Citation:
Bing-Qian WANG, Hui-Lin TAO, Ji-Lin TANG, Yi LI, Wei LIN, Yong-Fan ZHANG. Methane Oxidation on the Surfaces of Manganese Oxides: a First Principles Study[J]. Chinese Journal of Structural Chemistry,
2020, 39(8): 1405-1421.
doi:
10.14102/j.cnki.0254–5861.2011–2449
Methane Oxidation on the Surfaces of Manganese Oxides: a First Principles Study
Received Date:
08 May 2019 Accepted Date:
11 June 2020 Available Online:
01 August 2020
Fund Project:
Abstract:
A comprehensive density functional theory calculation was employed to investigate the possible reaction pathways and mechanisms of methane complete oxidation (CH4 + 2O2 → CO2 + 2H2O) on different manganese oxides including α-MnO2(100) and β-MnO2(111) surfaces. According to a coupling of the Mars-van Krevelen and Langmuir-Hinshelwood mechanism, the activation energy barrier and the reaction energy of each elementary surface reaction were determined. Our calculated results show that the detailed processes for methane oxidation on two surfaces are different due to the differences in the surface structure. The breaking of the last C–H bond of CH4 molecule is the rate-determining step with an activation barrier of 0.85 eV for α-MnO2(100) surface. By contrast, the overall reaction rate on β-MnO2(111) surface is limited by the dissociation of the second O2 molecule adsorbed on the vacancy site, and re-oxidation of the reduced β-MnO2(111) surface by the gaseous oxygen requires a much higher energy barrier of 1.44 eV. As a result, the α-MnO2(100) exhibits superior activity and durability in the methane oxidation reaction than β-MnO2(111) surface. The present study provides insight into understanding the structure-catalytic activity relationship of the catalysts based on manganese oxides towards the methane oxidation reaction.
With more attention paid to the problem of environmental pollution, natural gas (NG), which is comparatively clean, abundant and low-priced, provides an up-and-coming alternative to moderately replace petroleum and coal in chemical such as the conversion of methanol, syngas and ethylene and energy industries, for instance, the NG engine[1-4]. As the main component of NG, methane has an excellent unit calorific value, the more fuel efficient and less CO2 production than diesel or gasoline engines for its high H/C ratio[5]. On the other hand, as a greenhouse gas 20 times stronger than CO2, residual NG from agricultural operations, livestock, industrial production and motor vehicle emissions can cause environmental damage, which becomes one of the main sources of atmospheric pollutants[6-9]. Therefore, the emission of methane must be treated and the catalytic oxidation is one of the effective ways.
Since CH4 molecule exhibits a stable tetrahedron configuration and the high bonding energy (434 kJ/mol) of C–H bond, direct dissociation of methane at relatively low temperature is difficult[10, 11]. So, it is considered to be one of the feasible ways to combust methane by catalytic oxidation at relatively low temperature through high-efficiency catalysts. Many investigations have shown that catalysts which supported noble metals (such as notably palladium) have the excellent activity for complete oxidation of methane, but the high cost, metal loading and the easy sintering at high temperature enormously limit their practical effects. Due to the above defects, the relatively low performance of noble metal supported catalysts is obviously impractical for widespread use in industry[12-15].
A promising alternative to noble metal catalysts is the using of transition metal oxides (TMOs) including MnO2, Co3O4, Fe3O4, etc.[16-23], in which manganese oxides show good catalytic activity in methane oxidation. On the other hand, previous studies indicate that the activity of catalysts is closely related to their compositions, crystal structures, shape and pore structures[24]. From what has been discussed in experiments[25, 26], manganese oxides with various types (α-, β-, δ-MnO2, Mn2O3, Mn3O4, and so on) and morphological (rod-, wire-, tube-, and flower-like) structures are undoubtedly one of the primary choices for the development of economical and high activity TMOs catalysts. For instance, in a recent experimental study[26], the electrocatalytic properties of α-, β-, δ-MnO2 and amorphous-MnO2 were investigated for catalyzing both oxygen evolution reaction (OER) and oxygen reduction reaction (ORR), and the results show that the catalytic activities are strongly dependent on the crystallographic structure. The superior activity and durability for α-MnO2 were revealed, which can be attributed to the fact that there are many bridging oxygen atoms with high catalytic activity existing in the surface of α-MnO2. Currently, for the reaction mechanism of methane oxidation on the surface of TMOs, Mars-van Krevelen (MvK) mechanism that is mainly related to the lattice oxygen and Langmuir-Hinshelwood (L-H) mechanism that concerns about the adsorbed oxygen has been proposed as two possible mechanisms of the methane oxidation reaction, and the entire process is generally recognized as a coupling of the above two mechanisms[27-29]. Methane is believed to be oxidized by surface adsorbed oxygen molecule following the L-H mechanism while it is initialized by the activation process through the formation of lattice oxygen vacancies (MvK route)[27]. Due to the complicated reaction mechanism of methane oxidation on the TMO surface, how the surface structure affects the catalytic performance in methane oxidation for manganese oxides is not yet clear[30].
In order to better understand the structure-catalytic activity relationship for manganese oxides toward the methane oxidation reaction, in this paper, using the first-principles method based on the density-functional theory (DFT), we systematically studied the reaction mechanisms of complete oxidation of methane on the surfaces of two manganese oxides with different structure including α-MnO2 and β-MnO2. The reaction pathways, transition states and intermediates of the methane oxidation over two surfaces are identified by DFT calculations combined with periodic slab model, and their similarities and differences are further discussed. Our results show activities for methane oxidation are associated with the surface structures, and depend strongly on the amount and the stability of bridging oxygen atoms. This finding can provide insight into understanding the relationship between the surface structure and reactivity, which is important for designing and optimizing new manganese oxide-based catalysts.
2.
SURFACE MODEL AND COMPUTATIONAL DETAILS
Self-consistent periodic DFT calculations were carried out utilizing the Vienna ab initio simulation package (VASP)[31-33]. The interactions between core electrons and ions were described by the projector-augmented wave (PAW) method. The generalized gradient approximation Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional was employed[34], and the kinetic cut-off energy for the plane-wave expansion was set to 500 eV. The influences of van der Waals interactions were taken into account by using the dispersion corrected vdW-DF2 functional[35]. The Brillouinzone integrations were sampled using a (5 × 5 × 1) Monkhorst-Pack k-point grid. The effects of spin polarization were considered, and the dipole correction in the surface normal direction was applied. The convergence thresholds of the energy change and the maximum force were set to 10-5 eV and 0.02 eV/Å, respectively. To properly describe the electron correlation of partially occupied Mn 3d states, the PBE plus on-site repulsion U (PBE + U) method was adopted. Here, the PBE + U calculations were carried out following the simplified rotationally invariant form proposed by Dudarev to address the self-interaction energy, and the effective single parameter U-J of 4.0 eV was used for Mn atom[36]. The climbing image-nudged elastic-band (CI-NEB) method[37, 38] was used to search for transition states (TS) and minimum energy paths in the reactions of methane oxidation on the surfaces of α-MnO2 and β-MnO2. The vibrational frequency analysis was also performed to ensure that the predicted TS corresponded to the first-order saddle point in the reaction path. The vibrational frequencies were calculated from the diagonalization of the mass-weighted Hessian matrix constructed by the finite-difference process.
