯(图 24a)。Manyik的金属五元环机理并没有解释高选择性的1-己烯和没有C6烯烃生成的原因。1989年, Briggs[73]在此基础上对乙烯三聚单金属环机理进行了修改(图 24b),第3个乙烯分子是快速插入铬金属五元环中形成铬金属七元环中间体,而不是β-H转移乙烯插入,铬金属七元环再发生环内β-H迁移开环解释了1-己烯的选择性由金属环戊烷和金属环庚烷的相对稳定性决定,乙烯插入到铬金属五元环的速率大于其β-H还原消去的速率时, 并且金属七元环还原消去1-己烯的速率大于乙烯进一步插入成环速率时, 得到高选择性1-己烯。
图 24

4.3 乙烯四聚金属环机理
自从2004年Bollmann等[25]首次合成Cr-PNP乙烯四聚催化体系,研究人员也一直对乙烯四聚催化反应机理进行探索,并取得了一些进展。Overett等[74]采用氘标记技术研究了乙烯四聚合成1-辛烯的反应机理,认为乙烯选择性四聚机理与乙烯选择性金属环三聚机理类似。目前,已报道的乙烯四聚金属环催化机理有铬单核和双核两种。
4.3.1 乙烯四聚单核金属环机理
Overett等[74]认为乙烯四聚催化反应过程中可能存在两种反应机理。首先,形成金属七元环中间体后再插入一个乙烯分子形成了金属九元环,金属九元环发生还原消除形成1-辛烯,这种方式形成的1-辛烯比乙烯插入到铬金属七元环中间体而形成高碳数金属环化物容易。第2种推测的可能是金属七元环中间体上发生了β-H转移到同等位置的乙烯上生成一个己基乙烯结构,然后再发生消除反应生成1-辛烯,或继续进行乙烯插入使链继续增长。另外,乙烯四聚反应过程中还会形成甲基环戊烷和亚甲基环戊烷,两者物质的量比是1∶1。其生成机理主要是金属七元环重排生成环戊甲基氢化铬中间体,再发生β-H消除形成甲基环戊烷和亚甲基环戊烷。
图 25

Britovsek等[75]利用DFT方法研究了Cr-PNP催化体系乙烯齐聚机理,计算结果显示,乙烯与金属五元环双配位形成1-辛烯而与金属五元环单配位时形成1-己烯,金属五元环与乙烯分子的配位能力是决定1-己烯和1-辛烯选择性的关键。
图 26
Kwon等[76]采用DFT理论过渡态模型,计算了一类膦单环亚胺(P,N)配体铬催化剂,其中配体结构的变化控制着1-己烯和1-辛烯的选择性,当亚胺邻位取代基为甲基时产物主要为1-己烯,当亚胺邻位取代基为氢时,1-辛烯含量显著增加。邻位甲基的引入增加了膦单环亚胺配体催化体系的空间位阻,导致生成α-烯烃的选择性不同。通过理论研究方法预测催化剂的选择性,对铬系齐聚催化剂的研究有重要的指导意义。
图 27

Boelter等[77]提出了一种用于1-己烯和1-辛烯生产的改进机理,如图 28所示,该机理也充分说明了乙烯齐聚过程中形成的主要副产物。两个具有Crn中心的乙烯分子的氧化偶合产生环戊烷(B),当第三乙烯插入其中,从而生成环庚烷(C)。立体空间上大的配体阻碍了另外的乙烯插入到铬环庚烷中间体中,并使其更可能发生3, 7-氢化物转移(D),从而生成1-己烯。乙烯与铬环庚烷C缔合可得到E,它是1-辛烯和环状产物的常见中间体。乙烯插入中间体E可以导致形成铬环壬烷F,后者通过3, 9-氢化物转移生成1-辛烯,或者将β-H转移至乙烯可以导致Cr-乙基-己烯基G的形成。然后,环化导致形成配合物H,通过β-H提取将其分解为亚甲基环戊烷和甲基环戊烷以及乙烯和乙烷的环状产物。这种机理解释了为什么1-辛烯和环状产物的形成是相互联系的。
图 28

Agapie等[78]最近发表了一项同位素标记研究,证实乙烯三聚和四聚途径共用一个单体环庚烷中间体,解释为什么即使在增加乙烯压力下1-辛烯选择性限于约75%。研究认为选择性取决于乙烯插入和1-己烯从该中间体中消除的相对速率。
图 29

4.3.2 乙烯四聚双核金属环机理
由于九元环中间体的不稳定性,Pritz等[79]已经假定了替代四聚反应的机理,如图 30所示。首先,两个金属中心分别与乙烯分子配位,形成两个金属环戊烯,再结合形成双金属十元环,实现了1-辛烯的还原消除。在这一机理中,选择性由金属环戊烷的C—C偶合的相对速率或导致C—C偶合的步骤与乙烯插入的相对速率决定。并且此机理中的选择性可能会受到Cr浓度的影响,作为两种Cr物种相互耦合的竞争途径之一。然而该机理尚未得到证实,还需要进一步的验证。
图 30
4.4 活性金属中心氧化态
活性金属中心铬的氧化态在催化过程中可能会发生变化,活性中心包含Cr(Ⅰ)/Cr(Ⅲ)氧化态和Cr(Ⅱ)/Cr(Ⅳ)氧化态活性物种[80-84]。乙烯选择性齐聚反应中普遍认为活性中心价态与助催化剂的引入有关,助催化剂可使金属中心成为活性物种,乙烯的氧化加成和还原消除反应则使金属中心价态升高或降低,最后完成整个催化循环过程[85]。目前对乙烯选择性齐聚活性中心氧化态的研究并没有一致的结论。
