
Figure 1.
(a) Schematic illustration of the synthesis of Ru/TiO2-OV-x catalysts. (b) SEM image, (c) TEM image, (d) HRTEM image and (e) TEM element mapping images of Ru/TiO2-OV-50 catalyst.
Highly Efficient Photocatalytic CO2 Methanation over Ru-Doped TiO2 with Tunable Oxygen Vacancies
Zheyue Li , Di Wu , Wanbing Gong , Jiayi Li , Shuaikang Sang , Hengjie Liu , Ran Long , Yujie Xiong

The chemical conversion of carbon dioxide (CO2) and hydrogen (H2) into downstream fuels and chemicals is regarded as an efficient and promising technology to reduce atmospheric CO2 concentration and thus reliance on fossil resources.[1-3] Among them, the CO2 hydrogenation to high energy methane (CH4), i.e., the Sabatier reaction has great potential in modern chemical industries.[4,5] For instance, Guo et al. revealed the outstanding thermocatalytic CO2 hydrogenation activity and 98-100% CH4 selectivity can be obtained on the Ru nanoclusters/CeO2 catalyst, but the reaction temperature was still up to 190 ℃.[6] In this regard, the photocatalytic CO2 methanation using renewable solar energy has emerged as one of the most promising and green routes, yet remains a significant challenge because of the unsatisfactory reaction efficiency and CH4 selectivity.[7-12] Meanwhile, the design and development of high-efficient photocatalyst is urgently needed to overcome key problems such as thermodynamically inertia of CO2, slow kinetic process limitation and low light utilization efficiency.[13,14] So far, considerable progress has been made to develop CO2 methanation semiconductor photocatalysts, [15,16] including CeO2-, [17] TiO2-, [18] SrTiO3-, [19] and Ni2V2O7-[8] based catalysts. Although great progress has been made, the fabrication of highly active, selective and stable photocatalyst is still in urgent requirement for CO2 methanation under mild conditions.[20]
Among these semiconductor photocatalysts, TiO2-based catalysts have been intensively investigated due to their low cost, simple synthesis and chemical stability for the energy and environmental applications. However, the photocatalytic perfor-mance of reported TiO2-based catalysts can not meet specific requirements for the CO2 methanation because of their limited charge separation efficiency and low CO2 activation capabilities.[18,21] Fortunately, many strategies have been recently proposed to improve photocatalytic performance of TiO2-based catalysts such as heterojunction construction, [22,23] heteroatom doping, [24] metal cocatalyst[25-28] and defect engineering.[29-31] Considering the requirement for activating CO2 and H2, the construction of metal cocatalyst is an ideal strategy over the TiO2-based materials to improve the photocatalytic CO2 methanation by simultaneously promoting the charge separation and reactant activation. Among various metals that have been explored, Ru site has been considered as a promising candidate for low temperature CO2 methanation reactions.[8,32,33] For instance, Ye's group demonstrated Ru possessed an efficient ability to promote the photogenerated charge separation as well as the hydrogen activation and utilization for the CO2 photo-hydrogenation to CH4.[34] Besides, Cai et al. recently have showed the introduction of Ru species on CdS greatly promoted the activation of CO2 and the separation efficiency of photogenerated carriers, thus accelerating the conversion of CO2 to CH4.[35] Based on the above considerations, the Ru-TiO2 composite can be further used to improve photocatalytic CO2 methanation performance, yet has rarely been reported. In this respect, Zhou et al. reported the 1% Ru-TiO2-x nanocrystal with abundant oxygen vacancies exhibited a remarkable photocatalytic performance with a CH4 yield of 31.63 μmol g-1 h-1 due to synergistic effect of Ru and oxygen vacancies.[21] Lin et al. reported that the Ru/TiO(2-x)Nx catalyst exhibited superior light-assisted CO2 methanation performance, which was resulted from the combined effect of oxygen vacancies and electron-rich Ru sites.[18] Therefore, it is highly desirable to prepare Ru-TiO2 photocatalyst to catalyze CO2 methanation process, which can effectively improve the charge separation and reactant activation due to the synergistic effect of Ru and TiO2 with oxygen vacancies.
In this work, we report a facile and efficient strategy to synthesize Ru-doped TiO2 with abundant oxygen vacancies by using ascorbic acid as a reducing agent. The as-synthesized Ru/TiO2-OV-50 catalyst exhibits an outstanding activity, selectivity and stability for the photocatalytic hydrogenation of CO2 to CH4 with an 81.7 mmol·g-1·h-1 reaction rate and 100% CH4 selectivity at a 1.5 W·cm-2 light illumination. The experimental results show the enhanced photocatalytic CO2 methanation performance is due to the synergistic effect of Ru and TiO2 with tunable oxygen vacancies, which is mainly resulted from the light rather than the thermal effect. Finally, the CO2 methanation reaction pathway on this catalyst is also proposed using in situ spectroscopic techno-logy.
The synthetic process of Ru/TiO2-OV-x (x stands for the ascorbic acid amount) is schematically illustrated in Figure 1a. Briefly, the TiO2-OV-x precursors with tunable oxygen vacancies were first synthesized by a simple solvothermal method using ascorbic acid as a reducing agent. The as-synthesized TiO2-OV-x precursors exhibit rod-like shape from the transmission electron microscopy (TEM) images (Figure S1). Notably, the TiO2-OV-x nanorods be-come shorter in length and larger in diameter with the increase of ascorbic acid amount. Subsequently, an electrostatic adsorption-reduction strategy was used to prepare Ru/TiO2-OV-x, which was finally reduced at 400 ℃ for 2 h under a 10 vol% H2/Ar flow. The scanning electron microscopy (SEM) and TEM images (Figures S2, 3) show that the original morphology of TiO2-OV-x precursors is preserved well after the introduction of Ru species. For the typical Ru/TiO2-OV-50 catalyst, the rod-like shape with around 100-200 nm in length and 10-20 nm in diameter is well maintained after the reduction treatment (Figure 1b,c). High-resolution TEM (HRTEM) image (Figure 1d) shows a lattice spacing of 0.215 nm corresponding to the (002) lattice fringe of metallic Ru. The average size of Ru sites is around 2-3 nm (Figure S4). The corresponding TEM elemental mapping images (Figure 1e) reveal highly homogeneous distributions of Ru, Ti and O over the skeleton. For comparison, the Ru-doped commercial TiO2 nano-particles (NPs) catalyst (denoted as Ru/TiO2) was also prepared using the same procedure, and the morphology and structure are shown in Figures S1f, 2f and 3f.