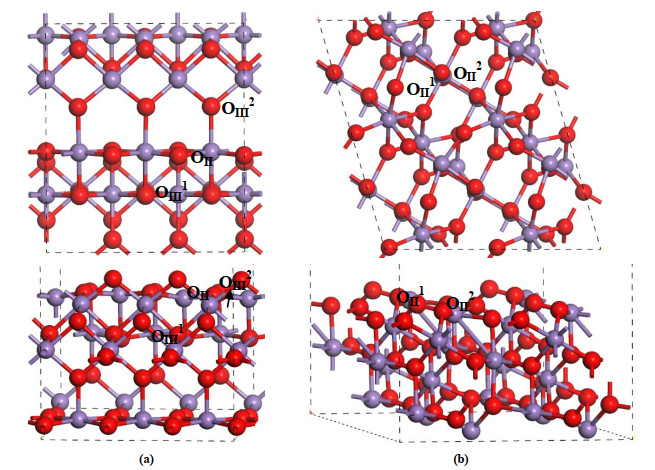
It is well known that due to the property of [MnO6] octahedra connected via sharing common corners or edges, manganese oxides have extremely diverse structures. Among them, α-MnO2 phase is constructed by double chains of [MnO6] octahedra, while β-MnO2 is built by a single chain of edge-sharing [MnO6] octahedra. Previous experimental results of high-resolution transmission electron microscopy (HRTEM) suggest that the (100) surface is the main exposed plane of α-MnO2[26]. After carefully examining the convergences of the surface structure and adsorption energy of key intermediate as functions of the thickness of slab, a periodic slab with a p(1 × 3) supercell including eighteen atomic layers was used to model the α-MnO2(100) surface. During geometry optimization, the top thirteen layers were fully relaxed whereas the bottom five layers were fixed to bulk positions. To avoid the interactions between the neighboring slabs, the vacuum between the adjacent slabs was set to 15 Å. As shown in Fig. 1a, there are two kinds of O atoms, namely the 2-fold-coordinated bridging OII atoms and 3-fold in-plane OIII2 atoms on the α-MnO2(100) surface. For β-MnO2, the (111) surface only terminated by bridging oxygen atoms OII1 and OII2 (see Fig. 1b) is also observed in experiments[39-41]. A twelve-layer slab with a p(2 × 2) supercell was employed for simulating the β-MnO2(111) surface, in which the top eight layers were fully relaxed in all directions and the atoms at the bottom four layers were fixed at bulk positions. Moreover, using the same calculation method, the optimized lattice parameters of α-MnO2 (β-MnO2) bulk are a = b = 9.841 (4.401) and c = 2.904 (2.919) Å, respectively, which are consistent with the experimental results of 9.815 (4.388) and 2.847 (2.860) Å[24]. The two clean surface energies of α-MnO2(100) and β-MnO2(111) were calculated to compare their thermodynamic stability. The surface energy (Esurf) is calculated by the following equation, where, $E_{slab }^N $ and
Figure 1.
Top and side views of the optimized structures of (a) α-MnO2(100) and (b) β-MnO2(111) surfaces. In the figures, the Mn and O atoms are denoted by violet and red spheres, and OII and OIII are used to represent the two- and three-coordinate oxygen, respectively
$E_{bulk } $ are the total surface energy of the N unit phase in the number of atoms and bulk phase, and A is the area of surface. Surface energy is inversely proportional to its thermodynamic stability. The calculated surface energies of α-MnO2(100) and β-MnO2(111) are 0.31 and 0.15 J/m2, respectively, indicating that the β-MnO2(111) clean surface has better thermodynamic stability.
3.
RESULTS AND DISCUSSION
Similar to complete oxidation of methane on the surface of other TMOs[16], the dissociation products of CH4 contain CH3*, CH2*, CH* and CO2 species. The elementary reaction steps for the methane oxidation on MnO2 surface are listed as follows (Herein, we use X* to represent X species adsorbed on the surface, and OV denotes the oxygen vacancy on the MnO2 substrate).
The above eaction mechanism is derived from a coupling mechanism that the MvK and L-H routes work together, in which steps (1) to (4) belong to MvK, and the next five steps are regarded as L-H.
3.1
Physisorption of methane (CH4 (g) → CH4*)
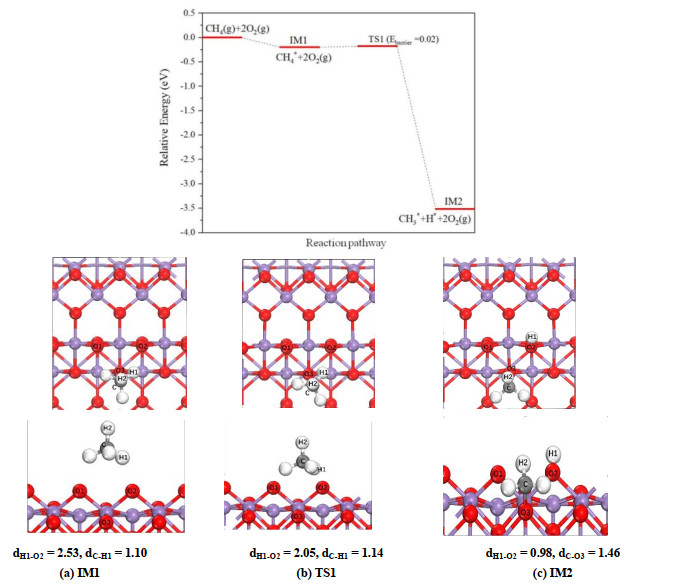
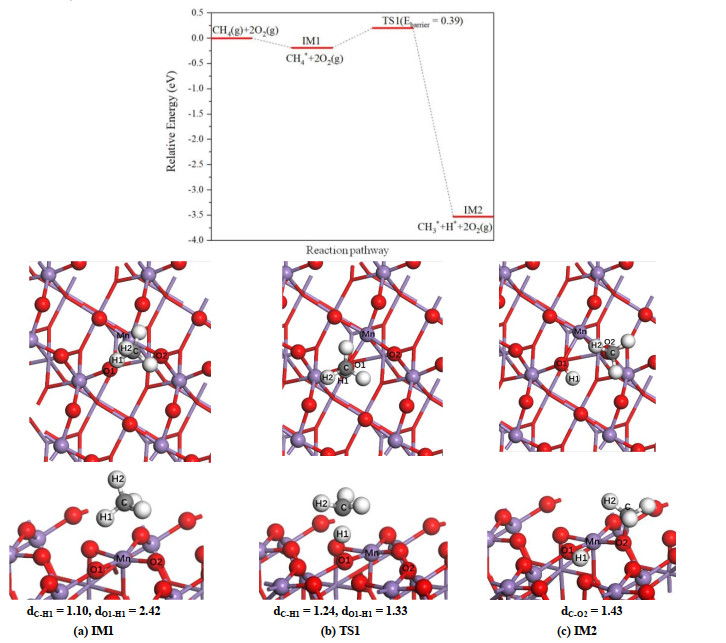
As the first step of the whole reaction, the methane molecule is physisorbed on the surface. The configurations of physisorption state (IM1) of methane molecules on α-MnO2(100) and β-MnO2(111) surfaces are determined, and the corresponding side and top views are shown in Fig. 2a and 3a, respectively. The methane exhibits an adsorption configuration that H(1) atom points to bridge oxygen with a distance about 2.50 Å on both surfaces. Over α-MnO2(100) surface, CH4 tends to be adsorbed near the bridging oxygen atom, while on β-MnO2(111) surface the methane prefers a site nearly above the five-fold coordinated manganese atom. The calculated adsorption energies for the physisorption of CH4 on two surfaces are similar, which are 0.20 and 0.19 eV, respectively.
Figure 2
Figure 2.
Potential energy profile as well as top and side views of configurations for breaking the first C–H bond of methane on the α-MnO2(100) surface. IM and TS represent intermediate and transition state, respectively. The unit of distance is Å. In the figures, the Mn, O, C, and H atoms are denoted by violet, red, dark gray, and white spheres, respectively
3.2
Breaking the first C–H bond (CH4* + O* → CH3* + HO*)
In the process of methane oxidation, one C–H bond of methane breaks at first, then CH3* and H* are formed and chemisorbed on the surface sites, respectively.
On α-MnO2(100) surface (Fig. 1a), the bridging oxygen at the top layer is the active adsorption site. As shown in Fig. 2b, in TS1 the H(1) atom is inclined to the bridging oxygen, and the C–H(1) bond is elongated from 1.10 to 1.14 Å. The breaking of C–H bond overcomes a small energy barrier (0.02 eV), and in IM2 state (Fig. 2c) the CH3 group is adsorbed on the three-fold coordinated O(3) atom. The calculated reaction energy of this step is –3.52 eV, corresponding to a strongly exothermic process.
