Table 1.
Selected Bond Lengths (Å) and Bond Angles (°) for Complex 1
Citation:
Yi-Feng WANG, Mou-Hai SHU. Synthesis and Crystal Structure of a Copper(II) Complex with Funnel-like Ligand Based on Calix[6]arene[J]. Chinese Journal of Structural Chemistry,
2022, 41(3): 220318.
doi:
10.14102/j.cnki.0254-5861.2011-3312

Synthesis and Crystal Structure of a Copper(II) Complex with Funnel-like Ligand Based on Calix[6]arene
English
Synthesis and Crystal Structure of a Copper(II) Complex with Funnel-like Ligand Based on Calix[6]arene
Abstract:
The preparation method of a funnel like ligand derived from calix[6]arene and tris(2-pyridyl-methyl)anime has been improved. The Cu(II) complex was obtained and characterized by ESI-MS, EPR, UV-Vis spectra and crystallographic analysis. In the complex, a guest acetonitrile molecule is introduced into the cavity of the ligand host via the coordination with Cu(II) center.
-
Key words:
- Cu(II) complex
- / crystal structure
- / calix[6]arene
- / host-guest interaction
-
1. INTRODUCTION
Metalloenzymes in organisms catalyze the biochemical reactions with high efficiency and selectivity. The unusual selective catalytic function is closely associated with the structural characteristics of metalloenzymes including the catalytic metal centers and the backbone of proteins via noncovalent interactions including intermolecular force, hydrogen bond, π-π stacking, hydrophobic interactions, etc. Chemists have designed and synthesized host molecules with defined, hydrophobic pockets to simulate the enzyme active centers. And these host molecules usually include the coordination polyhedrons[1-6], organic cages[7-12] and covalent molecules bearing the macrocyclic units like cyclodextrins, cucurbiturils, pillar[n]arenes and calixarenes[13-18]. As an important building unit, the calixarenes with a different number of benzene rings provide various inner cavity. The cavity size of calix[4]arene is smaller, while that of calix[8]arene is larger, but the structural rigidity of the latter is insufficient and the conformation is various, which makes the post modification quite complicated. Calix[6]arene has a certain rigidity to maintain a certain cavity size, and its high symmetry also provides the possibilities for the post modification. For example, the molecular host calix[6]-TMPA is synthesized by the reaction of the C3 symmetrically tri-methylated calix[6]arene with tripodal tris(pyridylmethyl)amine unit[19], and the host can recognize the linear organic ammonium[19] or CH3CN, C2H5OH, CH3CN, C2H5OH molecules via the coordination with Cu(II) ion[20]. In addition, the combined O2 within the molecular host is activated through the coordination to Cu(I) ion, thus realizing the oxidation reaction of CH2 to C=O[21].
It is found that there are six large tert-butyl groups at the open end in calix[6]arene, which hinders the entry of large guest molecules into the cavity[19, 22]. In view of this, we designed and synthesized the molecular host by removing three tert-butyl groups from calix[6]arene unit, and characterized the complex formed with Cu(I, II) and Zn(II) ions in the solution by electrospray ionization mass spectrometry[23]. Recently, we have improved the synthetic route and increased the preparation yield of the host molecule. Herein we report the improved synthetic method of the ligand, the preparation of the Cu(II) complex, and the characterizations by various techniques such as high resolution ESI-MS, EPR spectroscopy, UV-Vis spectrum and crystallographic structure analysis.
2. EXPERIMENTAL
2.1 Instruments and reagents
Dichloromethane and acetonitrile were dried by CaH2 and distilled before use. The other chemicals were used directly. Nuclear magnetic resonance spectra were recorded on Bruker AVANCE III HD 400 or 500 spectrometer at 293 K. ESI-MS was measured on a Waters Micro mass Q-TOF Premier system. The ultraviolet visible spectrum was measured on a Lambda 35 UV/Vis Spectrometer. Electron paramagnetic resonance data were obtained on a Bruker EMXplus-9.5/12 spectrometer at 100 K. 5, 17, 29-tri-tert-butyl-38, 40, 42-tri-methoxycalix[6]arene-37, 39, 41-triol[24, 25] and 2, 6-pyridine-dimethanol[26] were prepared following the literature methods.