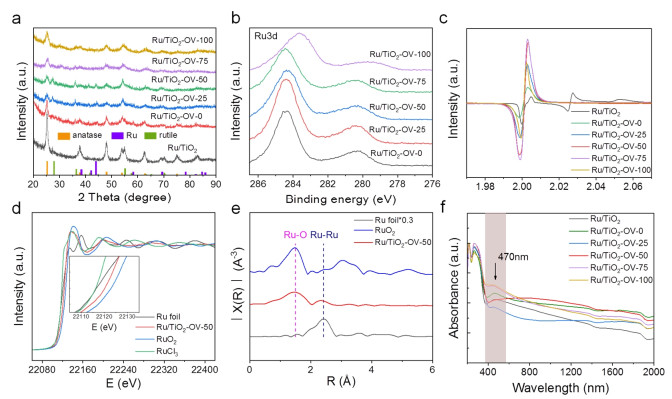
X-ray diffraction (XRD) patterns (Figure 2a) of Ru/TiO2-OV-x show the anatase and rutile mixed phase TiO2, which are different from the anatase phase of commercial TiO2 (Figure S5). Impressively, characteristic XRD peaks of Ru species are not clearly observed after Ru doping, indicating the high dispersion of Ru species (Figures 2a, S6). X-ray photoelectron spectroscopy (XPS) was performed to investigate the surface properties and chemical states of the catalysts. As shown in Figure S7, the XPS survey spectra of Ru/TiO2-OV-x show the presence of Ru, Ti and O elements. The Ru 3d spectra (Figure 2b) of Ru/TiO2-OV-x at ~280.4 and 284.5 eV, which are assigned to 3d5/2 and 3d3/2 of Ru0, respectively, indicating the transformation of Ru3+ into a lower valence state of Ru.[36] As the ascorbic acid amount increases, a shift to lower binding energy is observed. Such a negative shift indicates a higher electron density of Ru site, thus leading to a stronger ability to attach and activate CO2.[37,38] The Ti 2p spectra (Figure S8) show the peaks at ~458 and 464 eV corresponding to Ti 2p3/2 and Ti 2p1/2, respectively.[38] Notably, a negative shift in the binding energy is observed with the increase of ascorbic acid amount, which indicates the more oxygen vacancies are generated on the surface of catalyst.[39] For the O 1s spectra (Figure S9), the peaks at 529.4 and 531.0 eV correspond to lattice oxygen species and adsorbed oxygen species, respectively.[40-42] Furthermore, the electron paramagnetic resonance (EPR) was employed to investigate the oxygen vacancies, and the results in Figure 2c exhibit an obvious signal at g = 2.003 for the Ru/TiO2-OV-x catalysts, which is attributed to the oxygen vacancy signal.[43-46] It can be found that much higher intensity compared with Ru/TiO2 and Ru/TiO2-OV-0 confirmed the presence of abundant oxygen vacancies, which suggests the importance of ascorbic acid.[43] In addition, the contents of oxygen vacancies are accordingly increased after Ru doping due to the reductive treatment of H2. The fine structure information of optimum Ru/TiO2-OV-50 catalyst was further studied by X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements. The white-line intensity of Ru/TiO2-OV-50 is higher than that of Ru foil and lower than that of RuO2, indicating a partial positive charge of Ru species (Figure 2d).[47] The corresponding Fourier transform EXAFS spectrum (Figure 2e) of Ru/TiO2-OV-50 shows two obvious peaks at ~1.5 and 2.4 Å, which can be assigned to Ru-O and Ru-Ru coordination, respectively. According to the fitting results (Table S1), the coordination numbers of Ru-O and Ru-Ru are 1.41 and 3.95, respectively, which suggests the formation of low-coordination Ru clusters and NPs. The ultraviolet-visible-near infrared (UV-vis-IR) spectroscopy was used to investigate the light adsorption ability. Figure 2f shows the Ru/TiO2-OV-x samples can adsorb visible-near infrared light with a range of 400-2000 nm, and an adsorption peak at λ = 470 nm corresponds to the interband adsorption of Ru species. Compared with the absorption spectra of TiO2-OV-x samples (Figure S10), such excellent light absorption ability makes these candidates very suitable for photocatalytic CO2 hydrogenation reaction. The Ru contents are respectively determined to be 1.8, 1.4, 1.5, 1.6, 1.9 and 2.1 wt.% for Ru/TiO2, Ru/TiO2-OV-0, Ru/TiO2-OV-25, Ru/TiO2-OV-50, Ru/TiO2-OV-75 and Ru/TiO2-OV-100 catalysts by inductively coupled plasma-atomic emission spectrometry (ICP-AES) analysis, which indicates that the loading amount of Ru sites increases with the increase of oxygen vacancies. Furthermore, the specific surface area measurements at 77 K (Figures S11, 12) show that the Ru/TiO2-OV-50 gives a large specific surface area (152.0 m2/g), which is about 1.5 and 1.8 times higher than those of Ru/TiO2 (101.7 m2/g) and Ru-TiO2-OV-0 (83.6 m2/g), respectively. It can be found that the specific surface area increases first and then decreases with the increase of ascorbic acid amount. In a word, the Ru-doped TiO2 nanorods catalyst with tunable oxygen vacancies is successfully synthesized.