The result of β-MnO2(111) surface is displayed in Fig. 3. In TS1, the methane rotation becomes atop the O(1) atom, and the C–H(1) and O(1)–H(1) bond distances are 1.33 and 1.24 Å, respectively. In IM2 state the H(1) atom and methyl group are adsorbed on the O(1) and O(2) atoms, respectively. Since the methyl group may pass over Mn atom when moving to the O(2) atom in the reaction path, the possible intermediate that methyl group adsorbed on Mn is also considered. However, the calculated result indicates that such intermediate is not stable, in which CH3 group still migrates toward the O(2) atom. Although the calculated reaction energy (–3.53 eV) of β-MnO2(111) surface is similar to that of α-MnO2(100) surface, it needs to overcome a relatively higher activation barrier (0.39 eV) to break the first C–H bond of CH4 molecule.
3.3
Dissociation of methyl and formation of water (CH3* + HO* → CH2* + H2O*)
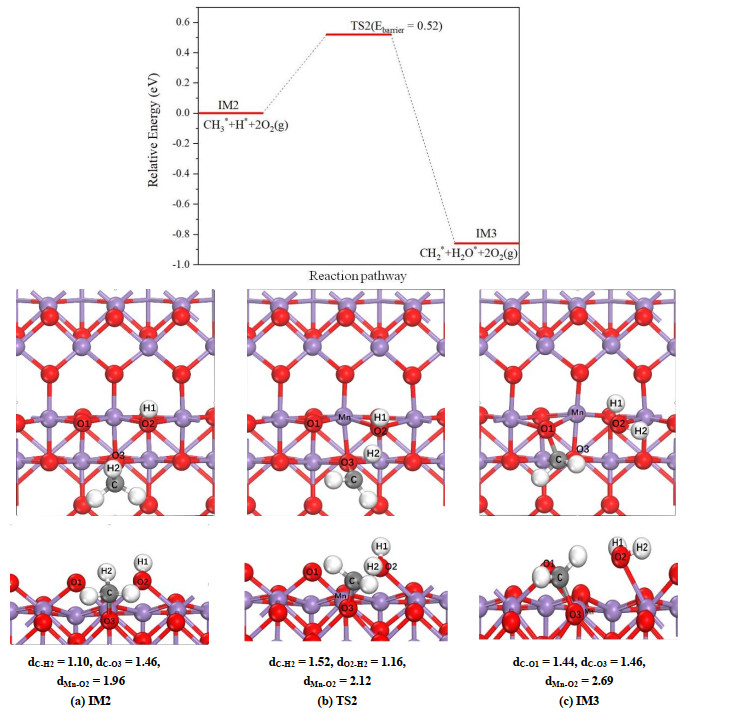
The cracking of one C–H bond of CH3* group leads to the formation of methylene (CH2*) species, and the associated H atom transfers to O atom of hydroxyl group formed in previous step. Since carbon atom has a tendency to form tetrahedral geometry, the CH2 species favors the bridge site between two neighboring surface oxygen atoms.
For α-MnO2(100) surface, as displayed in Fig. 4, the methyl group adsorbed on the O(3) atom rotates to a position that makes the H(2) atom is point to O(2) atom of hydroxyl group, and then the O(2)–H(2) bond is formed with the breaking of C–H(2) bond. Meanwhile, in IM3 a new C–O bond is formed between C and O(1) atoms, now the methylene is adsorbed on surface through two C–O adsorption bonds. During above process, the obvious weakening of the Mn–O(2) bond is observed, and the corresponding bond length increases from 1.96 (IM2) to 2.12 (TS2) and then to 2.69 Å (IM3), indicating that the H2O* group in IM3 can be desorbed from the surface easily. According to the predicted potential energy, this step requires to overcome an activation barrier of 0.52 eV, and the reaction energy is predicted to be –0.86 eV.
Figure 4
Figure 4.
Potential energy profile as well as top and side views of configurations for breaking one C–H bond of methyl group on α-MnO2(100) surface
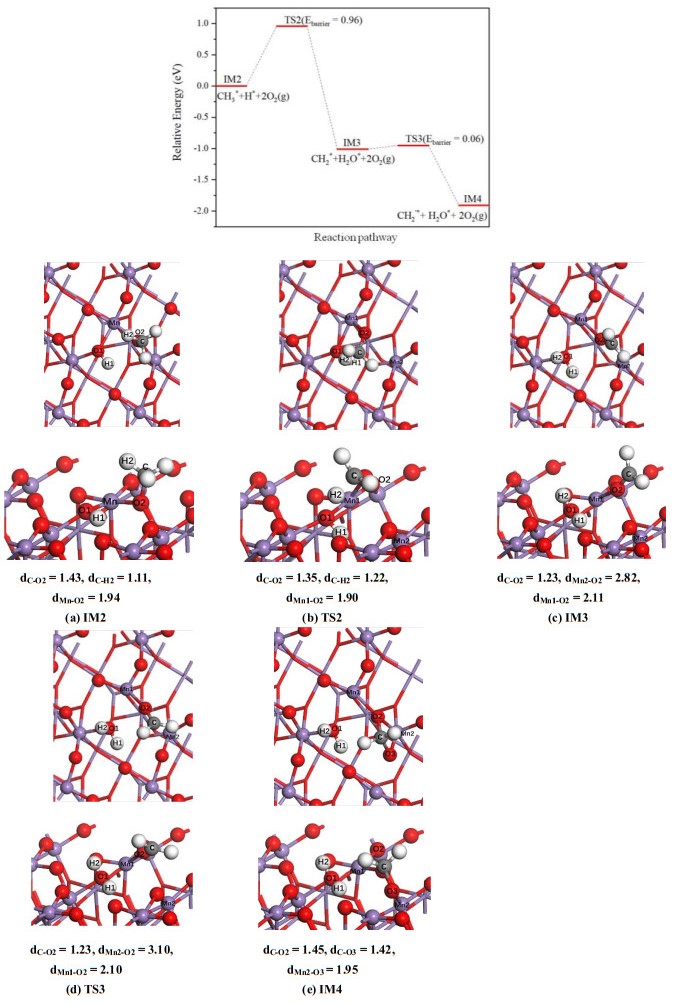
In the case of β-MnO2(111) surface (Fig. 5), the reaction for the dissociation of CH3* into CH2* group is more complicated, which is a two-step process. First, the methyl group adsorbed on O(2) site tilts toward hydroxyl group, then the C–H(2) bond is broken and a new O(1)–H(2) bond is formed, resulting in a metastable intermediate, IM3 (Fig. 5c). In this structure, CH2* group exhibits a planar configuration, containing one C=O double bond with a length of 1.23 Å. The calculated activation barrier of this step is 0.96 eV. In the second step, CH2* moves toward O(3) atom to form a new C–O(3) adsorption bond. So in IM4 structure, the carbon atom regains a tetrahedral coordination geometry. The small energy barrier (0.06 eV) of this step indicates that the transformation from IM3 to IM4 structure is very easy to happen. The reaction energy of the whole process is –1.91 eV. Since the activation barrier of β-MnO2(111) surface is obviously higher than that of α-MnO2(100) (0.96 vs. 0.52 eV), the dissociation of CH3* into CH2* group is more likely to occur on the α-MnO2(100) surface.
Figure 5
Figure 5.