2.2 Synthesis of 2-(hydroxymethyl)-6-(p-tosyloxymethyl)pyridine[27]
2, 6-Pyridinedimethanol (2.78 g, 0.020 mol), potassium iodine (0.700 g, 0.004 mol) and Ag2O (8.00 g, 0.030 mol) were suspended in dichloromethane (140 mL) in a flask. p-Toluenesulfonyl chloride (3.80 g, 0.20 mol) was added in portions under stirring at −20 ℃. After 40 min, the solid was filtered off and rinsed with dichloromethane. After removing solvent, 2-(hydroxymethyl)-6-(p-tosyloxymethyl)pyridine was purified by column chromatography on silica (200~300 mesh) with dichloromethane/hexane and ethyl acetate as the eluents. Yield 4.8 g (82%). The data of 1H NMR spectrum were consistent with those reported in the literature[23].
2.3 Synthesis of tris(6-hydroxymethyl-pyridin-2-methyl)amine[28]
2-(Hydroxymethyl)-6-(p-tosyloxymethyl)pyridine (7.33 g, 0.025 mol), ammonium acetate (0.635 g, 0.0083 mol), Na2CO3 (1.75 g, 0.017 mol) and dry acetonitrile (150 mL) were added in a flask. Then the suspension was stirred at 30 ℃ for three days. Another portion of Na2CO3 (0.560 g, 0.0073 mol) was added. After stirring for two days, the solids were filtered off and rinsed twice with acetonitrile. The volume of the filtrate was reduced to 10 mL. The precipitations formed upon the addition of 50 mL of ethyl ether, and isolated by filtration followed by washing with ethyl ether. Tris(6-hydroxymethyl-2-pyridylmethyl)amine was obtained as white microcrystals. Yield 2.80 g, 88%, m.p. 158~160 ℃. Data of 1H NMR spectrum match those reported in the literature[28].
2.4 Synthesis of tris(6-(4-methylbenzenesulfonyl-methyl)-pyridin-2-methyl)amine[22, 23]
KOH powder (2.80 g, 0.050 mol) was slowly added to a mixture of tris(6-hydroxymethyl-pyridin-2-methyl)anime (3.80 g, 10.0 mmol) and p-toluenesulfonyl chloride (6.27 g, 33.0 mmol) in dry dichloromethane (50 mL) at 0 ℃. After stirring at 25 ℃ for 6 hours, the insoluble matters were isolated by filtration and rinsed twice with CH2Cl2. The organic phase was washed with water and NaHCO3 solution and dried with anhydrous MgSO4, obtaining a yellow oil after the removal of the solvent. Tris(6-(4-methylbenzenesulfonylmethyl)-pyridin-2-methyl)anime was obtained as a pink oil by column chromatography on silica (eluent dichloromethane to 5% methanol in dichloromethane). Yield 7.10 g, 84%. The data of 1H NMR spectrum are close to those reported[23].
2.5 Synthesis of 11, 23, 35-tri(tert-butyl)-37, 39, 41-trimethoxycalix[6]arene-38, 40, 42-tri(6-methoxy-2-pyridin-methyl)anime (L)[22, 23]
5, 17, 29-Tri-tert-butyl-38, 40, 42-trimethoxycalix[6]arene-37, 39, 41-triol (424 mg, 0.50 mmol), potassium carbonate (690 mg, 5.0 mmol) and dry acetonitrile (100 mL) were introduced in a flask, to which a solution of tris(6-(4-methyl-benzene-sulfonyl-methyl-pyridin-2-methyl)anime (422 mg, 0.50 mmol) in acetonitrile (50 mL) was added using dropping funnel. The suspension was refluxed under stirring for two days. Acetonitrile was removed by using rotary evaporator, and the residual was extracted with dichloromethane. After removal of dichloromethane, the ligand was obtained by column chromatography on silica (eluent with CH2Cl2/CH3OH = 20/1), yield 417 mg (71%). 1H NMR (500 MHz, CDCl3) δ = 7.96 (d, J = 8.0 Hz, 3H), 7.86 (t, J = 8.0 Hz), 7.48 (d, J = 8.0 Hz, 3H), 7.25 (d, J = 8.0 Hz, 6H), 6.90 (d, J = 8.0 Hz, 6H), 6.90 (s, 6H), 6.82 (t, J = 8.0 Hz, 3H), 5.66 (s, 6H), 5.08 (s, 6H), 4.34 (d, J = 12.0 Hz, 6H), 3.44 (d, J = 12.0 Hz, 6H), 2.55 (s, 9H), 1.37 (s, 27H). 13C NMR (125 MHz, CDCl3) δ = 160.35, 155.08, 152.72, 149.13, 146.64, 138.67, 134.26, 132.80, 128.79, 124.21, 122.48, 119.89, 73.04, 61.70, 56.64, 34.20, 31.55, 30.44. ESI-MS: m/z for C78H85N4O6 calculated 1173.6469 [M + H]+, found 1173.6449.