The photocatalytic CO2 hydrogenation to CH4 was used as a model reaction to evaluate the catalytic performance of as-synthesized samples. As seen in Figure 3a, the Ru/TiO2-R catalyst shows a higher reaction rate (11.20 mmol·g-1·h-1) than Ru/TiO2-A (0.43 mmol·g-1·h-1), Ru/CeO2 (1.11 mmol·g-1·h-1), Ru/In2O3 (1.96 mmol·g-1·h-1) and Pd/TiO2 (2.62 μmol·g-1·h-1) at an irradiation intensity of 1.0 W·cm-2, which confirms the synergistic effect of Ru and TiO2 for the enhanced photocatalytic CO2 methanation performance. More importantly, an almost 100% CH4 selectivity is obtained on the Ru/TiO2 catalyst. Although it exhibits an excellent CH4 selectivity, low reaction rate still needs to be improved due to the absence of oxygen vacancies over commercial TiO2. For this purpose, a series of Ru/TiO2-OV-x catalysts with tunable oxygen vacancies were synthesized. The results in Figure 3b show that Ru/TiO2-OV-50 has an 81.7 mmol·g-1·h-1 reaction rate with a 100% CH4 selectivity, which is ~ 2.4 times higher than those of Ru/TiO2 (34.6 mmol·g-1·h-1) and Ru/TiO2-OV-0 (34.8 mmol·g-1·h-1) at an irradiation intensity of 1.5 W·cm-2. This indicates the oxygen vacancies of TiO2 nanorods can efficiently catalyze the photo-catalytic CO2 methanation reaction due to the generated active sites and better light response ability. In addition, the reaction rate increases first then decreases with the increase of ascorbic acid amount, suggesting the importance of tunable oxygen vacancies amount.
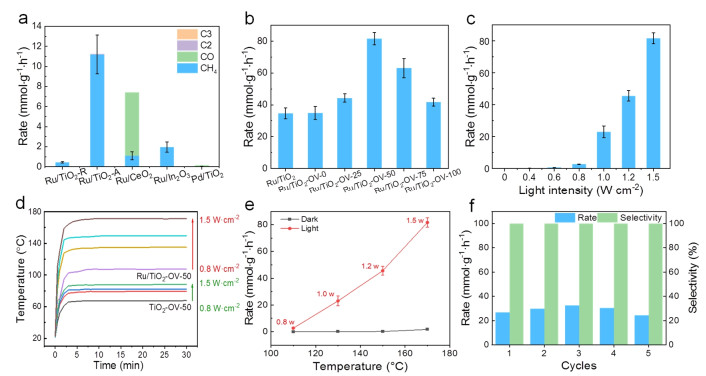
To optimize the reaction conditions and investigate the catalytic mechanism, a series of photocatalytic CO2 methanation reactions were conducted over the optimum Ru/TiO2-OV-50 catalyst. First, the influence of light intensity on catalytic performance was investigated. As shown in Figure 3c, no activity is observed in the absence of light irradiation, indicating the significance of light. The reaction rate is increased from 0.18 mmol·g-1·h-1 at 0.4 W·cm-2 to 45.6 mmol·g-1·h-1 at 1.2 W·cm-2, and then the reaction rate is further increased to 81.7 mmol·g-1·h-1 at 1.5 W·cm-2. It is worth noting that the catalyst not only possesses excellent catalytic activity, but also displays 100% CH4 selectivity. Considering the intensity distribution of the light at different frequencies, the apparent quantum efficiency is estimated to be 3.01% over Ru/TiO2-OV-50 under the optimum reaction conditions. Compared to reported results (Table S2), our catalytic perfor-mance exceeds most of the reported metal catalysts. Herein, the photothermal effect towards the gas-phase CO2 methanation reaction needs to be considered. Therefore, we monitored the surface temperature of the catalysts over time under light irradiation. The results show that the surface temperatures are rapidly increased in three to five minutes and then remain unchanged over the as-synthesized samples (Figures 3d, S13). Specifically, the surface temperatures over TiO2-OV-50 sample are 67.7, 79.2, 82.3 and 88.2 ℃ at 0.8, 1.0, 1.2 and 1.5 W·cm-2, respectively, which are much higher than those over commercial TiO2 and TiO2-OV-0 (Figure S13). It illustrates that the surface photothermal temperatures can be obviously increased in the presence of oxygen vacancies for the TiO2 catalysts. After Ru doping, the surface temperatures are further increased due to the excellent optical adsorption properties of Ru site. For instance, the surface temperatures over Ru/TiO2-OV-50 sample are 107.5, 135.1, 149.8 and 171.2 ℃ at 0.8, 1.0, 1.2 and 1.5 W·cm-2 (Figure 3d) respectively, indicating the excellent photothermal properties of metallic Ru site. To evaluate the contribution of thermal effect, we compared the catalytic performance with or without light irradiation. The results in Figure 3e reveal the reaction rates are 2.7, 23.0, 45.6 and 81.7 mmol·g-1·h-1 at 0.8, 1.0, 1.2 and 1.5 W cm-2, respectively, which are 82.3, 303.6, 263.4 and 48.1 times higher than those at the same reaction temperature under dark conditions, which confirm the significance of light other than thermal effect. In addition, we also explored the influence of wavelength on the catalytic performance of Ru/TiO2-OV-50. As shown in Tables S3, 4, the ultraviolet light gives almost no contribution to catalytic performance. The visible light, especially the wavelength between 420-600 nm, plays the most important role for this catalytic reaction. Subsequently, the effect of mixed gas ratio on catalytic activity was investigated, as shown in Figure S14. The reaction rate and CH4 selectivity increase with the increase of H2 to CO2 ratio, and a 100% CH4 selectivity is obtained when the H2 to CO2 ratio is greater than 1. When H2 to CO2 ratio is less than 1, the main product is CO. At last, the cycling test was examined at an irradiation intensity of 1.0 W·cm-2. The results in Figure 3f show the catalyst exhibits an excellent stability during five successive cycles. The XRD patterns of Ru/TiO2-OV-50 before and after 5 cycles are almost the same (Figure S15), implying its structural stability.