Potential energy profile as well as top and side views of configurations for breaking one C–H bond of methyl group on β-MnO2(111) surface
3.4
Desorption of H2O* and adsorption of O2 molecule (H2O* → H2O (g) + OV, O2(g) + OV → O2*)
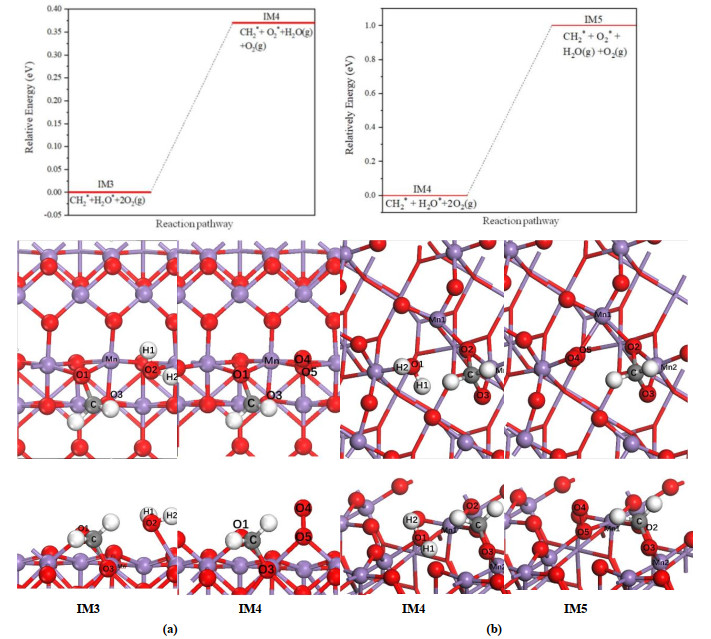
The H2O molecule yielded in previous step is desorbed from the surface, resulting in an oxygen defect on the substrate, and meanwhile O2 molecule in the gas phase is adsorbed on the surface to heal the oxygen vacancy. As shown in Fig. 6, this process has little influence on the configuration of CH2* group, and O2 is chemisorbed nearly vertically on both surfaces. After adsorption, the O–O bond length of O2 molecule is increased obviously from 1.21 to about 1.30 Å, in which the O(5) atom occupies the vancancy site. This preocess corresponds to an endothermic reaction, and positive reaction energies are obtained for both surfaces. However, comparing the results of two surfaces (0.37 vs. 1.00 eV), it seems that this step is more likely to be observed on α-MnO2(100) surface.
Figure 6
Figure 6.
Relative energy as well as top and side views of configurations for the adsorption of CH2 group and oxygen molecule on (a) α-MnO2 (100) and (b) β-MnO2(111) surfaces
After the adsorption of oxygen molecule on the surface, the further dissociation of CH2* species follows the L-H mechanism.
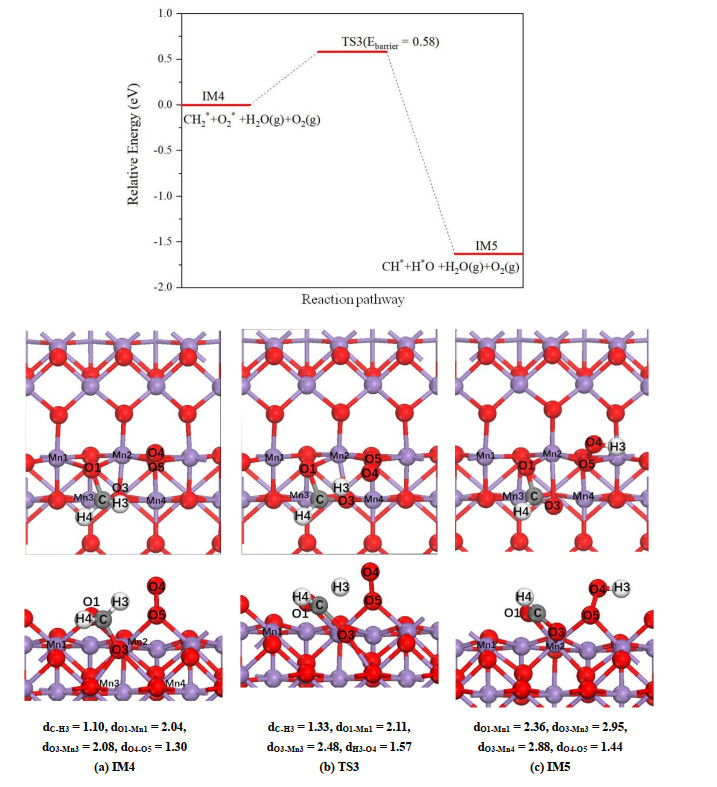
As presented in Fig. 7, on α-MnO2(100) surface the H(3) atom of methylene in IM4 will be transferred to the terminal O(4) atom to form hydroxyl. During this process CH2* has a tendency to close to the O(4) atom, which leads to the weakening of Mn(1)–O(1) bond, and the corresponding bond length is elongated from 2.04 (IM4) to 2.11 (TS) and then to 2.36 Å (IM5). It is noted that the weakening of those Mn–O bonds around O(3) atom is more pronounced, and in IM5 structure the Mn(3)–O(3) and Mn(4)–O(3) bonds are destroyed. In the final state (Fig. 7c), the remaining CH* group is attached to the surface by C–O(1) and C–O(3) bonds with the lengths of 1.24 and 1.33 Å, respectively. The breaking of C–H(3) bond in this step needs to overcome an activation barrier of 0.58 eV and the reaction energy of this process is calculated to be exothermic by 1.63 eV.
Figure 7
Figure 7.
Potential energy profile for dissociation of CH2* on α-MnO2(100) as well as the corresponding top and side views of the configurations
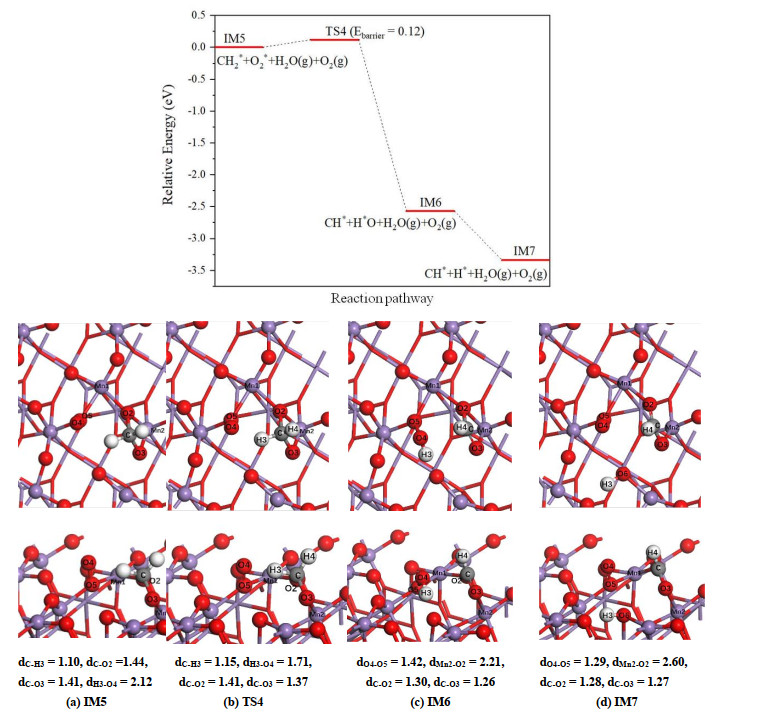
The dissociation of CH2* on β-MnO2(111) surface (Fig. 8) is different from the process occurring on the α-MnO2(100) surface. In this case, the H(3) atom first migrates from carbon to O(4) atom of the pre-adsorbed O2 molecule, and the breaking of C–H(3) bond only requires overcoming a small energy barrier of 0.12 eV. In IM6 structure, CH* species is adsorbed on the surface through two C–O bonds with an average bond length of 1.28 Å. However, IM6 can convert into IM7 structure that is about 0.77 eV lower in energy, in which the H(3) atom transfers from O(4) to the surface bridging O(6) atom. Therefore, unlike α-MnO2(100) surface, it is the surface oxygen to be hydroxylated on β-MnO2(111) surface rather than the terminal oxygen of pre-adsorbed O2 molecule. Moreover, the dissociation of CH2* on β-MnO2(111) corresponds to a strongly exothermic reaction with a reaction energy of –3.34 eV.
Figure 8
Figure 8.
Potential energy profile for dissociation of CH2* on β-MnO2 (111) as well as the corresponding top and side views of the configurations
3.6
Dissociation of CH* group and formations of CO2 as well as the second H2O (CH* + HOO* +O*→ CO2* + H2O*)
In this elementary reaction step, the only C–H bond of CH* group is destroyed, and then the final oxidation product of methane, namely the carbon dioxide is generated with the formation of the second water molecule.