Mcguinness等[86]将不同的铬化合物与催化性能较好的SNS/PNP配体反应,得到具有不同价态的金属活性中心催化剂,研究金属中心价态对催化剂催化性能的影响,如图 31所示。双核Cr/SNS配合物在AlR3/B(C6F5)3的作用生成高选择性的乙烯三聚产物,发现这些配体容易去质子化,产生单阴离子PNP和SNS配合物。并且认为Cr/SNS乙烯三聚催化体系中存在Cr(Ⅱ)/Cr(Ⅳ)和Cr(Ⅰ)/Cr(Ⅲ)两种催化循环。
图 31

随着理论化学研究的不断深入,常设计合适的的分子模型计算活性中心的氧化态。配体结构的不同也会导致金属氧化态的不同,在以往的研究中发现含有吡咯配体和氯原子辅助配体的铬催化体系的分子模型中,活性中心的氧化态可能是Cr(Ⅱ)/Cr(Ⅳ)[87-88]。而含有PNP配体或者咔唑配体的铬催化体系,Cr(Ⅰ)/Cr(Ⅲ)可能是活性中心的氧化态[89-90]。
刘柏平等[91]从头计算(ab initio)结合的方法研究了金属氧化态、取代基空间位阻对Cr/2, 2′-二甲基吡啶胺催化体系的Cr(Ⅰ)/Cr(Ⅲ)催化循环的乙烯三/四聚反应机理。金属环庚烷是决定选择性的中间体,控制选择性的关键是金属环庚烷扩张和金属环庚烷分解过渡态的相对能。研究证实了配体上甲基取代基的位置在乙烯选择性齐聚中发挥着重要作用,远端甲基的存在有利于金属环庚烷增长过程,表现出乙烯四聚选择性;而近端甲基极大地影响了金属环庚烷增长过程的电荷排布及乙烯插入能垒,因而体系表现出乙烯三聚选择性。
5. 结论与展望
乙烯选择性齐聚工艺二十多年来发展迅速,铬系催化剂因性能优异受到广泛重视和深入研究。以催化剂配体结构修饰和改性为主导的研究占据了乙烯选择性齐聚的大部分。膦胺配体和双膦配体具有调控简单、与Cr配位性能好等优点,借助配体构型、刚性、空间位阻以及配位咬合角的角度,能在催化体系周围营造出特殊环境并显著影响催化剂的催化性能。当前助催化剂MAO价格昂贵并且消耗量巨大,很难应用在大规模工业化生产中,学界和业界的科研人员正努力开发其他活化性能优异并且价格低廉的助催化剂。另外,大部分乙烯齐聚催化剂除了产生齐聚物外,还会伴随着副产物的生成,催化体系中通入H2在保证良好催化性能的前提下,还可以减少聚合物生成,但解决副产物生成的关键要从催化剂根本出发。乙烯选择性机理的研究为新型催化剂的开发提供了支持,氘代实验已经证明反应路径是按照金属环机理进行。关于乙烯四聚的反应路径,多数研究倾向于1-辛烯形成的单核机理,但不能否认双核机理的可能性。在理论研究方法方面,DFT、电子顺磁共振(EPR)等技术为反应机理研究提供了重要依据,可更有力地推动乙烯选择性齐聚机理的研究。
乙烯三聚生产1-己烯在工业生产中已经得到满意的选择性,然而乙烯四聚制1-辛烯的选择性不高且副产聚合物的生成等问题还没有彻底解决,较低的选择性会导致分离能耗成本高,较多的副产物更会造成反应器黏壁甚至管路堵塞,这些问题都阻碍了乙烯四聚工业化的进程。基于以上问题我们认为未来乙烯选择性齐聚的研究应主要集中在以下3个方面:1)开发不产生高相对分子质量聚乙烯副产物的催化体系,以解决黏釜堵塞管道等问题。2)优化助催化剂结构并提高助催化剂利用效率,制备可代替MAO的助催化剂降低催化成本。3)采用实验与理论结合的研究方法,对不同催化体系的活化、失活机理进行研究,指导新型高性能催化剂的设计和研发。相信随着科研人员的不懈努力和研究工作的不断推进,这些问题在不远的将来都能得到解决,而通过乙烯选择性齐聚生产更高碳数α-烯烃的研究也一定会获得突破。
-
-
[1]
WANG Z, LIU Q B, SUN W H. Recent advances in Ni-mediated ethylene chain growth: N imine-donor ligand effects on catalytic activity thermal stability and oligo-/polymer structure[J]. Coord Chem Rev, 2017, 350: 68-83. doi: 10.1016/j.ccr.2017.06.003
-
[2]
CHEN L, LI G, WANG Z. Ethylene oligomerization over nickel supported silica-alumina catalysts with high selectivity for C10+ products[J]. Catal, 2020, 10(2): 180. doi: 10.3390/catal10020180
-
[3]