2.6 Preparation of complex 1
The ligand L (23.5 mg, 0.0200 mmol) and copper perchlorate hydrate (8.20 mg, 0.0240 mmol) were suspended in acetonitrile (5 mL). Then a clear green solution was obtained after heating at 60 ℃ for 30 min. The solution was kept undisturbed to concentrate, obtaining green crystals qualified for crystallographic analysis within a week. Yield 24.0 mg (81% based on the ligand).
2.7 Crystallographic measurements and structure determination
Crystallographic data were obtained on a Bruker D8 VENTURE diffractometer equipped with a Helios MX multilayer monochromator Cu-Kα radiation (λ = 1.54178 Å). The data were collected and reduced by using the SMART and SAINT software. Structure was solved with SHELXL-2014[29] by using direct method, and refined with SHELXT-2014[30]. The H atoms were located at theoretical positions. Total 70 restraints including DFIX, DANG, EXYZ, EADP and so on were applied to refine the disordered perchlorates. The maximum residual density for the last refinement is 1.052, which is near the disordered perchlorate in 1 (2.10 Å from atom Cl(3)/Cl(3)′, and 0.68 Å from atom O(44)). To improve the data quality of the main structure of the complex, SQUEEZE method in PLATON was used to treat the disordered solvent molecules. The important bond parameters of 1 are reported in Table 1, and the detailed structural information can be found from the CIF file via CCDC No. 2097353.
Table 1

 DownLoad:
CSV
DownLoad:
CSV
Bond Dist. Bond Dist. Bond Dist. Cu(1)–N(1) 1.960(2) Cu(1)–N(3) 2.208(2) Cu(1)–N(5) 1.959(2) Cu(1)–N(2) 2.171(2) Cu(1)–N(4) 2.199(2) Angle (°) Angle (°) Angle (°) N(1)–Cu(1)–N(2) 82.00(9) N(2)–Cu(1)–N(3) 118.33(9) N(3)–Cu(1)–N(5) 98.66(9) N(1)–Cu(1)–N(3) 80.69(9) N(2)–Cu(1)–N(4) 117.34(8) N(4)–Cu(1)–N(5) 99.11(8) N(1)–Cu(1)–N(4) 80.26(9) N(2)–Cu(1)–N(5) 99.26(9) N(1)–Cu(1)–N(5) 178.74(10) N(3)–Cu(1)–N(4) 117.11(8) Crystal data for 1: space group C2/c, a = 61.352(11), b = 15.030(3), c = 46.854(8) Å, α = 90, β = 128.5880(10), γ = 90°, V = 35603.4(12) Å3, Z = 16, T = 17(2) K, Dc = 1.102 g/cm3, μ = 1.363 mm-1, F(000) = 12432, 190250 reflections measured (2.406≤θ ≤68.387º, −74≤h≤74, −18≤k≤18, −52≤l≤56), 32631 independent (Rint = 0.0586), the final R = 0.0617, wR = 0.1658, GOOF = 1.034, (Δρ)max = 1.052, (Δρ)min = −1.386 e/Å3.