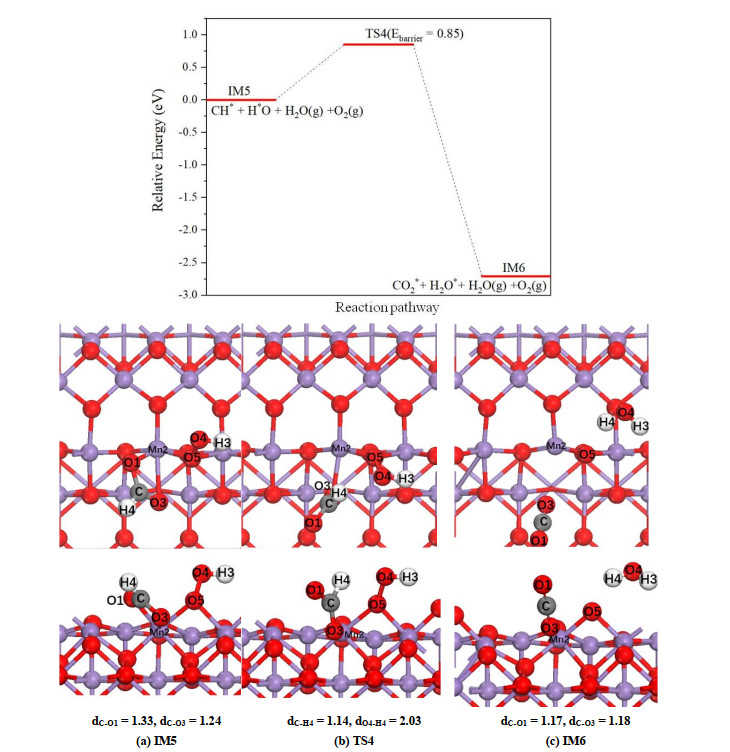
As shown in Fig. 9, for α-MnO2(100) surface, the shift of H(4) atom toward O(4) atom results in the weakening of the Mn(2)–O(1) bond, and in TS structure the Mn(2)–O(1) bond is completely broken. As the second H2O is formed, the remaining O(1)–C–O(3) structure is finally converted into linear CO2 molecule with C–O bond lengths of 1.17 and 1.18 Å, respectively. At the final state IM6, the generated CO2 and H2O are physisorbed on the surface after structural optimization. The energy barrier of this step is predicted to be 0.85 eV with a reaction energy of –2.71 eV. Since the release of CO2 takes away two surface oxygen atoms, i.e. the O(1) and O(3) atoms, two new oxygen vacancies will appear on the α-MnO2(100) surface.
Figure 9
Figure 9.
Potential energy profile for decomposition of CH* on α-MnO2(100) surface as well as the corresponding top and side views of the configurations
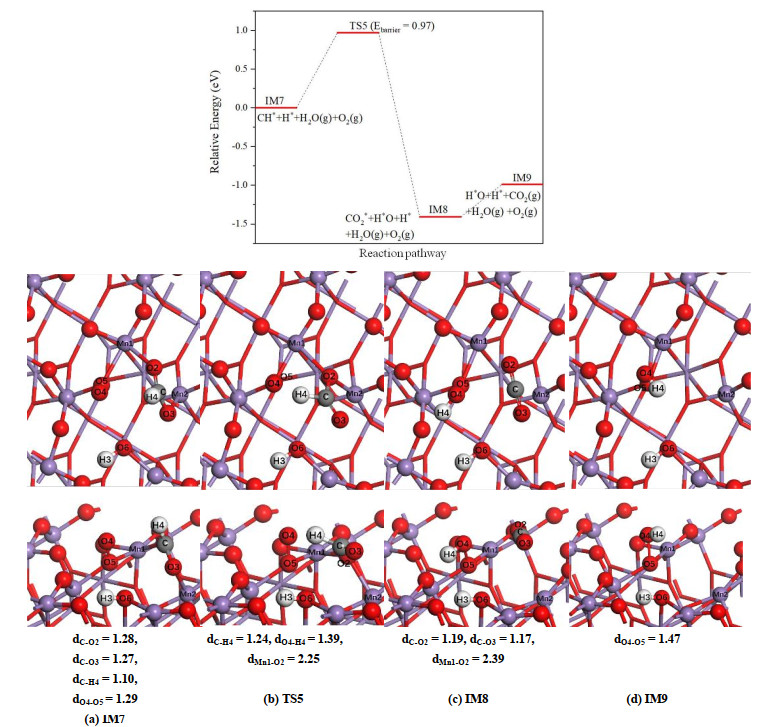
For β-MnO2(111) surface, because in the previous step the H(3) atom is selectively adsorbed to the adjacent bridging O(6) atom instead of the terminal O(4) atom, the productions of CO2 and the second H2O do not occur simultaneously, in which the carbon dioxide is yielded first. As displayed in Fig. 10, when CH* species is dissociated, the H(4) atom will transfer to the O(4) atom. Similar to α-MnO2(100) surface, this shift of H(4) atom causes the weakening or breaking of those Mn–O bonds around O(2) and O(3) atoms, and in IM8 the carbon dioxide is produced with two C–O distances of 1.19 and 1.17 Å, respectively. The CO2 molecule is interacted with the surface through Mn(1)–O(2) bond. Above process needs to overcome an energy barrier of 0.97 eV, and the calculated reaction energy is –1.41 eV. In IM9 structure, CO2 is desorbed from the surface, leaving two oxygen vacancies on the β-MnO2(111) surface. By comparing the relative energies of IM8 and IM9, CO2 molecule is weakly attached to the surface with an adsorption energy of 0.42 eV.
Figure 10
Figure 10.
Potential energy profile for the dissociation of CH* on β-MnO2(111) surface as well as the corresponding top and side views of the configurations
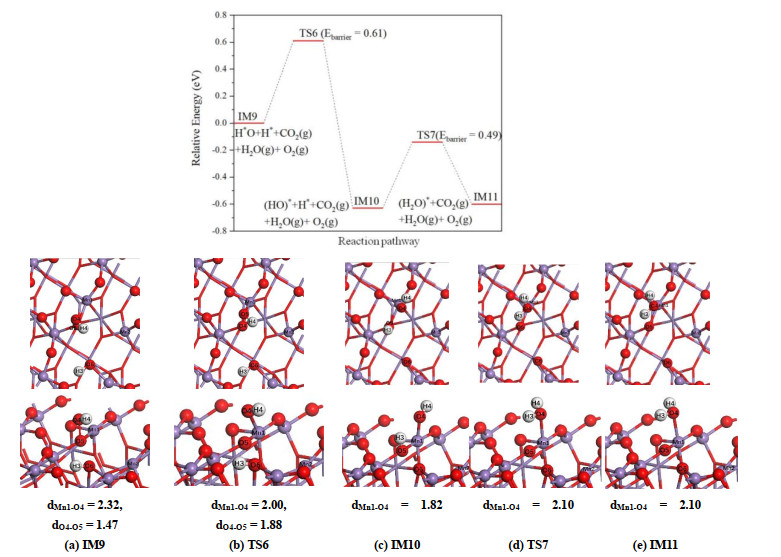
On β-MnO2(111) surface, the formation of the second H2O is complicated. In the initial state IM9 presented in Fig. 11a, the H(3) and H(4) are adsorbed on the bridging O(6) and terminal O(4) atoms, respectively. Our calculated results indicate that the configuration of forming H2O on the bridging O(6) is unstable, and the H(3) atom tends to move to O(4) atom. During this process, the hydroxyl attached to O(5) atom will move toward the Mn(1) atom, and in IM10 shown in Fig. 11c, the hydroxyl is adsorbed on the surface via the Mn(1)–O(4) adsorption bond. Meanwhile, the H(3) atom transfers to the O(5) atom. The formation of IM10 structure requires overcoming an activation barrier of 0.61 eV and this step is thermodynamically favored with a negative reaction energy of –0.63 eV. The further shift of H(3) atom to O(4) site results in the formation of the second H2O molecule. In the final state IM11, the water is adsorbed on Mn(1) site and the length of Mn(1)–O(4) adsorption bond is about 2.10 Å.