王俊, 刘锦义, 陈丽铎. 超支化双吡啶亚胺铬催化剂的合成及催化乙烯齐聚性能[J]. 应用化学, 2019,36,(7): 773-781. WANG J, LIU J Y, CHEN L D. Synthesis and ethylene oligomerization behavior of hyperbranched bispyridineimine chromium catalyst[J]. Chinese J Appl Chem, 2019, 36(7): 773-781.
-
[4]
LI J, ZHANG Q, HU X. 2-Acetyloxymethyl-substituted 5, 6, 7-trihydroquinolinyl-8-ylideneamine-Ni(Ⅱ) chlorides and their application in ethylene dimerization/trimerization[J]. Appl Organomet Chem, 2020, 34(1): e5254.
-
[5]
吴昊, 毛国梁, 王月含. 乙烯齐聚工艺-从非选择性齐聚到选择性齐聚[J]. 化工科技, 2019,27,(1): 83-90. WU H, MAO G L, WANG Y H. Ethylene oligomerization technology from non-selective oligomerization to selective oligomerization[J]. Sci Technol Chem Ind, 2019, 27(1): 83-90.
-
[6]
GRAUKE R, SCHEPPER R, RABEAH J. Impact of Al activators on structure and catalytic performance of Cr catalysts in homogeneous ethylene oligomerization-a multitechnique in situ/operando study[J]. Chem Cat Chem, 2020, 12: 1-2.
-
[7]
NEWLAND R J, SMITH A, SMITH D M. Accessing alkyl- and alkenyl-cyclopentanes from Cr-catalysed ethylene oligomerization using 2-phosphinophosphinine ligands[J]. Organometallics, 2018, 37(6): 1062-1073. doi: 10.1021/acs.organomet.8b00063
-
[8]
AZIMNAVAHSI L, MOHAMADNIA Z. Optimization of ethylenetrimerization using catalysts based on TiCl3/half-sandwich ligands[J]. Appl Organomet Chem, 2019, 33(2): e4666. doi: 10.1002/aoc.4666
-
[9]
FALLAHI M, AHMADI E, MOHAMADNIA Z. Effect of inorganic oxide supports on the activity of chromium-based catalysts in ethylene trimerization[J]. Appl Organomet Chem, 2019, 33(8): e4975.
-
[10]
ALSA'DOUN A W. Dimerization of ethylene to butene-1 catalyzed by Ti(OR')4-AlR3[J]. Cheminform, 1993, 25(7): 1-40.
-
[11]
LICCIULLI S, ALBAHILY K, FOMITCHEVA V. A chromium ethylidene complex as a potent catalyst for selective ethylene trimerization[J]. Angew Chem, 2011, 50(10): 2346-2355. doi: 10.1002/anie.201006953
-
[12]
FREEMAN J W, BUSTER J L, KNUDSEN R D. Olefin production: US 5856257[P]. 1999.
-
[13]
YOSHIDA T, YAMAMOTO T, OKADA H, et al. Catalyst for trimerization of ethylene and process for trimerizing ethylene using the catalyst: US 0035029[P]. 2002.