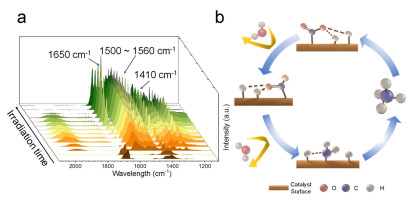
To confirm the carbon source of the produced CH4, isotope labelling experiment was carried out using 13CO2 as reactant for CO2 hydrogenation. The gas chromatography-mass spectroscopy (GC-MS) analysis (Figure S16) clearly confirms the origin of produced CH4. Meanwhile, the high CH4 selectivity is also confirmed by this isotope labelling experiment. Afterwards, the photocurrent and electrochemical impedance spectroscopy (EIS) results (Figure S17) show that the Ru/TiO2-OV-50 has a higher photocurrent density and smaller arc radius than commercial Ru/TiO2 and TiO2-OV-50 samples, which suggests a higher charge separation efficiency and lower charge-transfer resistance over Ru/TiO2-OV-50. Therefore, these results indicate the synergistic effect of Ru and TiO2 with the oxygen vacancies for the enhanced photocatalytic CO2 methanation performance. As for the role of oxygen vacancies, the transfer of photoexcited electrons TiO2 with oxygen vacancies to the electron-rich Ru NPs promotes the adsorption and dissociation of CO2 on the Ru sites. In addition, the oxygen vacancies can enhance the absorption of visible-near infrared light at a range of 400-2000 nm (Figure 2f). Finally, the existence of oxygen vacancies can effectively regulate the loading amount of Ru sites and thus specific surface area of catalysts. So far, two main pathways for CO2 methanation reaction have been proposed: (1) associative path, where oxygenate intermediates are first produced by reacting CO2 with H adatom (Had) species; (2) dissociative path, in which CO2 is first dissocia-ted to carbonyl and O adatom (Oad) species.[48] To explore the reaction pathway and mechanism in this work, in situ diffuse reflectance infrared Fourier transformations spectroscopy (DRIFTS) was therefore performed as shown in Figure 4a. Under CO2 atmosphere, the two obvious peaks at 1650 and 1410 cm-1 corresponding to formate are observed, [49] and the other weak peaks between 1560 and 1500 cm-1 corresponding to surface formates are also identified.[50] Notably, the peak at around 2000 cm-1 attributed to carbonyl species is not observed, indicating that the carbonyl species produced by dissociative path is not an essential intermediate.[48-56] In addition, the DRIFTS under a CO atmosphere shows the four peaks at 2172, 2115, 2060 and 1960 cm-1 are observed, which are assigned as the absorbed CO on the Ru NPs (Figure S14).[50] Thus, it can be inferred that the main reaction pathway is an associative path, as shown in Figure 4b. Briefly, CO2 and H2 are firstly adsorbed on the surface of catalyst to produce CO2 adsorption species and Had species, respectively. Then the C-O bond is broken and thus the C-H bond is concurrently formed with formate intermediate. Finally, the methane is obtained by further hydrogenation.[48] The proposed photocatalytic CO2 methanation pathway is different from that of the reported thermocatalytic process.[48-50,52,56]
In summary, the Ru-doped TiO2 photocatalysts with tunable oxygen vacancies have been successfully synthesized by using ascorbic acid as a reducing agent. In a photocatalytic CO2 methanation reaction, the optimal Ru/TiO2-OV-50 exhibited a remarkable performance with an 81.7 mmol·g-1·h-1 reaction rate and 100% CH4 selectivity under a 1.5 W·cm-2 light illumination, which was much better than that of commercial Ru/TiO2 and other reported catalysts. The detailed characterizations and experimental results revealed that the highly efficient photocatalytic CO2 methanation performance was mainly attributed to the synergistic effect of metallic Ru site and TiO2 with tunable oxygen vacancies. Meanwhile, the CO2 methanation performance was resulted from the light rather than the thermal effect. Moreover, based on the in situ spectroscopic analysis, we proposed the reaction pathway of CO2 methanation on this photocatalyst. It is hoped that this work would provide new insights for the design and synthesis of efficient photocatalysts for solar-driven CO2 transformation.
Lithium acetate, ascorbic acid, ruthenium chloride hydrate (RuCl3·xH2O) and commercial titanium dioxide (30 nm, anatase) were purchased from Aladdin Biochemical Technology Co., Ltd. N, N-dimethylformamide, acetic acid, tetrabutyl titanate and ammonia solution were purchased from Sinopharm Chemical Reagent Co., Ltd.
Synthesis of TiO2-OV-x. The titanium dioxides with tunable oxygen vacancies were synthesized via a simple solvothermal method. Firstly, 200 mg lithium acetate and x (x = 0, 25, 50, 75 or 100) mg ascorbic acid were successively added into a mixture of 6 mL N, N-dimethylformamide (DMF) and 4 mL acetic acid with continuous stirring until complete dissolution. Subsequently, 2 mL tetrabutyl titanate was added dropwise with stirring, and then the solution was transferred into a 100 mL Teflon-lined autoclave and heated at 200 ℃ for 20 h. The obtained precipitate was washed with deionized water and ethanol for three times and dried in an oven for 12 hours. The final product was denoted as TiO2-OV-x.