3. RESULTS AND DISCUSSION
3.1 Preparation of the ligand and the Cu(II) complex
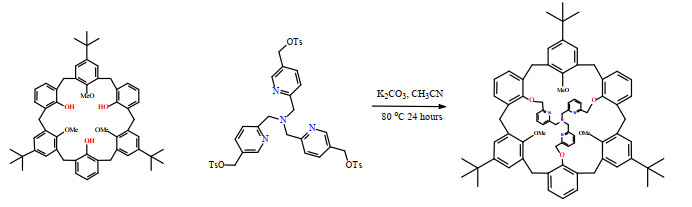
The ligand was synthesized with good yields by the coupling reaction between the C3 symmetrically trimethylated calix[6]arene with tripodal tris(pyridylmethyl)amine unit (Scheme 1). 1H and 13C NMR spectra (Figs. S1 and S2) of the ligand show that the ligand is pure. The simple pattern of the signals of the ligand indicates that the ligand is in a high symmetrical model in the solution. The main peak of the ESI-MS of the ligand is located at m/z 1173.6449 (100%), which is from the species (M + H)+. All the observations confirm that the ligand is as per the design. The Cu(II) complex was obtained by the reaction of copper(II) perchlorate with the ligand in acetonitrile, and green crystals were acquired by slow evaporation of the solvent. Many efforts have been made to determine the crystal structure of the ligand, no ideal data set was obtained due to the quick efflorescence of the crystals.
Scheme 1
 Scheme 1. Synthesis of the ligand L
Scheme 1. Synthesis of the ligand L3.2 Structure description of the complex
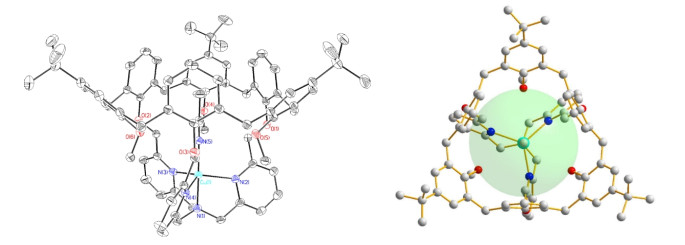
There are two copper(II) centers in the minimum symmetric unit of 1 {[Cu(C78H85N4O6)(CH3CN)]⋅(ClO4)2⋅solvents}. Due to the fact that the structures of two Cu(II)-centers are similar, only the detailed structural information of Cu(1) atom is described. As shown in Fig. 1, the Cu(1) center ion is complexed by four nitrogen atoms from the TMPA cone of the ligand, and a nitrogen atom from the guest acetonitrile forming a trigonal bi-pyramidal geometry. The N(2), N(3) and N(4) atoms from the pyridine units of the ligand are located in the equatorial plane, and N(1), N(5) atoms located at the axial sites. The Cu–N bond lengths within the equatorial plane vary from 2.171(2) to 2.208(2) Å, and those at the axial positions are 1.960(2) and 1.959(2) Å. The apical compression trigonal bi-pyramidal geometric construction of the Cu(II) center is similar to the observation in the previous results[20]. The bond angles change from 80.26(9) to 178.74(10)°. The τ value of the CuN5 unit is 1.007 by literature method[31]. Therefore, the coordination geometry is quite close to an ideal trigonal bi-pyramidal geometry. Interestingly, the guest molecule acetonitrile was introduced into the cavity of the funnel-like ligand via the coordination with the central Cu(II) ion.
Figure 1
 Figure 1. Coordination environments (side view and top view) of the center ions in complex 1
Figure 1. Coordination environments (side view and top view) of the center ions in complex 1In the funnel-like molecular host based on calix[6]arene, the tert-butyl on calixarene prevents the larger molecules such as tert-butylanime from entering the cavity of calix-[6]arene[22]. The diameter of the open end of calix[6]arene is about 4.4 Å in the crystal structure of Cu(II) complex[20], while the opening diameter of calix[6]arene in complex 1 is enlarged to 6.8 Å, which provides an opportunity for introducing larger guest molecules into the cavity of the calix-[6]arene and exploring the chemical reaction of guest molecules in the host cavity.
There are hydrogen bonds between oxygen atom of perchlorates and hydrogen atom in the crystal. The hydrogen bond data of 1 are given in Table 2.
Table 2
Table 2. Hydrogen Bonds for Complex 1 (Å and °)DownLoad:
CSV
D–H···A d(D–H) d(H···A) d(D···A) ∠DHA C(1)–H(1B)···O(32) 0.99(1) 2.572 3.203(2) 122 C(1)–H(1B)···O(34) 0.99(1) 2.369 3.320(2) 161 C(8)–H(8B)···O(22′) 0.99(1) 2.294 2.991(2) 127 C(81)–H(81B)···O(22′)a 0.99(1) 2.055 2.952(2) 150 C(88)–H(88A)···O(31) 0.99(1) 2.500 3.167(2) 125 C(95)–H(95A)···O(53) 0.99(1) 2.251 3.107(2) 144 C(95)–H(95B)···O(24)a 0.99(1) 2.624 3.377(2) 133 C(95)–H(95B)···O(23′)a 0.99(1) 2.250 3.123(2) 146 C97–H97A···O23′a 0.95(1) 2.590 3.177(2) 120 Symmetry code: (a) –x+3/2, y+1/2, –z+3/2 3.3 ESI-MS, EPR and UV-Vis spectra of the complex
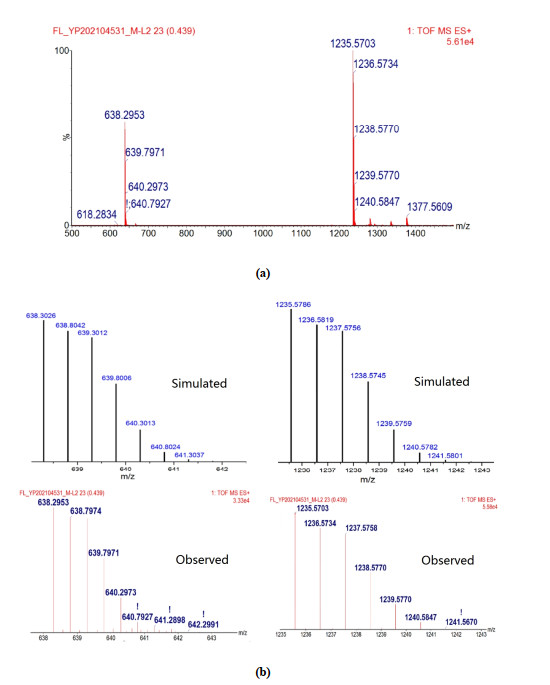
The ESI-MS of complex 1 is shown in Fig. 2a. The main peak in the image is at m/z = 1235.5703 (100%), corresponding to the species [LCu(I)]+, indicating that the complex is reduced to Cu(I) under ESI conditions, and similar result have been reported in the literature[32]. Two peaks observed at m/z = 638.2953 (60%) and 1377.5609 (8%) correspond to [LCu(II)(CH3CN)]2+ and [LCu(II)(CH3CN)(ClO4)]+, respectively. The isotope distributions of the peaks at m/z = 1235.5703 and 638.2953 are given in Fig. 2b, and the agreement between the observed and the calculated model is excellent and ensured the assignments.
Figure 2
 Figure 2. (a) Electrospray ionization mass spectrum (ESI-MS) of complex 1, (b) Isotopic contributions of the main peaks
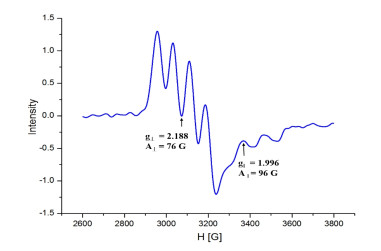
Figure 2. (a) Electrospray ionization mass spectrum (ESI-MS) of complex 1, (b) Isotopic contributions of the main peaksEPR spectrum (X band) of complex [LCu(CH3CN)](ClO4)2 was measured in frozen CH3CN at 100 K, as shown in Fig. 3. The related parameters were obtained from the EPR spectrum of the complex as follows, g‖ = 1.996, g⊥ = 2.188, A‖ = 75, A⊥ = 109, and these parameters are similar to the reported ones[20].
Figure 3
 Figure 3. EPR spectrum (X-band) of complex 1 in frozen CH3CN (100 K)
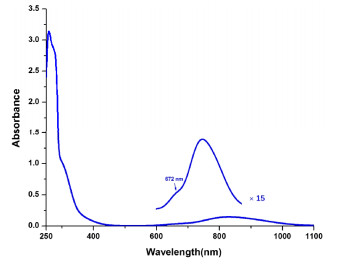
Figure 3. EPR spectrum (X-band) of complex 1 in frozen CH3CN (100 K)As shown in Fig. 4, two d–d transitions at 672, 829 nm and a LMCT at 306 nm were observed from the UV-Vis spectrum of complex 1 measured in CH3CN solution at room temperature, which is similar to the reported result[20]. The observation
Figure 4
 Figure 4. UV-Vis spectrum of complex 1 in CH3CN (293 K)
Figure 4. UV-Vis spectrum of complex 1 in CH3CN (293 K)shows that the removal of the three tert-butyl groups have slight influence on the electronic properties of the ligand.
4. CONCLUSION
In summary, the preparation method of the molecular host based on calix[6]arene and tris(2-pyridylmethyl)anime has been improved. The Cu(II) complex was obtained and characterized by HRMS, EPR, UV-Vis spectra as well as crystallographic analysis technique. The guest acetonitrile molecule was introduced into the cavity of the ligand host by the coordination with Cu(II) ion.
-
-
[1]
Hong, C. M.; Bergman, R. G.; Raymond, K. N.; Toste, F. D. Self-assembled tetrahedral hosts as supramolecular catalysts. Acc. Chem. Res. 2018, 51, 2447−2455. doi: 10.1021/acs.accounts.8b00328
-
[2]
Zarra, S.; Wood, D. M.; Roberts, D. A.; Nitschke, J. R. Molecular containers in complex chemical systems. Chem. Soc. Rev. 2015, 44, 419−432. doi: 10.1039/C4CS00165F
-
[3]
Li, X.; Wu, J.; He, C.; Meng, Q.; Duan, C. Asymmetric catalysis within the chiral confined space of metal-organic architectures. Small 2019, 15, 1804770. doi: 10.1002/smll.201804770
-
[4]
Brown, C. J.; Toste, D.; Bergman, R. G.; Raymond, K. N. Supramolecular catalysis in metal-ligand cluster hosts. Chem. Rev. 2015, 115, 3012−3035. doi: 10.1021/cr4001226
-
[5]
Tan, C.; Chu, D.; Tang, X.; Liu, Y.; Xuan, W.; Cui, Y. Supramolecular coordination cages for asymmetric catalysis. Chem. Eur. J. 2019, 25, 662−672. doi: 10.1002/chem.201802817
-
[6]
Percástegui, E. G.; Ronson, T. K.; Nitschke, J. R. Design and applications of water-soluble coordination cages. Chem. Rev. 2020, 120, 13480−13544. doi: 10.1021/acs.chemrev.0c00672
-
[7]
Hasell, T.; Cooper, A. I. Porous organic cages: soluble, modular and molecular pores. Nat. Rev. Mater. 2016. 16053.
-
[8]
Mastalerz, M. Porous shape-persistent organic cage compounds of different size, geometry, and function. Acc. Chem. Res. 2018, 51, 2411−2422. doi: 10.1021/acs.accounts.8b00298
-
[9]
Liu, W.; Bobbala, S.; Stern, C. L.; Hornick, J. E.; Liu, Y.; Enciso, A. E.; Scott, E. A.; Stoddart, J. F. XCage: a tricyclic octacationic receptor forperylene diimide with picomolar affinity in water. J. Am. Chem. Soc. 2020, 142, 3165−3173. doi: 10.1021/jacs.9b12982
-
[10]
Li, H.; Zhang, H.; Lammer, A. D.; Wang, M.; Li, X.; Lynch, V. M.; Sessler, J. L. Quantitative self-assembly of a purely organic three-dimensional catenane in water. Nat. Chem. 2015, 7, 1003−1008. doi: 10.1038/nchem.2392
-
[11]
Hasell, T.; Wu, X.; Jones, J. T. A.; Bacsa, J.; Steiner, A.; Mitra, T.; Trewin, A.; Adams, D. J.; Cooper, A. I. Triply interlocked covalent organic cages. Nat. Chem. 2010, 2, 750−755. doi: 10.1038/nchem.739
-
[12]
Zhang, Z. Q.; Ren, Q. X.; Tian, W. F.; Sun, W. H.; Cao, X. P.; Shi, Z. F.; Chow, H. F.; Kuck, D. Synthesis of enantiopure hydrocarbon cages based on an optically resolved C3-symmetric triaminotribenzotriquinacene. Org. Lett. 2021, 23, 1478−1483. doi: 10.1021/acs.orglett.1c00176
-
[13]
Liu, Z.; Nalluri, S. K. M.; Stoddart, J. F. Surveying macrocyclic chemistry: from flexible crown ethers to rigid cyclophanes. Chem. Soc. Rev. 2017, 46, 2459−2478. doi: 10.1039/C7CS00185A
-
[14]
Ogoshi, T.; Yamagishi, T. A.; Nakamoto, Y. Pillar-shaped macrocyclic hosts pillar[n]arenes: new key players for supramolecular chemistry. Chem. Rev. 2016, 116, 7937−8002. doi: 10.1021/acs.chemrev.5b00765
-
[15]
He, Q.; Vargas-Zúñiga, G. I.; Kim, S. H.; Kim, S. K.; Sessler, J. L. Macrocycles as ion pair receptors. Chem. Rev. 2019, 119, 9753−9835. doi: 10.1021/acs.chemrev.8b00734
-
[16]
Jana, A.; Bähring, S.; Ishida, M.; Goeb, S.; Canevet, D.; Sallé, M.; Jeppesen, J. O.; Sessler, J. L. Functionalised tetrathiafulvalene-(TTF-) macrocycles: recent trends in applied supramolecular chemistry. Chem. Soc. Rev. 2018, 47, 5614−5645. doi: 10.1039/C8CS00035B
-
[17]
He, C.; Sheng, T. P.; Dai, F. R.; Chen, Z. N. Sulfonylcalix[4]arene-based coordination supercontainers. Chin. J. Struct. Chem. 2020, 39, 2077−2084.
-
[18]
Sheng, T. P.; Dai, F. R.; Zheng, G. Z.; Chen, Z. N. Synthesis, crystal structure and gas adsorption properties of metal-organic supercontainer based 2, 6-naphthalenedicarboxylate linker. Chin. J. Struct. Chem. 2021, 40, 311−316.
-
[19]
Zeng, X.; Coquière, D.; Alenda, A.; Garrier, E.; Prangé, T.; Li, Y.; Reinaud, O.; Jabin, I. Efficient synthesis of calix[6]tmpa: a new calix[6]-azacryptand with unique conformational and host-guest properties. Chem. Eur. J. 2006, 12, 6393−6402. doi: 10.1002/chem.200600278
-
[20]
Izzet, G.; Zeng, X.; Akdas, H.; Marrot, J.; Reinaud, O. Drastic effects of the second coordination sphere on neutral vs. anionic guest binding to a biomimetic Cu(II) center embedded in a calix[6]aza-cryptand. Chem. Commun. 2007, 8, 810−812.
-
[21]
Thiabaud, G.; Guillemot, G.; Schmitz-Afonso, I.; Colasson, B.; Reinaud, O. Solid-state chemistry at an isolated copper(I) center with O2. Angew. Chem. Int. Ed. 2009, 48, 7383−7386. doi: 10.1002/anie.200902691
-
[22]
Tu, C. L.; Zheng, C.; Chen, Y.; Shu, M. H. Synthesis and reversible recognition for organic ammoniums of a molecular receptor based on calix[6]-arene. Chem. J. Chin. Univ. 2007, 28, 1917−1919.
-
[23]
Ai, T. T.; Li, Q. Q.; Liang, T.; Tu, C. L; Shu, M. H. Syntheses and electrospray mass spectroscopy of functionalized N4-calix[6]arene and complexes with copper(I, II) and zinc salts. Chin. J. Inorg. Chem. 2008, 24, 1159−1163.
-
[24]
Gutsche, C. D.; Dhawan, B.; Leonis, M.; Stewart, D. p-tert-Butylcalix[6]arene. Org. Synth. 1990, 68, 238−238. doi: 10.15227/orgsyn.068.0238
-
[25]
Janssen, R. G.; Vervoom, W.; Reinhoudt, D.; Casnati, A.; Freriks, M.; Pochini, A.; Ugozzoli, F.; Ungaro, R.; Nieto, P. M.; Carramolino, M. Procedure for the selective alkylation of calix[6]arenas at the lower rim. Synthesis 1994, 380−385.
-
[26]
Newcomb, M.; Timko, J. M.; Walba, D. M.; Cram, D. J. Host-guest complexation. 3. Organization of pyridyl binding sites. J. Am. Chem. Soc. 1977, 99, 6392−6398. doi: 10.1021/ja00461a035
-
[27]
Almaliti, J.; Al-Hamashi, A. A.; Negmeldin, A. T.; Hanigan, C. L.; Perera, L.; Pflum, M. K. H.; Casero, R. A.; Tillekeratne, L. M. V. Largazole analogues embodying radical changes in the depsipeptide ring: development of a more selective and highly potent analogue. J. Med. Chem. 2016, 59, 10642−10660. doi: 10.1021/acs.jmedchem.6b01271
-
[28]
He, Z.; Chaimungkalanont, P. J.; Craig, D. C.; Colbran, S. B. Copper(II) complexes of 6-hydroxymethyl-substituted tris(2-pyridylmethyl)amine ligands. J. Chem. Soc. Dalton Trans. 2000, 9, 1419−1429.
-
[29]
Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3−8. doi: 10.1107/S2053229614024218
-
[30]
Sheldrick, G. M. SHELXT-integrated space-group and crystal structure determination. Acta Crystallogr. A 2015, 71, 3−8. doi: 10.1107/S2053273314026370
-
[31]
Addison, A. W.; Rao, T. N.; Reedijk, J.; van Rijn, J.; Verschoor, G. C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1, 7-bis(N-methylbenzimidazol-2′-yl)-2, 6-dithiaheptane] copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 1349−1356.
-
[32]
Shu, M. H.; Sun, W. Y.; Duan, C. Y.; Fu, Y. J.; Zhang, W. J.; Tang, W. X. The influence of the linkage mode between bipyridine units in oligobipyridine ligands on the formation of copper-(I) and-(II) helicates. J. Chem. Soc. Dalton Trans. 1999, 729−734.
-
[1]
-
Figure 1 Coordination environments (side view and top view) of the center ions in complex 1
Figure 2 (a) Electrospray ionization mass spectrum (ESI-MS) of complex 1, (b) Isotopic contributions of the main peaks
Table 1. Selected Bond Lengths (Å) and Bond Angles (°) for Complex 1
Bond Dist. Bond Dist. Bond Dist. Cu(1)–N(1) 1.960(2) Cu(1)–N(3) 2.208(2) Cu(1)–N(5) 1.959(2) Cu(1)–N(2) 2.171(2) Cu(1)–N(4) 2.199(2) Angle (°) Angle (°) Angle (°) N(1)–Cu(1)–N(2) 82.00(9) N(2)–Cu(1)–N(3) 118.33(9) N(3)–Cu(1)–N(5) 98.66(9) N(1)–Cu(1)–N(3) 80.69(9) N(2)–Cu(1)–N(4) 117.34(8) N(4)–Cu(1)–N(5) 99.11(8) N(1)–Cu(1)–N(4) 80.26(9) N(2)–Cu(1)–N(5) 99.26(9) N(1)–Cu(1)–N(5) 178.74(10) N(3)–Cu(1)–N(4) 117.11(8)  下载: 导出CSV
下载: 导出CSV
Table 2. Hydrogen Bonds for Complex 1 (Å and °)
D–H···A d(D–H) d(H···A) d(D···A) ∠DHA C(1)–H(1B)···O(32) 0.99(1) 2.572 3.203(2) 122 C(1)–H(1B)···O(34) 0.99(1) 2.369 3.320(2) 161 C(8)–H(8B)···O(22′) 0.99(1) 2.294 2.991(2) 127 C(81)–H(81B)···O(22′)a 0.99(1) 2.055 2.952(2) 150 C(88)–H(88A)···O(31) 0.99(1) 2.500 3.167(2) 125 C(95)–H(95A)···O(53) 0.99(1) 2.251 3.107(2) 144 C(95)–H(95B)···O(24)a 0.99(1) 2.624 3.377(2) 133 C(95)–H(95B)···O(23′)a 0.99(1) 2.250 3.123(2) 146 C97–H97A···O23′a 0.95(1) 2.590 3.177(2) 120 Symmetry code: (a) –x+3/2, y+1/2, –z+3/2
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 5
- 文章访问数: 1671
- HTML全文浏览量: 154