-
[14]
ZHANG J, LI A, HOR T S A. Crystallographic revelation of the role of AlMe3(in MAO) in Cr[NNN] pyrazolyl catalyzed ethylene trimerization[J]. Organometallics, 2009, 28(10): 2935-2937. doi: 10.1021/om9002347
-
[15]
李雅丽. 南非Sasol公司成功投运全球首套乙烯四聚工业化生产装置[J]. 石油化工技术与经济, 2014,30,(2): 61-61. LI Y L. South Africa Sasol company successfully put into operation the world's first ethylene tetramerization industrial production equipment[J]. Technol Econom Petrochem, 2014, 30(2): 61-61.
-
[16]
WANG Z, SOLAN G A, SUN W H. Carbocyclic-fused N, N, N-pincer ligands as ring-strain adjustable supports for iron and cobalt catalysts in ethylene oligo-/polymerization[J]. Coord Chem Rev, 2018, 363(6): 92-108.
-
[17]
GONG M, LIU Z, LI Y. Selective co-oligomerization of ethylene and 1-hexene by chromium-PNP catalysts: a DFT study[J]. Organometallics, 2016, 35(7): 972-981. doi: 10.1021/acs.organomet.5b01029
-
[18]
YUAN S F, YAN Y, SUN W H. Recent advancements in N-ligated group 4 molecular catalysts for the (co)polymerization of ethylene[J]. Coord Chem Rev, 2020, 411: 213254-213269. doi: 10.1016/j.ccr.2020.213254
-
[19]
SVEJDA S A, BROOKHART M. Ethylene oligomerization and propylene dimerization using cationic (α-diimine)nickel(Ⅱ) catalysts[J]. Organometallics, 2017, 18(1): 65-74.
-
[20]
BARIASHIR C, HUANG C, SUN W H. Recent advances in homogeneous chromium catalyst design for ethylene tri-, tetra-, oligo- and polymerization[J]. Coord Chem Rev, 2019, 385: 208-229. doi: 10.1016/j.ccr.2019.01.019
-
[21]
WU Q, WANG W, XU G. Bulky iminophosphine-based nickel and palladium catalysts bearing 2, 6-dibenzhydryl groups for ethylene oligo-/polymerization[J]. Appl Organomet Chem, 2020, 34: e5428.
-
[22]
WANG J, LIU J, CHEN L. Preparation of chromium catalysts bearing bispyridylamine and its performance in ethylene oligomerization[J]. Trans Met Chem, 2019, 44(7): 681-688. doi: 10.1007/s11243-019-00333-3
-
[23]
CHEN L, HUO H, WANG J. Ethylene oligomerization studies utilizing nickel complexes bearing pyridine-imine ligands[J]. Inorg Chim Acta, 2019, 491: 67-75. doi: 10.1016/j.ica.2019.04.001
-
[24]
ZHANG L, WEI W, JIANG T. Efficient chromium-based catalysts for ethylene tri-/tetramerization switched by silicon-bridged/N, P-based ancillary ligands: a structural, catalytic and DFT study[J]. Appl Petrochem Res, 2017, 7: 5011-5088.
-
[25]
BOLLMANN A, BLANN K, DIXON J T. Ethylene tetramerization: a new route to produce 1-octene in exceptionally high selectivities[J]. J Am Chem Soc, 2004, 126(45): 14712-14713. doi: 10.1021/ja045602n
-
[26]
ZHOU Y, WU H, XU S. Highly active chromium-based selective ethylene tri-/tetramerization catalysts supported by PNPO. phosphazane ligands[J]. Dalton Trans, 2015, 44(20): 9545-9550. doi: 10.1039/C5DT00801H
-
[27]
TOBIAS , DIXON J T, HAUMANN M. Trimerization and tetramerization of ethylene in continuous gas-phase reaction using a Cr-based supported liquid phase catalyst[J]. React Chem Eng, 2019, 4(1): 131-140. doi: 10.1039/C8RE00179K
-
[28]
ZHANG L, MENG X, CHEN Y. Chromium-based ethylene tetramerization catalysts supported by silicon-bridged diphosphine ligands: further combination of high activity and selectivity[J]. ChemCatChem, 2017, 9(1): 76-79. doi: 10.1002/cctc.201600941
-
[29]
FERREIRA J, ZILZ R, BOEIRA I S. Chromium complexes based on thiophene-imine ligands for ethylene oligomerization[J]. Appl Organomet Chem, 2019, 33(3): e4697. doi: 10.1002/aoc.4697
-
[30]
ALFEROV K, BELOV G P, MENG Y. Chromium catalysts for selective ethylene oligomerization to 1-hexene and 1-octene: recent results[J]. Appl Catal A-Gen, 2017, 542: 71-124. doi: 10.1016/j.apcata.2017.05.014
-
[31]
刘清云, 高榕, 侯俊先. P.N.P三齿铬配合物合成和结构及其催化乙烯齐聚与聚合[J]. 有机化学, 2013,33,(4): 808-814. LIU Q Y, GAO R, HOU J X. Tridentate P.N.P chromium complexes: synthesis, characterization and their ethylene oligomerization and polymerization[J]. Chinese J Org Chem, 2013, 33(4): 808-814.