Synthesis of Ru/TiO2-OV-x. The Ru-doped titanium dioxides were synthesized by an electrostatic adsorption-reduction method. For a typical synthesis of Ru/TiO2-OV-50, 100 mg TiO2-OV-50 was first dissolved in a mixture solution containing 30 mL deionized water and 7.5 mL ammonia. Then, 11.9 mg RuCl3·xH2O was dissolved in another mixture solution containing 3 mL deionized water and 0.75 mL ammonia. After that, two solutions were mixed and stirred for 4 h. The obtained precursor was washed with deionized water and dried in an oven overnight. Subsequently, the precursor powder was reduced at 400 ℃ for 2 h with a heating rate of 5 ℃ min-1 under a 10 vol% H2/Ar flow to obtain Ru/TiO2-OV-50. The other Ru-doped titanium dioxide catalysts (Ru/TiO2-OV-x) were prepared by the same procedure, except that different titanium dioxides were used.
Scanning electron microscopy (SEM) images were taken on a ZEISS GeminiSEM 450 Schottky field-emission scanning electron microscope operated at 5 kV. TEM images and the corresponding EDS mapping analysis images were taken on a JEOL JEM-2100 Plus field-emission high-resolution transmission electron microscope operated at 200 kV. Powder XRD patterns were recorded by using a Philips X'Pert Pro Super X-ray diffractometer with Cu-Kα radiation (λ = 1.5418 Å). UV-vis-NIR diffuse reflectance data were recorded in the spectral region of 240-800 nm with a Shimadzu SolidSpec-3700 spectrophotometer. XPS was recorded on an X-ray photoelectron spectrometer (Thermo ESCALAB 250, USA). The specific surface areas were determined by BET measurement (JK-BK200C, JWGB SCI. & TECH) using a nitrogen adsorption-desorption isotherm at 77 K. X-ray absorption fine structure (XAFS) measurements were measured at the BL14W1 beamline in Shanghai Synchrotron Radiation Facility and Singapore Synchrotron Light Source. In situ DRIFTS measure-ments were performed using a Bruker IFS 66v Fourier-transform spectrometer equipped with a Harrick diffuse reflectance accessory at the Infrared Spectroscopy and Microspectroscopy Endstation (BL01B) in the NSRL. EPR spectra were collected using a JEOL JES-FA200 electron spin resonance spectrometer at 140 K (9.062 GHz). The Ru contents were measured by ICP-AES. The photocurrent curves and EIS experiments were carried out on a CHI660E electrochemical workstation.
In a typical measurement, 5 mg of catalyst was first dissolved in 300 μL deionized water and dispersed evenly on the surface of a FTO glass slice (15mm × 30mm × 2.2mm). Then the glass slice was placed into a 32 mL custom-made quartz tube. The custom-made photoreactor was continuously degassed and purged with a mixture of CO2 and H2 (3:1) three times. After the photoreactor was sealed, the reaction was conducted for 1.5 h under the irradiation of a 300 W Xenon lamp (Beijing Perfectlight, PLS-SXE300D). After reaction, the amounts of CO and CH4 were determined using a flame ionization detector (FID) of the gas chromatograph (GC, 7890A, Agilent). Other products such as C2H4 were determined by another gas chromatograph (GC, 7890B, Agilent) equipped with thermal conductivity detector and flame ionization detector (FID).
Gao, P.; Li, S.; Bu, X.; Dang, S.; Liu, Z.; Wang, H.; Zhong, L.; Qiu, M.; Yang, C.; Cai, J.; Wei, W.; Sun, Y. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017, 9, 1019-1024. doi: 10.1038/nchem.2794
Wang, L.; Zhang, W.; Zheng, X.; Chen, Y.; Wu, W.; Qiu, J.; Zhao, X.; Zhao, X.; Dai, Y.; Zeng, J. Incorporating nitrogen atoms into cobalt nanosheets as a strategy to boost catalytic activity toward CO2 hydrogenation. Nat. Energy 2017, 2, 869-876. doi: 10.1038/s41560-017-0015-x
Cai, M.; Wu, Z.; Li, Z.; Wang, L.; Sun, W.; Tountas, A. A.; Li, C.; Wang, S.; Feng, K.; Xu, A. -B.; Tang, S.; Tavasoli, A.; Peng, M.; Liu, W.; Helmy, A. S.; He, L.; Ozin, G. A.; Zhang, X. Greenhouse-inspired supra-photothermal CO2 catalysis. Nat. Energy 2021, 6, 807-814. doi: 10.1038/s41560-021-00867-w
Barrio, J.; Mateo, D.; Albero, J.; García, H.; Shalom, M. A heterogeneous carbon nitride-nickel photocatalyst for efficient low-temperature CO2 methana-tion. Adv. Energy Mater. 2019, 9, 1902738. doi: 10.1002/aenm.201902738
Kattel, S.; Liu, P.; Chen, J. G. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 2017, 139, 9739-9754. doi: 10.1021/jacs.7b05362
Guo, Y.; Mei, S.; Yuan, K.; Wang, D. -J.; Liu, H. -C.; Yan, C. -H.; Zhang, Y. -W. Low-temperature CO2 methanation over CeO2-supported Ru single atoms, nanoclusters, and nanoparticles competitively tuned by strong metal-support interactions and H-spillover effect. ACS Catal. 2018, 8, 6203-6215. doi: 10.1021/acscatal.7b04469
Wan, L.; Zhou, Q.; Wang, X.; Wood, T. E.; Wang, L.; Duchesne, P. N.; Guo, J.; Yan, X.; Xia, M.; Li, Y. F.; Jelle, A. A.; Ulmer, U.; Jia, J.; Li, T.; Sun, W.; Ozin, G. A. Cu2O nanocubes with mixed oxidation-state facets for (photo)catalytic hydrogenation of carbon dioxide. Nat. Catal. 2019, 2, 889-898. doi: 10.1038/s41929-019-0338-z
Chen, Y.; Zhang, Y.; Fan, G.; Song, L.; Jia, G.; Huang, H.; Ouyang, S.; Ye, J.; Li, Z.; Zou, Z. Cooperative catalysis coupling photo-/photothermal effect to drive Sabatier reaction with unprecedented conversion and selectivity. Joule 2021, 5, 3235-3251. doi: 10.1016/j.joule.2021.11.009
Han, S.; Li, B.; Huang, L.; Xi, H.; Ding, Z.; Long, J. Construction of ZnIn2S4-CdIn2S4 microspheres for efficient photo-catalytic reduction of CO2 with visible light. Chin. J. Struct. Chem. 2022, 41, 2201007-2201013.