Figure 11
Figure 11.
Potential energy profile for the transformation of H atom and the formation of the second H2O on β-MnO2(111) surface as well as the corresponding top and side views of the configurations
3.7
Adsorption and dissociation of the second oxygen molecule (O2(g) + 2OV → 2O*)
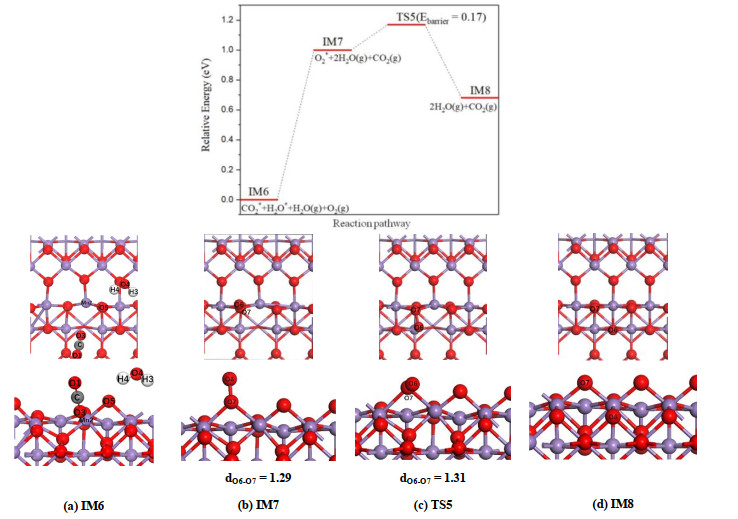
As mentioned above, two new oxygen vacancies are created after releasing the carbon dioxide. Therefore, it is necessary to adsorb and dissociate the second O2 molecule to recover the original perfect surface. On α-MnO2(100) surface, the CO2 and H2O molecules generated in the previous step are physisorbed on the surface with adsorption energies of 0.35 and 0.42 eV, respectively. Starting from IM7 structure shown in Fig. 12b, the second oxygen molecule can be dissociated easily on α-MnO2(100) surface, which requires an energy barrier of 0.17 eV.
Figure 12
Figure 12.
Potential energy profile for the desorptions of H2O and CO2 and the dissociation of the second O2 molecules on α-MnO2(100) surface as well as the corresponding top and side views of the configurations
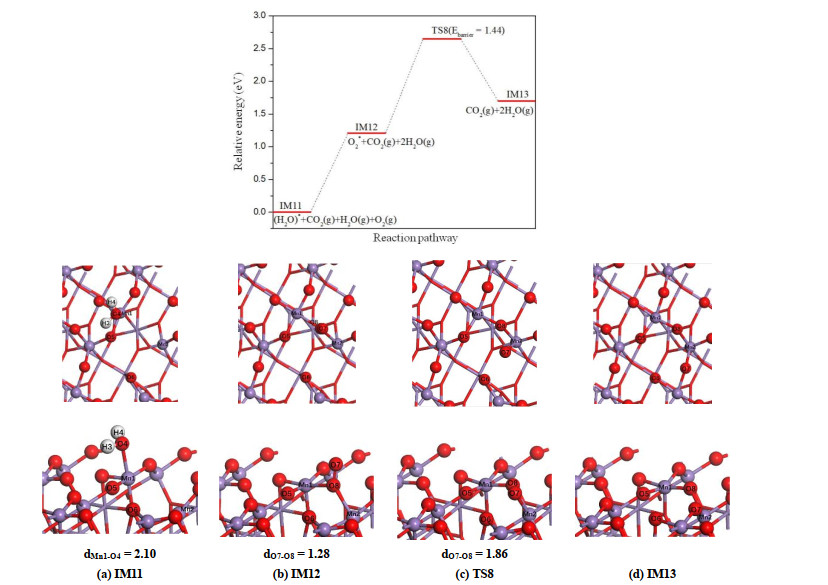
Different from α-MnO2(100) surface, the second water molecule is chemisorbed on β-MnO2(111) surface, and the large adsorption energy (1.21 eV) indicates that H2O is not easily desorbed. Additionally, according to the energy profile shown in Fig. 13, the subsequent dissociation of the second oxygen molecule on β-MnO2(111) surface requires a large energy barrier (1.44 eV). Hence, it seems that the recovering of β-MnO2(111) surface is more difficult with respect to the α-MnO2(100) surface.
Figure 13
Figure 13.
Potential energy profile for the desorption of H2O and the dissociation of the second O2 molecules on β-MnO2(111) surface as well as the corresponding top and side views of the configurations
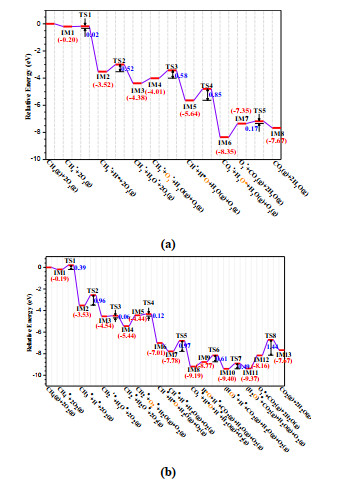
For clarity, based on the above results of each elementary step, Fig. 14 summarizes the whole potential energy profiles, and the difference between reaction mechanisms for the methane oxidation on two surfaces can be identified. It is clear that the elementary reaction steps for the formation of the first water molecule on two surfaces are similar. However, unlike the β-MnO2(111) surface, besides the bridging oxygen atoms, the additional three-fold coordinated O atoms on α-MnO2(100) surface also participate in the adsorption of CH3* and CH2* species. Through comparing the energy barriers of TS2 (0.52 vs. 0.96 eV), the formation of H2O* on α-MnO2(100) is easier than on the β-MnO2(111) surface. In addition, α-MnO2(100) is better to the desorption of water molecule with respect to β-MnO2(111) surface, which can be confirmed by comparing the formation energy of oxygen vacancy. The vacancy formation energies (Ef) of two surfaces are calculated according to following equation
Figure 14.
Overall potential energy profile for the CH4 oxidation on (a) α-MnO2(100) and (b) β-MnO2(111) surface. For clarity, on horizontal axis the oxygen atom originated from the first adsorbed O2 molecule is highlighted in orange
where, Edefect and Eperfect are the total energies of surfaces with and without bridging oxygen vacancy, and μO is the chemical potential of oxygen atom that is obtained from the energy (EO2) of O2 molecule by μO = 1/2EO2. The Ef value of α-MnO2(100) surface (0.85 eV) is smaller than that of β-MnO2(111) (1.52 eV). Therefore, the bridging oxygen on α-MnO2(100) is more easily to be removed, and this is beneficial for the desorption of the first water molecule. After the first oxygen molecule is adsorbed on the defect site, the subsequent steps for the further oxidation of CH2* species on two substrates are quite different. The corresponding procedure is more straightforward on α-MnO2(100) surface, in which two hydrogen atoms of CH2* are successively transferred to the terminal oxygen atom of pre-adsorbed O2 molecule, resulting in the formation of the second H2O. It is noted that the loss of H will enhance the strengths of two C–O adsorption bonds, and correspondingly the lengths of C–O bonds are decreased, which eventually causes the generation of carbon dioxide. Because the energy barrier associated with TS4 is relatively high (0.85 eV, Fig. 14a), the breaking of the last C–H bond of methane is the most difficult step on α-MnO2(100) surface. By contrast, a more complicated procedure is involved in the oxidation of methylene on β-MnO2(111) due to its more bridging oxygen atoms on the surface. When the first C–H bond of CH2* is destroyed, the H atom first forms O–H bond with oxygen atom of pre-adsorbed O2 molecule (IM6 in Fig. 14b), and then it moves to a surface bridging oxygen to yield a more stable intermediate IM7. Breaking the next C–H bond makes the generation of CO2*, and meanwhile the corresponding H atom transfers to oxygen of pre-adsorbed O2. This process needs to overcome an energy barrier of 0.97 eV, still higher than the value (0.85 eV) of α-MnO2 (100). Since two H atoms are adsorbed on different sites, the succeeding formation of the second water involves the migration of OH group toward Mn atom. Consequently, unlike α-MnO2(100), the second H2O is chemisorbed on the surface metal site (IM11 in Fig. 14b). Therefore, the five-fold coordinated Mn atoms at the top layer visibly take part in the methane oxidation on β-MnO2(111) surface. In the last stage, the adsorption and dissociation of the second O2 molecule are required to recover the surface. The calculated energy barriers (0.17 vs. 1.44 eV) indicate that β-MnO2(111) surface is more difficultly restored than α-MnO2(100). It is noted that, from Fig. 14, on α-MnO2(100) surface the breaking of the last C–H bond of methane is the rate-limiting step, while the dissociation of the second O2 molecule is the rate-determining step on β-MnO2(111) surface.