-
[32]
JIANG T, ZHANG S, JIANG X. The effect of N-aryl bisphosphineamine ligands on the selective ethylene tetramerization[J]. J Mol Catal A Chem, 2008, 279(1): 90-93. doi: 10.1016/j.molcata.2007.10.009
-
[33]
CLOETE N, VISSER H G, ENGELBRECHT I. Ethylene tri- and tetramerization: a steric parameter selectivity switch from X-ray crystallography and computational analysis[J]. Inorg Chem, 2013, 52(5): 2268-2270. doi: 10.1021/ic302578a
-
[34]
NIFANTEV I E, VINOGRADOV A A, VINOGRADOV A A. 5, 6-Dihydrodibenzo[c, e] [1, 2] azaphosphinine-based PNP ligands, Cr(0) coordination, and Cr(Ⅲ) precatalysts for ethylene oligomerization[J]. Organometallics, 2018, 37(16): 2660-2664. doi: 10.1021/acs.organomet.8b00427
-
[35]
WANG J, GAO R, ZHANG N. Novel dendritic PNP chromium complexes: synthesis, characterization, and performance on ethylene oligomerization[J]. Helv Chim Acta, 2017, 100(12): e1700162. doi: 10.1002/hlca.201700162
-
[36]
ALBAHILY K, GAMBAROTTA S, DUCHATEAU R. Ethylene oligomerization promoted by a silylated-SNS chromium system[J]. Organometallics, 2011, 30(17): 4655-4664. doi: 10.1021/om200505a
-
[37]
ALAM F, ZHANG L, JIANG T. Catalytic systems based on chromium(Ⅲ) silylated-diphosphinoamines for selective ethylene tri-/tetramerization[J]. ACS Catal, 2018, 8(11): 10836-10845. doi: 10.1021/acscatal.8b02698
-
[38]
刘睿, 钟向宏, 刘振宇. N-四氢糠基PNP配体/铬催化体系及其乙烯选择性齐聚性能[J]. 有机化学, 2017,37,(9): 2315-2321. LIU R, ZHOU X H, LIU Z Y. Selective ethylene oligomerization catalyzed by the chromium complex bearing N-tetrahydrofurfuryl PNP ligand[J]. Chinese J Org Chem, 2017, 37(9): 2315-2321.
-
[39]
HÄRZSCHEL S, KVHN F E, ROSENTHAL U. Comparative study of new chromium-based catalysts for the selective tri- and tetramerization of ethylene[J]. Catal Sci Technol, 2015, 5(3): 1678-1682. doi: 10.1039/C4CY01441C
-
[40]
STENNETT T E, HEY T W, WASS D F. N, N-diphospholylamines-a new family of ligands for highly active chromium-based selective ethene oligomerisation catalysts[J]. ChemCatChem, 2013, 5(10): 2946-2954. doi: 10.1002/cctc.201300306
-
[41]
ZHOU Y, WU H, ZHANG J. Highly active chromium-based selective ethylene tri-/tetramerization catalysts supported by PNPO phosphazane ligands[J]. Dalton Trans, 2015, 44(20): 9545-9550. doi: 10.1039/C5DT00801H
-
[42]
JI X, SONG L, ZHANG C. Highly active chromium-based selective ethylene tri-/tetramerization catalysts supported by N, N-diphospholylamines[J]. Inorg Chim Acta, 2017, 466: 177-121.
-
[43]
封智超, 毛国梁, 吴韦. 基于5-氨基邻甲酚的膦配体的合成及在乙烯齐聚中的应用[J]. 有机化学, 2018,38,(3): 698-704. FENG Z C, MAO G L, WU W. Synthesis of phosphine ligands based on 5-amino-o-cresol and its application in ethylene oligomerization[J]. Chinese J Org Chem, 2018, 38(3): 698-704.