Wang, Z.; Hong, J.; Ng, S. -F.; Liu, W.; Huang, J.; Chen, P.; Ong, W. -J. Recent progress of perovskite oxide in emerging photocatalysis landscape: water splitting, CO2 reduction, and N2 fixation. Acta Phys. Chim. Sin. 2021, 37, 2011033.
Li, N.; Peng, J.; Shi, Z.; Zhang, P.; Li, X. Charge transfer and orbital reconstruction of non-noble transition metal single-atoms anchored on Ti2CTx-MXenes for highly selective CO2 electrochemical reduction. Chin. J. Catal. 2022, 43, 1906-1917. doi: 10.1016/S1872-2067(21)64018-4
Xu, Z. -T., X.; Xie, K. Enhanced CO2 electrolysis with metal-oxide interface structures. Chin. J. Struct. Chem. 2021, 40, 31-41.
Wang, C.; Sun, Z.; Zheng, Y.; Hu, Y. H. Recent progress in visible light photocatalytic conversion of carbon dioxide. J. Mater. Chem. A 2019, 7, 865-887. doi: 10.1039/C8TA09865D
He, K.; Shen, R.; Hao, L.; Li, Y.; Zhang, P.; Jiang, J.; Xin, L. Advances in nanostructured silicon carbide photocatalysts. Acta Phys. Chim. Sin. 2022, 38, 2201021.
Chai, Y.; Chen, Y.; Wang, B.; Jiang, J.; Liu, Y.; Shen, J.; Wang, X.; Zhang, Z. Sn2+ and Cu2+ self-codoped Cu2ZnSnS4 nanosheets switching from p-type to n-type semiconductors for visible-light-driven CO2 reduction. ACS Sustain. Chem. Eng. 2022, 10, 8825-8834. doi: 10.1021/acssuschemeng.2c01564
Shen, R.; Hao, L.; Ng, Y. H.; Zhang, P.; Arramel, A.; Li, Y.; Li, X. Heterogeneous N-coordinated single-atom photocatalysts and electrocatalysts. Chin. J. Catal. 2022, 43, 2453-2483. doi: 10.1016/S1872-2067(22)64104-4
Quan, F.; Zhan, G.; Mao, C.; Ai, Z.; Jia, F.; Zhang, L.; Gu, H.; Liu, S. Efficient light-driven CO2 hydrogenation on Ru/CeO2 catalysts. Catal. Sci. Technol. 2018, 8, 6503-6510. doi: 10.1039/C8CY01787E
Lin, L.; Wang, K.; Yang, K.; Chen, X.; Fu, X.; Dai, W. The visible-light-assisted thermocatalytic methanation of CO2 over Ru/TiO(2-x)Nx. Appl. Catal., B. 2017, 204, 440-455. doi: 10.1016/j.apcatb.2016.11.054
Mateo, D.; Albero, J.; Garcia, H. Titanium-perovskite-supported RuO2 nanoparticles for photocatalytic CO2 methanation. Joule 2019, 3, 1949-1962. doi: 10.1016/j.joule.2019.06.001
Sun, Z.; Talreja, N.; Tao, H.; Texter, J.; Muhler, M.; Strunk, J.; Chen, J. Catalysis of carbon dioxide photoreduction on nanosheets: fundamentals and challenges. Angew. Chem. Int. Ed. 2018, 57, 7610-7627. doi: 10.1002/anie.201710509
Zhou, Y.; Zhang, Q.; Shi, X.; Song, Q.; Zhou, C.; Jiang, D. Photocatalytic reduction of CO2 into CH4 over Ru-doped TiO2: synergy of Ru and oxygen vacancies. J. Colloid Interf. Sci. 2022, 608, 2809-2819. doi: 10.1016/j.jcis.2021.11.011
Liu, Y.; Yu, F.; Wang, F.; Bai, S.; He, G. Construction of Z-scheme In2S3-TiO2 for CO2 reduction under concentrated natural sunlight. Chin. J. Struct. Chem. 2022, 41, 2201034-2201039.
Su, B.; Huang, H.; Ding, Z.; Roeffaers, M. B. J.; Wang, S.; Long, J. S-scheme CoTiO3/Cd9.51Zn0.49S10 heterostructures for visible-light driven photo-catalytic CO2 reduction. J. Mater. Sci. Technol. 2022, 124, 164-170. doi: 10.1016/j.jmst.2022.01.030
Wu, Z.; Guo, S.; Kong, L. -H.; Geng, A. -F.; Wang, Y. -J.; Wang, P.; Yao, S.; Chen, K. -K.; Zhang, Z. -M. Doping [Ru(bpy)3]2+ into metal-organic framework to facilitate the separation and reuse of noble-metal photosensitizer during CO2 photoreduction. Chin. J. Catal. 2021, 42, 1790-1797. doi: 10.1016/S1872-2067(21)63820-2
Chai, S.; Men, Y.; Wang, J.; Liu, S.; Song, Q.; An, W.; Kolb, G. Boosting CO2 methanation activity on Ru/TiO2 catalysts by exposing (001) facets of anatase TiO2. J. CO2 Util. 2019, 33, 242-252. doi: 10.1016/j.jcou.2019.05.031
Abe, T.; Tanizawa, M.; Watanabe, K.; Taguchi, A. CO2 methanation property of Ru nanoparticle-loaded TiO2 prepared by a polygonal barrel-sputtering method. Energy Environ. Sci. 2009, 2, 315-321. doi: 10.1039/b817740f
Kar, P.; Farsinezhad, S.; Mahdi, N.; Zhang, Y.; Obuekwe, U.; Sharma, H.; Shen, J.; Semagina, N.; Shankar, K. Enhanced CH4 yield by photocatalytic CO2 reduction using TiO2 nanotube arrays grafted with Au, Ru, and ZnPd nanoparticles. Nano Res. 2016, 9, 3478-3493. doi: 10.1007/s12274-016-1225-4
Li, X.; Yu, J.; Jaroniec, M.; Chen, X. Cocatalysts for selective photo-reduction of CO2 into solar fuels. Chem. Rev. 2019, 119, 3962-4179. doi: 10.1021/acs.chemrev.8b00400
Liu, S.; Li, Y.; Ding, K.; Chen, W.; Zhang, Y.; Lin, W. Mechanism on carbon vacancies in polymeric carbon nitride for CO2 photoreduction. Chin. J. Struct. Chem. 2020, 39, 2068-2076.