4.
CONCLUSION
In this work, we performed extensive DFT calculations to investigate the complete oxidation mechanism of methane (CH4 + 2O2 → CO2 + 2H2O) on α-MnO2(100) and β-MnO2(111) surfaces. Although the whole reaction procedure can be briefly described as breaking four C–H bonds step by step, the detailed processes occurring on two surfaces are different because of obvious differences in the surface structure. This can be reflected clearly by the reaction paths after the adsorption of the first oxygen molecule, especially the migration of OH group to Mn site observed on the β-MnO2(111) surface. On α-MnO2(100) surface the cracking of CH* group, i.e. CH* → C* + H*, is the rate-determining step with an activation barrier of 0.85 eV. However, for β-MnO2(111) surface, the overall reaction rate is limited by the dissociation of the second oxygen molecule adsorbed on the vacancy site, and this step requires a much higher energy barrier of 1.44 eV. Therefore, the catalytic activity of α-MnO2(100) is significantly better than the β-MnO2(111) surface. Additionally, the difficulty to destroy O–O bond of the second O2 molecule also implies that the regeneration of the pristine β-MnO2(111) surface is relatively hard. In summary, our results reveal that α-MnO2(100) exhibits superior activity and durability in the methane oxidation reaction than β-MnO2(111) surface. The present work can provide important information for developing new catalysts of the methane oxidation based on manganese oxides.
[1]
Li, J. H.; Liang, X.; Xu, S. C.; Hao, J. M. Catalytic performance of manganese cobalt oxides on methane combustion at low temperature. Appl. Catal. B-Environ.2009, 90, 307–312. doi: 10.1016/j.apcatb.2009.03.027
[2]
Matthiesen, J.; Smith, R. S.; Kay, B. D. Measuring diffusivity in supercooled liquid nanoscale films using inert gas permeation. II. Diffusion of Ar, Kr, Xe, and CH4 through methanol. J. Chem. Phys.2010, 133, 174505–11. doi: 10.1063/1.3497648
[3]
Wada, A.; Mochizuki, N.; Hiraoka, K. Methanol formation from electron-irradiated mixed H2O/CH4 ice at 10 K. Astrophys. J.2006, 644, 300–306. doi: 10.1086/503380
[4]
Wu, J. J.; Qin, S.; Hu, C. W. Na2WO4/Co-Mn/SiO2 catalyst for the simultaneous production of ethylene and syngas from CH4. Catal. Lett.2007, 118, 285–289. doi: 10.1007/s10562-007-9192-8
[5]
Ercolino, G.; Grzybek, G.; Stelmachowski, P.; Specchia, S.; Kotarba, A.; Specchia, V. Pd/Co3O4-based catalysts prepared by solution combustion synthesis for residual methane oxidation in lean conditions. Catal. Today2015, 257, 66–71. doi: 10.1016/j.cattod.2015.03.006
[6]
Lashof, D. A.; Ahuja, D. R. Relative contributions of greenhouse gas emissions to global warming. Nature1990, 344, 529–531. doi: 10.1038/344529a0
[7]
Rubin, E. S.; Cooper, R. N.; Frosch, R. A.; Lee, T. H.; Marland, G.; Rosenfeld, A. H.; Stine, D. D. Realistic mitigation options for global warming. Science1992, 257, 148–266. doi: 10.1126/science.257.5067.148
[8]
Alvarez, R. A.; Pacala, S. W.; Winebrake, J. J.; Chameides, W. L.; Hamburg, S. P. Greater focus needed on methane leakage from natural gas infrastructure. Proc. Natl. Acad. Sci. USA2012, 109, 6435–6440. doi: 10.1073/pnas.1202407109
[9]
Schmale, J.; Shindell, D.; von Schneidemesser, E.; Chabay, I.; Lawrence, M. Clean up our skies. Nature2014, 515, 335–337. doi: 10.1038/515335a
[10]
Gelin, P.; Primet, M. Complete oxidation of methane at low temperature over noble metal based catalysts: a review. Appl. Catal. B-Environ.2002, 39, 1–37. doi: 10.1016/S0926-3373(02)00076-0
[11]
Zarur, A. J.; Ying, J. Y. Reverse microemulsion synthesis of nanostructured complex oxides for catalytic combustion. Nature2000, 403, 65–67. doi: 10.1038/47450
[12]
Zou, X. L.; Rui, Z. B.; Ji, H. B. Core-shell NiO@PdO nanoparticles supported on alumina as an advanced catalyst for methane oxidation. Acs. Catal.2017, 7, 1615–1625. doi: 10.1021/acscatal.6b03105
[13]
Beck, I. E.; Bukhtiyarov, V. I.; Pakharukov, I. Y.; Zaikovsky, V. I.; Kriventsov, V. V.; Parmon, V. N. Platinum nanoparticles on Al2O3: correlation between the particle size and activity in total methane oxidation. J. Catal.2009, 268, 60–67. doi: 10.1016/j.jcat.2009.09.001
[14]
Xie, S. H.; Liu, Y. X.; Deng, J. G.; Zhao, X. T.; Yang, J.; Zhang, K. F.; Han, Z.; Arandiyan, H.; Dai, H. X. Effect of transition metal doping on the catalytic performance of Au-Pd/3DOM Mn2O3 for the oxidation of methane and o-xylene. Appl. Catal. B-Environ.2017, 206, 221–232. doi: 10.1016/j.apcatb.2017.01.030
[15]
Willis, J. J.; Goodman, E. D.; Wu, L.; Riscoe, A. R.; Martins, P.; Tassone, C. J.; Cargnello, M. Systematic identification of promoters for methane oxidation catalysts using size and composition-controlled Pd-based bimetallic nanocrystals. J. Am. Chem. Soc.2017, 139, 11989–11997. doi: 10.1021/jacs.7b06260
[16]
Tao, F. F.; Shan, J. J.; Nguyen, L.; Wang, Z.; Zhang, S.; Zhang, L.; Wu, Z.; Huang, W.; Zeng, S.; Hu, P. Understanding complete oxidation of methane on spinel oxides at a molecular level. Nat. Commun.2015, 6, 7798–10. doi: 10.1038/ncomms8798
[17]
Chin, Y. H.; Buda, C.; Neurock, M.; Iglesia, E. Consequences of metal-oxide interconversion for C–H bond activation during CH4 reactions on Pd catalysts. J. Am. Chem. Soc.2013, 135, 15425–15442. doi: 10.1021/ja405004m
[18]
Weng, X. F.; Ren, H. J.; Chen, M. S.; Wan, H. L. Effect of surface oxygen on the activation of methane on palladium and platinum surfaces. Acs. Catal.2014, 4, 2598–2604. doi: 10.1021/cs500510x
[19]
Mahara, Y.; Ohyama, J.; Tojo, T.; Murata, K.; Ishikawa, H.; Satsuma, A. Enhanced activity for methane combustion over a Pd/Co/Al2O3 catalyst prepared by a galvanic deposition method. Catal. Sci. Technol.2016, 6, 4773–4776. doi: 10.1039/C6CY00650G
[20]
Abbasi, R.; Huang, G. Y.; Istratescu, G. M.; Wu, L.; Hayes, R. E. Methane oxidation over Pt, Pt: Pd, and Pd based catalysts: effects of pre-treatment. Can. J. Chem. Eng.2015, 93, 1474–1482. doi: 10.1002/cjce.22229
[21]
Zhang, Z. S.; Li, J. W.; Yi, T.; Sun, L. W.; Zhang, Y. B.; Hu, X. F.; Cui, W. H.; Yang, X. G. Surface density of synthetically tuned spinel oxides of Co3+ and Ni3+ with enhanced catalytic activity for methane oxidation. Chin. J. Catal.2018, 39, 1228–1239. doi: 10.1016/S1872-2067(18)63055-4
[22]