-
[44]
KIM S, KIM T, CHUNG J. Bimetallic ethylene tetramerization catalysts derived from chiral DPPDME ligands: syntheses, structural characterizations, and catalytic performance of [(DPPDME)CrCl3]2 (DPPDME=S, S- and R, R-chiraphos and meso-achiraphos)[J]. Organometallics, 2010, 29(22): 5805-5811. doi: 10.1021/om100400b
-
[45]
CHEREDILIN D N, SHELOUMOV A M, SENIN A A. Catalytic properties of chromium complexes based on 1, 2-bis(diphenylphosphino)benzene in the ethylene oligomerization reaction[J]. Petrol Chem, 2019, 59(1): 72-78. doi: 10.1134/S0965544119130036
-
[46]
BOELTER S D, DAVIES D R, KLOSIN J. Phospholane-based ligands for chromium-catalyzed ethylene tri- and tetramerization[J]. Organometallics, 2020, 39(7): 967-987. doi: 10.1021/acs.organomet.9b00721
-
[47]
ZHANG C, SONG L, WU H. Ethylene tri-/tetramerization catalysts supported by diphosphinothiophene ligands[J]. Dalton Trans, 2017, 46(26): 8399-8404. doi: 10.1039/C7DT01060E
-
[48]
ZHANG J, WANG X, ZHANG X. Switchable ethylene tri-/tetramerization with high activity: subtle effect presented by backbone-substituent of carbon-bridged diphosphine ligands[J]. ACS Catal, 2016, 3(10): 2311-2317.
-
[49]
LEE H S, JOE Y, PARK H. Chromium catalysts for ethylene trimerization/tetramerization functionalized with ortho-fluorinated arylphosphine ligand[J]. Catal Commun, 2019, 121: 15-18. doi: 10.1016/j.catcom.2018.12.010
-
[50]
郑明芳, 吴红飞, 张军. 含桥联多膦配体的双核Cr(Ⅲ)乙烯齐聚催化剂[J]. 石油化工, 2018,47,(9): 924-928. ZHENG M F, WU H F, ZHANG J. Ethylene oligomerization catalyzed by binuclear Cr catalyst based on a bridged phosphine ligand[J]. Pet Technol, 2018, 47(9): 924-928.
-
[51]
于部伟, 蒋岩, 牟玉强. 氢气在Cr催化剂催化乙烯齐聚中的作用[J]. 精细石油化工, 2019,36,(6): 11-13. YU B W, JIANG Y, MOU Y Q. The role of hydrogen in Cr catalyst catalyzed oligomerization of ethylene[J]. Speciality Pet, 2019, 36(6): 11-13.
-
[52]
时鹏飞, 曹晨刚, 姜涛. 氢气对铬系催化剂催化乙烯四聚制1-辛烯的影响[J]. 石油化工, 2015,44,(8): 948-952. SHI P F, CAO C G, JIANG T. Effect of hydrogen on ehylene tetramerization to 1-octene with Cr catalyst[J]. Petrochem Technol, 2015, 44(8): 948-952.
-
[53]
HAGEN H, KRETSCHMER W P, BUREN F R V. Selective ethylene trimerization: a study into the mechanism and the reduction of PE formation[J]. J Mol Catal A-C, 2006, 248(1): 237-247.
-
[54]
徐珂, 栗同林, 郑明芳, 等. 乙烯齐聚生产α-烯烃工艺中去除催化剂和聚乙烯的方法: 中国, 107151195 A[P]. 2016.XU K, LI T L, ZHENG M F, et al. Method for removing catalyst and polyethylene in process for producing α-olefin by ethylene oligomerization: CN, 107151195 A[P]. 2016.
-
[55]
JIANG T, ZHANG L, GAO J. Hydrogen: efficient promoter for PNP/Cr(Ⅲ)/MAO catalyzed ethylene tetramerization toward 1-octene[J]. Appl Pet R, 2016, 6(4): 1-5. doi: 10.1007/s13203-016-0151-4?view=classic
-
[56]
BAHRI-LALEH N, KARIMI M, KALANTARI Z. H2 effect in Chevron-Phillips ethylene trimerization catalytic system: an experimental and theoretical investigation[J]. Polym Bull, 2017, 75(8): 3555-3565.
-
[57]
LIU L, LIU Z, CHENG R. Unraveling the effects of H2, N substituents and secondary ligands on Cr/PNP-catalyzed ethylene selective oligomerization[J]. Organometallics, 2018, 37(21): 3893-3900. doi: 10.1021/acs.organomet.8b00578
-
[58]
STENNETT T E, HADDOW M F, WASS D F. Avoiding MAO: alternative activation methods in selective ethylene oligomerization[J]. Organometallics, 2012, 31(19): 6960-6965. doi: 10.1021/om300739m
-
[59]
MCGUINNESS D S, RUCKLIDGE A J, TOOZE R P. Cocatalyst influence in selective oligomerization: effect on activity, catalyst stability, and 1-hexene/1-octene selectivity in the ethylene trimerization and tetramerization reaction[J]. Organometallics, 2007, 26(10): 2561-2569. doi: 10.1021/om070029c
-
[60]
HIRSCHER N A, AGAPIE T. Stoichiometrically activated catalysts for ethylene tetramerization using diphosphinoamine-ligated Cr tris(hydrocarbyl) complexes[J]. Organometallics, 2017, 36(21): 4107-4110. doi: 10.1021/acs.organomet.7b00706
-
[61]
MCGUINNESS D S, BROWN D B, TOOZE R P. Ethylene trimerization with CrPNP and CrSNS complexes: effect of ligand structure, metal oxidation state, and role of activator on catalysis[J]. Organometallics, 2006, 25(15): 3605-3610. doi: 10.1021/om0601091
-
[62]
KIM T H, LEE H M, JEONG M S. Methylaluminoxane-free chromium catalytic system for ethylene tetramerization[J]. ACS Omega, 2017, 2(3): 765-773. doi: 10.1021/acsomega.6b00506
-
[63]
KIM T H, LEE H M, PARK H S. MAO-free and extremely active catalytic system for ethylene tetramerization[J]. Appl Organomet Chem, 2019, 33(4): e4829. doi: 10.1002/aoc.4829
-
[64]
HIRSCHER N A, PEREZ S D, AGAPIE T. Robust chromium precursors for catalysis: isolation and structure of a single-component ethylene tetramerization precatalyst[J]. J Am Chem Soc, 2019, 141(14): 6022-6029. doi: 10.1021/jacs.9b01387
-
[65]
YANG Y, LIU Z, LIU B P. Selective ethylene tri-/tetramerization by in situ-formed chromium catalysts stabilized by N, P-based ancillary ligand systems[J]. ACS Catal, 2013, 3(10): 2353-2361. doi: 10.1021/cs4004968
-
[66]
AGAPIE T, SCHOFER S J, LABINGER J A. Mechanistic studies of the ethylene trimerization reaction with chromium diphosphine catalysts: experimental evidence for a mechanism involving metallacyclic intermediates[J]. J Am Chem Soc, 2004, 126(5): 1304-1305. doi: 10.1021/ja038968t
-
[67]
AGAPIE T, LABINGER J A, BERCAW J E. Mechanistic studies of olefin and alkyne trimerization with chromium catalysts: deuterium labeling and studies of regiochemistry using a model chromacyclopentane complex[J]. J Am Chem Soc, 2007, 129(46): 14281-14295. doi: 10.1021/ja073493h
-
[68]
ARLMAN E J, COSSEE P. Ziegler-Natta catalysis Ⅲ stereospecific polymerization of propene with the catalyst X system. TiCl3AlEt3[J]. J Catal, 1964, 3(1): 99-104. doi: 10.1016/0021-9517(64)90097-1
-
[69]
ALLEGRA G. Discussion on mechanism of polymerization of α-olefins with Ziegler-Natta catalysts[J]. Macromol Chem Phys, 1971, 145(1): 235-246. doi: 10.1002/macp.1971.021450119
-
[70]
SUTTIL J A, MCGUINNESS D S. Mechanism of ethylene dimerization catalyzed by Ti(OR') 4/AlR 3[J]. Organometallics, 2012, 31(19): 7004-7010. doi: 10.1021/om3008508
-
[71]
BELOV G P, DZHABIEV T S, KOLESNIKOV I M. Activation of C-H and C-C bonds in ethylene and piperylene catalytic reactions[J]. J Mol Catal, 1982, 14(1): 105-112. doi: 10.1016/0304-5102(82)80053-9
-
[72]
MANYIK R M, WALKER W E, WILSON T P. A soluble chromium-based catalyst for ethylene trimerization and polymerization[J]. J Catal, 1977, 47(2): 197-209. doi: 10.1016/0021-9517(77)90167-1
-
[73]
BRIGGS J R. The selective trimerization of ethylene to hex-1-ene[J]. J Chem Soc Chem Commun, 1989, 11(11): 674-675.
-
[74]
OVERETT M, BLANN K, BOLLMANN A. Mechanistic investigations of the ethylene tetramerisation reaction[J]. J Am Chem Soc, 2005, 127(30): 10723-10730. doi: 10.1021/ja052327b
-
[75]
BRITOVSEK G J, MCGUINNESS D S, WIERENGA T S. Single- and double-coordination mechanism in ethylene tri- and tetramerization with Cr/PNP catalysts[J]. ACS Catal, 2015, 5(7): 4152-4166. doi: 10.1021/acscatal.5b00989
-
[76]
KWON D, FULLER J T, KILGORE U J. Computational transition-state design provides experimentally verified Cr(P, N) catalysts for control of ethylene trimerization and tetramerization[J]. ACS Catal, 2018, 8(2): 1138-1142. doi: 10.1021/acscatal.7b04026
-
[77]
BOELTER S D, DAVIES D R, MARGL P M. Phospholane-based ligands for chromium-catalyzed ethylene tri- and tetramerization[J]. Organometallics, 2020, 39(7): 976-987. doi: 10.1021/acs.organomet.9b00722
-
[78]
HIRSCHER N A, LABINGER J A, AGAPIE T. Isotopic labelling in ethylene oligomerization: addressing the issue of 1-octene vs. 1-hexene selectivity[J]. Dalton Trans, 2019, 48(1): 40-44. doi: 10.1039/C8DT04509G
-
[79]
PEITZ S, ALURI B, PEULECKE N. An alternative mechanistic concept for homogeneous selective ethylene oligomerization of chromium-based catalysts: binuclear metallacycles as a reason for 1-octene selectivity?[J]. Chem Eur J, 2010, 16(26): 7670-7676. doi: 10.1002/chem.201000750
-
[80]
JABRI A, MASON C, SIM Y. Isolation of single-component trimerization and polymerization chromium catalysts: the role of the metal oxidation state[J]. Angew Chem Int Ed, 2008, 47(50): 9717-9721. doi: 10.1002/anie.200803434
-
[81]
VIDYARATNE I, NIKIFOROV G B, GORELSKY S I. Isolation of a self-activating ethylene trimerization catalyst[J]. Angew Chem Int Ed, 2009, 48(35): 6552-6556. doi: 10.1002/anie.200900957
-
[82]
ALBAHILY K, SHAIKH Y, SEBASTIAO E. Vinyl oxidative coupling as a synthetic route to catalytically active monovalent chromium[J]. J Am Chem Soc, 2011, 133(16): 6388-6395. doi: 10.1021/ja201003j
-
[83]
CARTER E, CAVELL K J, GABRIELLI W F. Formation of[Cr(CO)x(Ph2PN(iPr)PPh2)]+ structural isomers by reaction of triethylaluminum with a chromium N, N-bis(diarylphosphino)amine complex[Cr(CO)4(Ph2PN(iPr)PPh2)]+: an EPR and DFT investigation[J]. Organometallics, 2013, 32(6): 1924-1931. doi: 10.1021/om400029y
-
[84]
RUCKLIDGE A J, MCGUINNESS D S, TOOZE R P. Ethylene tetramerization with cationic chromium(Ⅰ) complexes[J]. Organometallics, 2007, 26(10): 2782-2787. doi: 10.1021/om0701975
-
[85]
宋闯, 毛国梁, 刘振华. 均相Cr系催化剂催化乙烯选择性齐聚反应机理研究进展[J]. 有机化学, 2016,36,(9): 2105-2120. SONG C, MAO G L, LIU Z H. Advances in mechanistic research of ethylene selective oligomerization catalyzed by homogeneous chromium-based catalysts[J]. Chinese J Org Chem, 2016, 36(9): 2105-2120.
-
[86]
MCGUINNESS , DAVID S. Olefin oligomerization via metallacycles: dimerization, trimerization, tetramerization, and beyond[J]. Chem Rev, 2011, 111(3): 2321-2341. doi: 10.1021/cr100217q
-
[87]
WERNER J V R, CRONJōG , STEYNBERG J P. A DFT study toward the mechanism of chromium-catalyzed ethylene trimerization[J]. Organometallics, 2004, 23(6): 1207-1222. doi: 10.1021/om0306269
-
[88]
BHADURI S, MUKHOPADHYAY S, KULKARNI S A. Density functional studies on chromium catalyzed ethylene trimerization[J]. J Organomet Chem, 2009, 694(9/10): 1297-1307.
-
[89]
KLEMPSl C, PAYET E, MAGNA L, et al. PCNCP ligands in the chromium-catalyzed oligomerization of ethylene: tri- versus tetramerization[J]. 2009, 15(33): 8259-8268.
-
[90]
BUDZELAAR P H M. Ethene trimerization at CrI/CrIII-a density functional theory (DFT) study[J]. Can J Chem, 2009, 87(7): 832-837. doi: 10.1139/V09-022
-
[91]
LIU L, LIU Z, TANG S. What triggered the switching from ethylene-selective trimerization into tetramerization over the Cr/(2, 2'-dipicolylamine) catalysts?[J]. ACS Catal, 2019, 9(11): 10519-10527. doi: 10.1021/acscatal.9b03340
-
[1]
-

-

 下载:
下载:





























 下载:
下载:






























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 80
- 文章访问数: 4332
- HTML全文浏览量: 658