Wang, R.; Yang, P.; Wang, S.; Wang, X. Distorted carbon nitride nano-sheets with activated n→π* transition and preferred textural properties for photocatalytic CO2 reduction. J. Catal. 2021, 402, 166-176. doi: 10.1016/j.jcat.2021.08.025
Li, D.; Huang, Y.; Li, S.; Wang, C.; Li, Y.; Zhang, X.; Liu, Y. Thermal coupled photoconductivity as a tool to understand the photothermal catalytic reduction of CO2. Chin. J. Catal. 2020, 41, 154-160. doi: 10.1016/S1872-2067(19)63475-3
Zhou, J.; Gao, Z.; Xiang, G.; Zhai, T.; Liu, Z.; Zhao, W.; Liang, X.; Wang, L. Interfacial compatibility critically controls Ru/TiO2 metal-support interaction modes in CO2 hydrogenation. Nat. Commun. 2022, 13, 327.
Panagiotopoulou, P. Methanation of CO2 over alkali-promoted Ru/TiO2 catalysts: Ⅱ. Effect of alkali additives on the reaction pathway. Appl. Catal., B 2018, 236, 162-170. doi: 10.1016/j.apcatb.2018.05.028
Li, M.; Li, P.; Chang, K.; Wang, T.; Liu, L.; Kang, Q.; Ouyang, S.; Ye, J. Highly efficient and stable photocatalytic reduction of CO2 to CH4 over Ru loaded NaTaO3. Chem. Commun. 2015, 51, 7645-7648. doi: 10.1039/C5CC01124H
Cai, S.; Zhang, M.; Li, J.; Chen, J.; Jia, H. Anchoring single-atom Ru on CdS with enhanced CO2 capture and charge accumulation for high selectivity of photothermocatalytic CO2 reduction to solar fuels. Solar RRL 2021, 5, 2000313. doi: 10.1002/solr.202000313
Lin, Y.; Tian, Z.; Zhang, L.; Ma, J.; Jiang, Z.; Deibert, B. J.; Ge, R.; Chen, L. Chromium-ruthenium oxide solid solution electrocatalyst for highly efficient oxygen evolution reaction in acidic media. Nat. Commun. 2019, 10, 162. doi: 10.1038/s41467-018-08144-3
Jarzembska, K.; Seal, S.; Woźniak, K.; Szadkowska, A.; Bieniek, M.; Grela, K. X-ray photoelectron spectroscopy and reactivity studies of a series of ruthenium catalysts. ChemCatChem 2009, 1, 144-151. doi: 10.1002/cctc.200900052
Morgan, D. J. Resolving ruthenium: XPS studies of common ruthenium materials. Surf. Interf. Anal. 2015, 47, 1072-1079. doi: 10.1002/sia.5852
Marchal, C.; Cottineau, T.; Méndez-Medrano, M. G.; Colbeau‐Justin, C.; Caps, V.; Keller, V. Au/TiO2-gC3N4 nanocomposites for enhanced photocatalytic H2 production from water under visible light irradiation with very low quantities of sacrificial agents. Adv. Energy Mater. 2018, 8, 1702142. doi: 10.1002/aenm.201702142
Wu, M.; Zhang, J.; Liu, C.; Gong, Y.; Wang, R.; He, B.; Wang, H. Rational design and fabrication of noble‐metal‐free NixP cocatalyst embedded 3D N-TiO2/g-C3N4 heterojunctions with enhanced photocatalytic hydrogen evolution. ChemCatChem 2018, 10, 3069-3077. doi: 10.1002/cctc.201800197
Eom, J. -Y.; Lim, S. -J.; Lee, S. -M.; Ryu, W. -H.; Kwon, H. -S. Black titanium oxide nanoarray electrodes for high rate Li-ion microbatteries. J. Mater. Chem. A 2015, 3, 11183-11188. doi: 10.1039/C5TA01718A
Ge, H.; Zhang, B.; Liang, H.; Zhang, M.; Fang, K.; Chen, Y.; Qin, Y. Photocatalytic conversion of CO2 into light olefins over TiO2 nanotube confined Cu clusters with high ratio of Cu+. Appl. Catal., B 2020, 263, 118133. doi: 10.1016/j.apcatb.2019.118133
Yin, G.; Huang, X.; Chen, T.; Zhao, W.; Bi, Q.; Xu, J.; Han, Y.; Huang, F. Hydrogenated blue titania for efficient solar to chemical conversions: preparation, characterization, and reaction mechanism of CO2 reduction. ACS Catal. 2018, 8, 1009-1017. doi: 10.1021/acscatal.7b03473
Zhou, Z.; Li, X.; Li, J.; You, Z. Promoting CO2 methanation performance of Ru/TiO2 through Co-activity of exposing (001) facets and oxygen vacancies of TiO2. Mater. Sci. Semicon. Proc. 2022, 146, 106677. doi: 10.1016/j.mssp.2022.106677
Cheng, S.; Gao, Y. -J.; Yan, Y. -L.; Gao, X.; Zhang, S. -H.; Zhuang, G. -L.; Deng, S. -W.; Wei, Z. -Z.; Zhong, X.; Wang, J. -G. Oxygen vacancy enhancing mechanism of nitrogen reduction reaction property in Ru/TiO2. J. Energy Chem. 2019, 39, 144-151. doi: 10.1016/j.jechem.2019.01.020
Chen, S.; Abdel-Mageed, A. M.; Li, D.; Bansmann, J.; Cisneros, S.; Biskupek, J.; Huang, W.; Behm, R. J. Morphology‐engineered highly active and stable Ru/TiO2 catalysts for selective CO methanation. Angew. Chem. Int. Ed. 2019, 58, 10732-10736. doi: 10.1002/anie.201903882
Du, J.; Huang, Y.; Huang, Z.; Wu, G.; Wu, B.; Han, X.; Chen, C.; Zheng, X.; Cui, P.; Wu, Y. Reversing the catalytic selectivity of single-atom Ru via support amorphization. JACS Au 2022, 2, 1078-1083. doi: 10.1021/jacsau.2c00192
Miao, B.; Ma, S. S. K.; Wang, X.; Su, H.; Chan, S. H. Catalysis mecha-nisms of CO2 and CO methanation. Catal. Sci. Technol. 2016, 6, 4048-4058. doi: 10.1039/C6CY00478D
Gupta, N.; Kamble, V.; Kartha, V.; Iyer, R.; Thampi, K. R.; Gratzel, M. FTIR spectroscopic study of the interaction of CO2 and CO2 + H2 over partially oxidized RuTiO2 catalyst. J. Catal. 1994, 146, 173-184. doi: 10.1016/0021-9517(94)90020-5
Abdel-Mageed, A. M.; Widmann, D.; Olesen, S. E.; Chorkendorff, I.; Biskupek, J.; Behm, R. J. Selective CO methanation on Ru/TiO2 catalysts: role and influence of metal-support interactions. ACS Catal. 2015, 5, 6753-6763. doi: 10.1021/acscatal.5b01520
Aldana, P. A. U.; Ocampo, F.; Kobl, K.; Louis, B.; Thibault-Starzyk, F.; Daturi, M.; Bazin, P.; Thomas, S.; Roger, A. C. Catalytic CO2 valorization into CH4 on Ni-based ceria-zirconia. Reaction mechanism by operando IR spectroscopy. Catal. Today 2013, 215, 201-207. doi: 10.1016/j.cattod.2013.02.019
Dalla Betta, R.; Shelef, M. Heterogeneous methanation: in situ infrared spectroscopic study of RuAl2O3 during the hydrogenation of CO. J. Catal. 1977, 48, 111-119. doi: 10.1016/0021-9517(77)90082-3
Eckle, S.; Anfang, H. -G.; Behm, R. J. R. Reaction intermediates and side products in the methanation of CO and CO2 over supported Ru catalysts in H2-rich reformate gases. J. Phys. Chem. C 2011, 115, 1361-1367. doi: 10.1021/jp108106t
Eckle, S.; Denkwitz, Y.; Behm, R. J. Activity, selectivity, and adsorbed reaction intermediates/reaction side products in the selective methanation of CO in reformate gases on supported Ru catalysts. J. Catal. 2010, 269, 255-268. doi: 10.1016/j.jcat.2009.10.025
Prairie, M. R.; Renken, A.; Highfield, J. G.; Thampi, K. R.; Grätzel, M. A fourier transform infrared spectroscopic study of CO2 methanation on supported ruthenium. J. Catal. 1991, 129, 130-144. doi: 10.1016/0021-9517(91)90017-X
Zhang, S. -T.; Yan, H.; Wei, M.; Evans, D. G.; Duan, X. Hydrogenation mechanism of carbon dioxide and carbon monoxide on Ru(0001) surface: a density functional theory study. RSC Adv. 2014, 4, 30241-30249. doi: 10.1039/C4RA01655F

Figure 1 (a) Schematic illustration of the synthesis of Ru/TiO2-OV-x catalysts. (b) SEM image, (c) TEM image, (d) HRTEM image and (e) TEM element mapping images of Ru/TiO2-OV-50 catalyst.

Figure 2 (a) XRD patterns and (b) high-resolution Ru 3d XPS spectra of Ru/TiO2-OV-x catalysts. (c) EPR spectra of Ru/TiO2 and Ru/TiO2-OV-x catalysts. (d) XANES spectra and (e) EXAFS spectra of Ru foil, RuO2 and Ru/TiO2-OV-50 catalysts. (f) UV-vis-IR spectra of Ru/TiO2 and Ru/TiO2-OV-x catalysts.

Figure 3 (a) Photocatalytic CO2 methanation over different catalysts at an irradiation intensity of 1.0 W·cm-2 (Ru/TiO2-R: rutile; Ru/TiO2-A: anatase). (b) Photocatalytic CO2 methanation over Ru/TiO2 and Ru/TiO2-OV-x catalysts at an irradiation intensity of 1.5 W·cm-2. (c) Photocatalytic CO2 methanation over Ru/TiO2-OV-50 catalyst under different light intensities. (d) The photothermal temperature monitoring for the TiO2-OV-50 and Ru/TiO2-OV-50 catalysts. (e) The CO2 methanation over Ru/TiO2-OV-50 catalyst with and without light. (f) Stability test for photocatalytic CO2 methanation over Ru/TiO2-OV-50 catalyst under 1.0 W·cm-2.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