Zhao, C. C.; Zhao, Y. H.; Li, S. G.; Sun, Y. H. Effect of Pd doping on CH4 reactivity over Co3O4 catalysts from density-functional theory calculations. Chin. J. Catal.2017, 38, 813–820. doi: 10.1016/S1872-2067(17)62817-1
[23]
Liu, R. Y.; Yang, M. H.; Huang, C. J.; Weng, W. Z.; Wan, H. L. Partial oxidation of methane to syngas over mesoporous Co-Al2O3 catalysts. Chin. J. Catal.2013, 34, 146–151. doi: 10.1016/S1872-2067(11)60481-6
[24]
Wang, F.; Dai, H.; Deng, J.; Bai, G.; Ji, K.; Liu, Y. Manganese oxides with rod-, wire-, tube-, and flower-like morphologies: highly effective catalysts for the removal of toluene. Environ. Sci. Technol.2012, 46, 4034–4041. doi: 10.1021/es204038j
[25]
Robinson, D. M.; Go, Y. B.; Mui, M.; Gardner, G.; Zhang, Z.; Mastrogiovanni, D.; Garfunkel, E.; Li, J.; Greenblatt, M.; Dismukes, G. C. Photochemical water oxidation by crystalline polymorphs of manganese oxides: structural requirements for catalysis. J. Am. Chem. Soc.2013, 135, 3494–3501. doi: 10.1021/ja310286h
[26]
Meng, Y.; Song, W.; Huang, H.; Ren, Z.; Chen, S. Y.; Suib, S. L. Structure-property relationship of bifunctional MnO2 nanostructures: highly efficient, ultra-stable electrochemical water oxidation and oxygen reduction reaction catalysts identified in alkaline media. J. Am. Chem. Soc.2014, 136, 11452–11464. doi: 10.1021/ja505186m
[27]
Hu, W. D.; Lan, J. G.; Guo, Y.; Cao, X. M.; Hu, P. Origin of efficient catalytic combustion of methane over Co3O4(110): active low-coordination lattice oxygen and cooperation of multiple active sites. Acs Catal.2016, 6, 5508–5519. doi: 10.1021/acscatal.6b01080
[28]
Zasada, F.; Piskorz, W.; Janas, J.; Grybos, J.; Indyka, P.; Sojka, Z. Reactive oxygen species on the (100) facet of cobalt spinel nanocatalyst and their relevance in 16O2/ 18O2 isotopic exchange, deN2O, and deCH4 processes-a theoretical and experimental account. Acs. Catal.2015, 5, 6879–6892. doi: 10.1021/acscatal.5b01900
[29]
Zasada, F.; Piskorz, W.; Sojka, Z. Cobalt spinel at various redox conditions: DFT plus U investigations into the structure and surface thermodynamics of the (100) facet. J. Phys. Chem. C2015, 119, 19180–19191. doi: 10.1021/acs.jpcc.5b05136
[30]
Liotta, L. F.; Wu, H. J.; Pantaleo, G.; Venezia, A. M. Co3O4 nanocrystals and Co3O4-MOx binary oxides for CO, CH4 and VOC oxidation at low temperatures: a review. Catal. Sci. Technol.2013, 3, 3085–3102. doi: 10.1039/c3cy00193h
[31]
Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B1993, 48, 13115–13118. doi: 10.1103/PhysRevB.48.13115
[32]
Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B1996, 54, 11169–11186. doi: 10.1103/PhysRevB.54.11169
[33]
Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci.1996, 6, 15–50. doi: 10.1016/0927-0256(96)00008-0
[34]
Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett.1996, 77, 3865–3868. doi: 10.1103/PhysRevLett.77.3865
[35]
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem.2006, 27, 1787–1799. doi: 10.1002/jcc.20495
[36]
Krcha, M. D.; Janik, M. J. Examination of oxygen vacancy formation in Mn-doped CeO2(111) using DFT plus U and the hybrid functional HSE06. Langmuir2013, 29, 10120–10131. doi: 10.1021/la401747n
[37]
Sheppard, D.; Xiao, P.; Chemelewski, W.; Johnson, D. D.; Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys.2012, 136, 074103–8. doi: 10.1063/1.3684549
[38]
Sheppard, D.; Terrell, R.; Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys.2008, 128, 134106–10. doi: 10.1063/1.2841941
[39]
Wang, J. F.; Deng, L. J.; Zhu, G.; Kang, L. P.; Lei, Z. B.; Liu, Z. H. Fluoride anions-assisted hydrothermal preparation and growth process of beta-MnO2 with bipyramid prism morphology. Crystengcomm2013, 15, 6682–6689. doi: 10.1039/c3ce40608c
[40]
Yao, W. T.; Odegard, G. M.; Huang, Z. N.; Yuan, Y. F.; Asayesh-Ardakani, H.; Sharifi-Asl, S.; Cheng, M.; Song, B.; Deivanayagam, R.; Long, F.; Friedrich, C. R.; Amine, K.; Lu, J.; Shahbazian-Yassar, R. Cations controlled growth of beta-MnO2 crystals with tunable facets for electrochemical energy storage. Nano Energy2018, 48, 301–311. doi: 10.1016/j.nanoen.2018.03.057
[41]
Su, D. W.; Ahn, H. J.; Wang, G. X. β-MnO2 nanorods with exposed tunnel structures as high-performance cathode materials for sodium-ion batteries. Npg Asia Mater.2013, 5, e70–7. doi: 10.1038/am.2013.56
Figure 1
Top and side views of the optimized structures of (a) α-MnO2(100) and (b) β-MnO2(111) surfaces. In the figures, the Mn and O atoms are denoted by violet and red spheres, and OII and OIII are used to represent the two- and three-coordinate oxygen, respectively
Figure 2
Potential energy profile as well as top and side views of configurations for breaking the first C–H bond of methane on the α-MnO2(100) surface. IM and TS represent intermediate and transition state, respectively. The unit of distance is Å. In the figures, the Mn, O, C, and H atoms are denoted by violet, red, dark gray, and white spheres, respectively
Figure 6
Relative energy as well as top and side views of configurations for the adsorption of CH2 group and oxygen molecule on (a) α-MnO2 (100) and (b) β-MnO2(111) surfaces
Figure 9
Potential energy profile for decomposition of CH* on α-MnO2(100) surface as well as the corresponding top and side views of the configurations
Figure 10
Potential energy profile for the dissociation of CH* on β-MnO2(111) surface as well as the corresponding top and side views of the configurations
Figure 11
Potential energy profile for the transformation of H atom and the formation of the second H2O on β-MnO2(111) surface as well as the corresponding top and side views of the configurations
Figure 12
Potential energy profile for the desorptions of H2O and CO2 and the dissociation of the second O2 molecules on α-MnO2(100) surface as well as the corresponding top and side views of the configurations
Figure 13
Potential energy profile for the desorption of H2O and the dissociation of the second O2 molecules on β-MnO2(111) surface as well as the corresponding top and side views of the configurations
Figure 14
Overall potential energy profile for the CH4 oxidation on (a) α-MnO2(100) and (b) β-MnO2(111) surface. For clarity, on horizontal axis the oxygen atom originated from the first adsorbed O2 molecule is highlighted in orange

 DownLoad:
DownLoad:

 下载:
下载:















