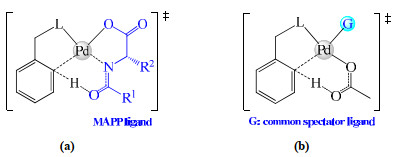
Figure 1.
Two types of ligands affecting C−H bond cleavage. (a) MAPP serving as a reacting ligand; (b) group G serving as a spectator ligand

Transition-metal-catalyzed C–H activation is one of the most widely investigated topics in modern organometallic chemistry, because it allows the direct conversion of C−H bonds into C−C or C−X bonds without the necessity of using pre-functionalized substrates[1]. Among many transition metals, palladium has been believed to be versatile in C−H activation[2, 3], and numerous C−H functionalizations mediated by palladium catalysts have been reported in the literature, such as C−H/C−X coupling[4, 5], C−H/C−H coupling[6, 7], dehydrogenative cycloaddition[8, 9], and so on. Current interests in the field of C−H activation include the control of regio- and stereo-selectivity, exploration of bifunctional ligands and metal-ligand cooperative systems, and application of C−H activation in total synthesis[10, 11].
Ligands have proved to influence C−H functionalizations, and in some cases they were capable of improving the reaction efficiency and/or rendering the selectivity. A ligand can act as either of two roles, namely the reacting ligand and the spectator ligand (Fig. 1). A ligand which is responsible for deprotonating the target C−H bond is a reacting ligand, while that merely coordinating with metal serves as a spectator ligand. The most widely used palladium catalyst is palladium acetate (Pd(OAc)2)[12]. Yu's group established a kind of mono-N-protected amino acid (MAPP) ligands and demonstrated that they not only stabilized monomeric palladium complexes but also acted as proton abstractors for C−H cleavage[13, 14]. In some cases, employment of external pyridine- or phosphorus-based ligands is crucial for Pd(OAc)2-catalyzed reactions[15, 16]. Recently, chiral N-heterocyclic carbene (NHC) ligands have drawn attention of organic chemists in enantioselective C−H functionalizations[17]. On the above examples, MAPP ligands serve as reacting ligands responsible for scissoring the target C−H bond (Fig. 1a), while the other ligands of interest are simply spectator ligands (Fig. 1b).
Despite plenty of mechanistic studies on transition-metal-catalyzed C−H activation in the literature[18-21], systematical investigation on the ligand effects, in a broader sense, has been rather rare. For example, the following questions have been unsolved: (1) What are the quantitative effects of spectator ligands on thermodynamic and kinetic parameters of C−H activation? (2) What are the unique geometrical and electronic characters of C−H activation in the presence of different spectator ligands?
In this work, we attempt to characterize the effects of an array of simple spectator ligands on palladium-catalyzed C− H activation on the basis of density functional theory (DFT) calculations. Our strategy is that a palladium complex similar to Pd(OAc)2 is chosen as the parent catalyst, and then, each of the spectator ligands is introduced to palladium by replacing one of the original ligands. Through comparison of the located mechanisms and energetics, the ligand effects can be measured.
All calculations were finished in the Gaussian 09 computational program[22]. Geometries were optimized with the B3LYP method[23], employing the double-ζ valence polarized DZVP basis set for all atoms[24, 25]. The default self-consistent reaction field polarizable continuum model was used with dichloroethane as solvent[26]. All the optimized geometries were subsequently subjected to frequency analyses, in order to calculate Gibbs free energies (298.15 K and 1 atm) and examine the number and motion of imaginary frequencies. To further refine the electronic energies obtained, single-point calculations were performed by using the B3LYP method with Grimm's D3 correction[27] and the Def2TZVPP basis set[28] for all atoms, on the basis of the same solvation model. The combination of the B3LYP method and a dispersion-corrected method has been widely used in mechanistic calculations[29, 30]. The 3D geometries were made by the ChemCraft software.
Fig. 2 shows the designed parent Pd(Ⅱ) catalyst, dimethylether-coordinated palladium acetate labeled as CAT in this paper, which has three ligands, namely a κ2-OAc ligand, a κ1-OAc ligand, and a dimethyl ether (OMe2) ligand. Replacement of the κ1-OAc ligand in CAT with another univalent ligand (X) or replacement of the OMe2 ligand in CAT with another neutral ligand (Y) can generate an array of palladium catalysts with different spectator ligands. The κ2-OAc ligand in CAT (and the other catalysts) is treated as the reacting ligand responsible for C−H bond cleavage. The C−H activation mechanisms and energetics mediated by different palladium catalysts will be compared in the following sections.
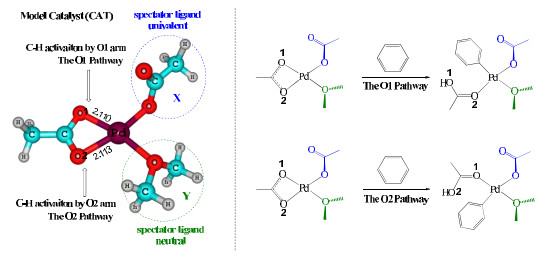
We first designed two C−H activation reactions of benzene catalyzed by CAT as given in Fig. 2, which differ in the position of the C−H activation site. If the C−H bond cleavage of benzene is induced by the O1 arm of κ2-OAc, the reaction channel is named as the O1 pathway; similarly, the other reaction channel is named as the O2 pathway. The detailed mechanism and the corresponding free-energy variations are shown in Fig. 3. Only the structures on the O2 pathway are given for simplicity, because those on the O1 pathway are very similar.
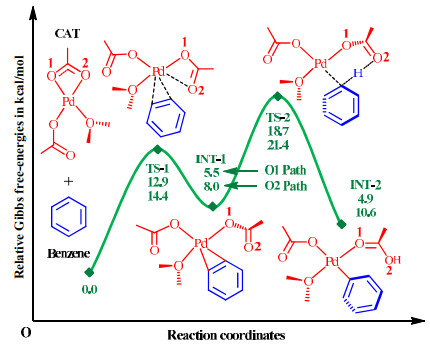
The located mechanism consists of two elementary steps, involving two transition states. In the first step, one of the two O arms of κ2-OAc is removed by benzene through a ligand substitution via transition state TS-1. The interaction between palladium and benzene in INT-1 is a η2-type coordination. In the second step, the target C−H bond is deprotonated by the adjacent OAc group via transition state TS-2, which affords intermediate INT-2 with a normal Pd−C6H5 coordinate bond.
The first step involves a free-energy barrier of 12.9 (O1 pathway) or 14.4 kcal/mol (O2 pathway), and INT-1 lies at 5.5 or 8.0 kcal/mol above the initial point. The free-energy barrier of the C−H activation step is calculated to be 13.2 or 13.4 kcal/mol. According to the steady-state approximation, however, the free-energy difference between TS-2 and the initial point should be assigned as the rate-determining activation barrier, which is computed to be 18.7 or 21.4 kcal/mol. The overall reaction needs to absorb free energies by 4.9 or 10.6 kcal/mol, indicating that it is unspontaneous under the standard condition. However, INT-2 only serves as an intermediate in the overall reaction rather than the final product, and subsequent transformations after INT-2 could release lots of free energies to achieve the overall spontaneity. It should be mentioned that benzene could also replace the OMe2 ligand in CAT and the C−H activation step would be induced by the original κ1-OAc group. Such a C−H activation pathway has also been characterized in our calculations, and the main results are provided in the Supporting Information. In fact, in this work we attempt to explore the effects of diverse spectator ligands in comparison to the κ1-OAc or OMe2 ligand, and hence, the mechanistic pattern given in Fig. 3 will be more beneficial for fulfilling our research goal.
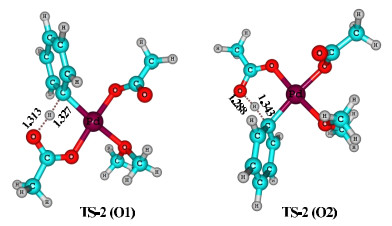
The three-dimensional structures of the proton-transfer transition states (TS-2) are given in Fig. 4. The located structures are consistent with the so-called concerted metalation-deprotonation (CMD) mechanism[18-20] and have a typical six-center cyclic structure, with H···O and H···C distances in the ranges of 1.288~1.313 and 1.327~1.343 Å, respectively. For the O2 pathway, the C···H distance is obviously longer than the O···H distance, while the two distances are relatively close for the O1 pathway, indicating that the proton-transfer transition state occurs later on the O2 pathway. Judging from the computed free-energy values and transition-state geometries, C−H activation should have more difficulty taking place at the position trans to the original κ1-OAc ligand (O2 pathway) in comparison to that cis to the original κ1-OAc ligand (O1 pathway).
In order to unveil the electronic effects of the spectator ligand X (univalent type) on C−H activation, we designed seven palladium catalysts by replacing the κ1-OAc ligand in CAT with a different univalent ligand, including methoxyl (OCH3), amide (NH2), methyl (CH3), phenyl (Ph), cyanide (CN), fluorine (F), and chlorine (Cl). The structures and compound names of these catalysts are defined in Fig. 5.
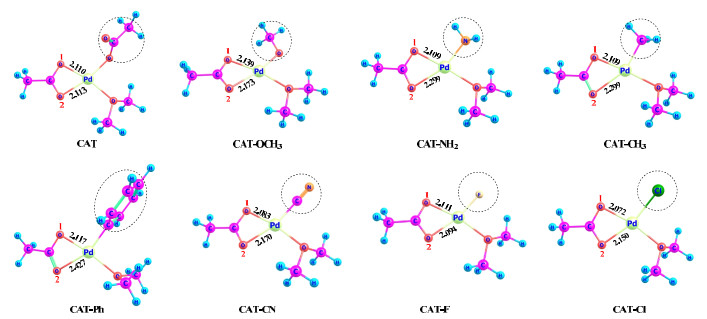
It is shown that the two Pd−O bonds coming from the κ2-OAc ligand are not of equal length, which is related to the nature of the introduced ligand X. Except for CAT−F, the Pd−O2 distance (trans to X) is longer than the Pd−O1 distance (cis to X); especially, the two Pd−O distances differ by about 0.3 Å for both CAT−CH3 and CAT−Ph. This phenomenon is attributed to the concept of trans effect or trans influence in coordination chemistry[31], which states that a σ-donating or/and π-accepting ligand tends to weaken its trans coordinate bond. The trans effect is normally more significant than the corresponding cis effect. As a strongly σ-donating ligand, both methyl and phenyl could exert a very significant trans effect.
Although the C−H activation mechanisms mediated by these catalysts are similar to that using CAT as the catalyst, the energetic parameters vary widely with the nature of the spectator ligand. The computed rate-determining activation barriers (the difference between TS-2 and the initial point, like Fig. 3) and overall free-energy changes are collected in Table 1. We first discuss the kinetic effects exerted by the seven ligands. It is shown that CAT−F and CAT−Cl have free-energy barriers (19.1~19.5 and 21.1~21.2 kcal/mol) very close to CAT, and hence, the poorly σ-donating ligands F and Cl appear not to hinder the C−H activation in progress. The catalysts CAT−OCH3, CAT−NH2, CAT−Ph, and CAT−CH3 all make the C−H activation process harder. For the O1 pathway, the free-energy barrier increases from 20.7 to 25.1 kcal/mol in the order of CAT−NH2, CAT−OCH3, CAT−Ph and CAT−CH3. For the O2 pathway, the free-energy barrier increases from 25.3 to 35.8 kcal/mol in the order of CAT−OCH3, CAT−NH2, CAT−Ph and CAT−CH3. The O1 pathway is obviously more favorable both kinetically and thermodynamically than the O2 one for all of the catalysts. The severely retarding effect caused by both CH3 and Ph should result from their strongly σ-donating ability and significant trans effect. Generally, the free-energy barrier of C−H activation increases in the order of κ1-OAc ≈ Cl ≈ F < OCH3 < NH2 < Ph < CH3. As an σ-donating and π-accepting ligand, the CN ligand displays a special behavior. The O1 pathway involves the lowest barrier of 18.3 kcal/mol, while the O2 pathway suffers from a high barrier of 25.2 kcal/mol, indicating that this ligand can promote the C−H activation at its cis position but may hinder the reaction at its trans position.
 DownLoad:
CSV
DownLoad:
CSV
| Catalysts | Pathway | ∆G | ∆G⧧ |
| CAT | O1 | 4.9 | 18.7 |
| O2 | 10.6 | 21.4 | |
| CAT−OCH3 | O1 | 8.4 | 22.1 |
| O2 | 19.2 | 25.3 | |
| CAT−NH2 | O1 | 6.7 | 20.7 |
| O2 | 25.3 | 27.3 | |
| CAT−CH3 | O1 | 11.5 | 25.1 |
| O2 | 35.6 | 35.8 | |
| CAT−Ph | O1 | 9.8 | 23.7 |
| O2 | 33.7 | 32.8 | |
| CAT−CN | O1 | 0.5 | 18.3 |
| O2 | 18.6 | 25.2 | |
| CAT−F | O1 | 5.0 | 19.5 |
| O2 | 10.7 | 21.2 | |
| CAT−Cl | O1 | 2.0 | 19.1 |
| O2 | 10.5 | 21.1 |
The thermodynamic parameters display a similar trend. In comparison to CAT, the introduction of OCH3, NH2, Ph or CH3 makes the C−H activation process more endothermal in terms of free energies, especially for Ph or CH3. The effect on the O2 pathway is more evident than that on the O1 pathway. In contrast, CAT−F and CAT−Cl have very similar thermodynamic parameters to CAT. As an exception, the O1 pathway mediated by CAT−CN is near free-energy neutral (ΔG = 0.5 kcal/mol), while the O2 pathway is strongly endothermal by 18.6 kcal/mol.
The transition-state geometries are provided in Fig. 6. As the free-energy barrier increases, in general, the difference of the C···H and O···H distances becomes larger and the transition state occurs later. For example, the transition state on the O2 pathway occurs later than that on the O1 pathway for all of the catalytic systems, because the C···H distance is obviously longer than the O···H distance for the O2 pathway. Note that the imaginary frequencies of TS-2-CH3 and TS-2-Ph on the O2 pathway are only –187.0 and –493.2 i cm-1, respectively, since they occur quite late on the reaction coordinates.
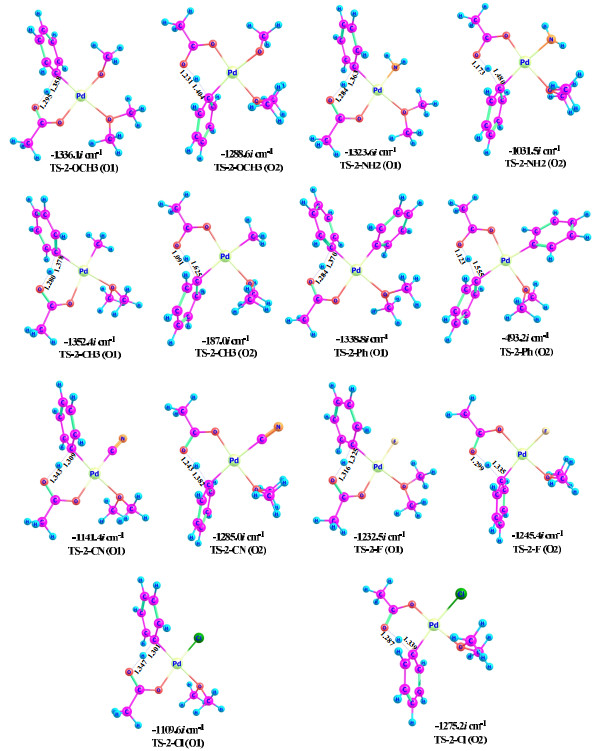
In summary, σ-donating ligands, such as OCH3, NH2, Ph, and CH3, could hinder the C−H activation in progress, and the effects of CH3 and Ph are evident. As the trans effect is more significant than the cis one and the small CH3 group causes a large difference, they should mainly be electronic rather than steric effects.
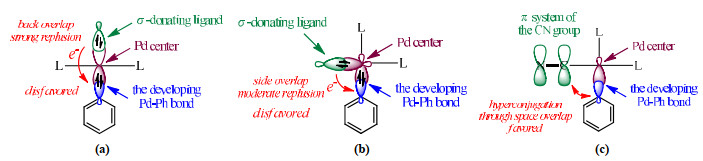
Fig. 7 provides a chemical model to explain the observed ligand effects. The effects of the σ-donating ligand can be rationalized by parts (a) and (b). The lone-pair orbital of this ligand could overlap with the fragment orbital having the largest component of the developing Pd···Ph bond. A small amount of electron density flows to the bonding region of Pd···Ph, and the consequent electron accumulation would increase electrostatic repulsion to hinder the C−H activation and Pd−C bond formation. It is understandable that the back overlap of the ligand is more effective than the side overlap, and hence, the trans effect must be more evident than the cis effect. For the CN ligand residing at the cis position, the developing Pd···Ph σ-lobe could be in conjugation with the π system of CN, through space overlap, to stabilize the Pd···Ph bonding.
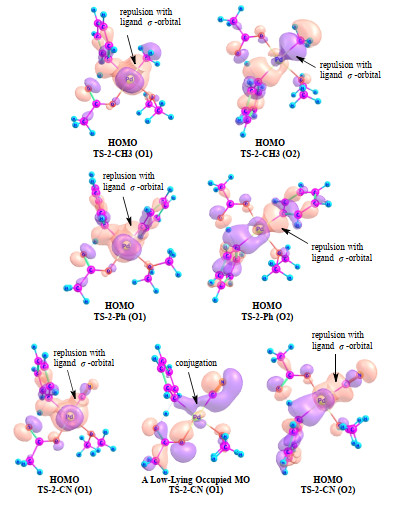
The proposed chemical model is supported by the calculated Kohn-Sham orbitals of some typical transition states (Fig. 8). The HOMOs of both TS-2-CH3 and TS-2-Ph indicate an overlap repulsion between the Pd···Ph and Pd−C bonds for both the O1 and O2 pathways. The HOMOs of TS-2-CN have similar feathers. For the O1 pathway, however, TS-2-CN has a low-lying orbital exhibiting a hyper-conjugation between the Pd···C σ-lobe and the C≡N π-lobe. The characters of these selected orbitals are generally in line with the proposed chemical model.
In order to explore the effects of the spectator ligand Y (neutral type) on C−H activation, we designed another seven palladium catalysts by replacing the OMe2 ligand in CAT with a different neutral ligand, including amine (NMe3), phosphine (PMe3), dimethyl sulfide (SMe2), ethene (CH2=CH2), ethyne (CH≡CH), N-heterocyclic carbene (NHC), and isonitrile (CNR). The structures and compound names of these catalysts are defined in Fig. 9.
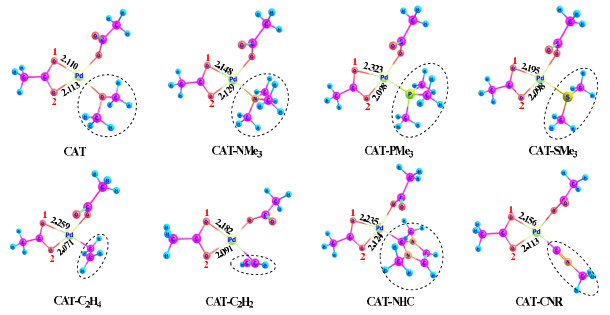
The neutral ligand Y exerts a trans effect on the O1 pathway, since it resides at the position trans to the O1 arm of κ2-OAc. The computed rate-determining activation barriers and overall free-energy changes are collected in Table 2. These ligands each hinder the C−H activation process as compared to the weakly σ-donating ligand OMe2, but their effects are less evident than those caused by the univalent ligands. In comparison to CAT for the O2 pathway, the NMe3 ligand substantially increases the barrier height by 6.6 kcal/mol, while all of the other ligands, except CNR, result in a small increase of the barrier height (by 1.4~3.3 kcal/mol). The different effects of NMe3 and PMe3 on the O2 pathway should be caused by steric effect rather than electronic effect, since the Pd−N and N−C bonds are shorter than Pd−P and P−C, which makes the phenyl and NMe3 groups more crowded. For the O1 pathway, however, CAT−PMe3 involves a high barrier of 28.4 kcal/mol and CAT−NMe3 shows a much lower barrier of 23.6 kcal/mol because PMe3 has stronger σ-donating ability than NMe3, and the former ligand could exert a more significant trans effect on the O1 pathway to hinder the C−H activation in progress. The different behaviors of PMe3 and NMe3 on the O1 and O2 pathways indicate the combined electronic and steric effects to be at work. For catalysts CAT−SMe2, CAT−C2H4, and CAT−C2H2, the free-energy barriers of the O1 and O2 pathways are close to each other.
DownLoad:
CSV
| Catalysts | Pathway | ∆G | ∆G⧧ |
| CAT | O1 | 4.9 | 18.7 |
| O2 | 10.6 | 21.4 | |
| CAT−NMe3 | O1 | 14.0 | 23.6 |
| O2 | 14.6 | 28.0 | |
| CAT−PMe3 | O1 | 24.2 | 28.4 |
| O2 | 9.6 | 24.7 | |
| CAT−SMe2 | O1 | 14.1 | 23.7 |
| O2 | 9.8 | 23.6 | |
| CAT−C2H4 | O1 | 11.0 | 22.1 |
| O2 | 7.8 | 23.2 | |
| CAT−C2H2 | O1 | 9.1 | 22.0 |
| O2 | 7.9 | 22.8 | |
| CAT−NHC | O1 | 24.4 | 29.3 |
| O2 | 12.2 | 23.4 | |
| CAT−CNR | O1 | 15.0 | 25.5 |
| O2 | 7.1 | 20.5 |
It seems that the catalyst CAT−NHC severely hinders the C−H activation on the O1 pathway, with the free-energy barrier of 29.3 kcal/mol that is even higher than that of CAT−PMe3, and hence, the trans effect of the NHC ligand should be significant. The thermodynamic aspect displays a similar trend. These indicate that the employment of a chiral NHC ligand in palladium-catalyzed C−H functionalizations could deliver the stereo-selectivity but might lower the reactivity towards C−H activation. Notably, as a σ-donating and π-accepting ligand, the CNR ligand appears to behave similarly to the CN ligand discussed before, since it could promote the O2 pathway (cis to CNR) but would hinder the O1 pathway (trans to CNR).
In order to save space, some of the transition-state geometries and typical molecular orbitals are given in the supporting information. For NMe3 and PMe3 systems, the calculated Kohn-Sham orbitals of the transition states indicate an overlap repulsion between the Pd···Ph and Pd−C bonds for both O1 and O2 pathways. The transition state of the NHC system on the O1 pathway has a similar high-lying molecular orbital. The transition state of the CNR system on the O2 pathway is featured by a conjugation between the π-bond of CNR and the developing Pd···Ph σ-lobe.
Dehydrogenative Heck reactions of arenes with alkenes furnish arylalkenes by direct C−H/C−H coupling. It was well established that the favorable pathway consists of three successive processes, including C−H activation of the arene, migratory insertion of the alkene, and β-H elimination (see Fig. 10, left part). This generally accepted mechanism demonstrates that dehydrogenation of the alkene is normally achieved by β-H elimination rather than C−H activation. Theoretically, the same arene-alkene cross-coupling could be realized through a different pathway, involving two C−H activation events and a reductive elimination step (Fig. 10, right part). On this pathway, after the C−H activation at the arene, the alkene would also undergo a C−H activation step to form an intermediate containing both the Pd−alkenyl and Pd−aryl bonds. Both two mechanisms shown in Fig. 10 are the typical PdⅡ/Pd0/PdⅡ catalytic cycles.
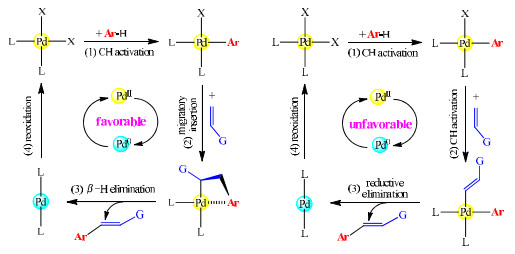
On the basis of the conclusions drawn in this work, we can explain why the conventional mechanism is plausible and the other mechanism is not. The C−H activation at the arene leading to an arylpalladium intermediate is always the first process on the whole catalytic pathway, but the following transformations are different. On the newly proposed pathway, the alkene is engaged in a C−H activation step instead of a migratory insertion step. However, the second C−H activation step at the same palladium center should be difficult due to the fact that the aryl ligand formed from the first C−H activation would hinder the C−H activation in progress. The strongly retarding effect of the phenyl ligand on C−H activation, as observed before, makes this pathway unlikely to be competitive.
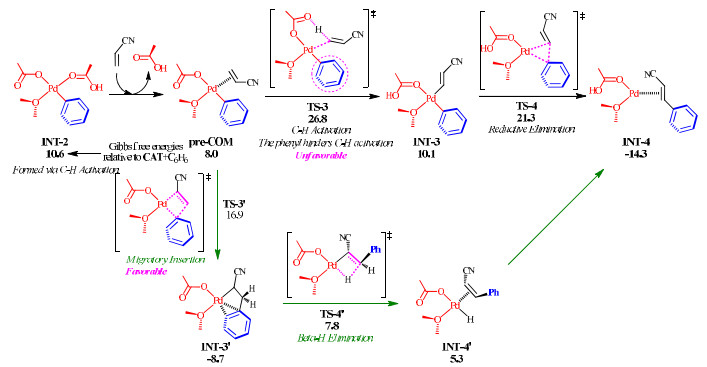
In order to support the above assumption, we performed DFT calculations on the two pathways (Fig. 11). The following discussion starts from INT-2, which is the intermediate product of the C−H activation of benzene with CAT. Using acrylonitrile as the alkene partner, we first characterize the conventional mechanism. The ligand exchange between acrylonitrile and acetic acid generates the precursor complex pre-COM. The cross-coupling between acrylonitrile and benzene results from the migratory insertion step via TS-3′, which affords INT-3′. Then, one of the two hydrogen atoms on the methylene group is transferred to palladium by the β-H elimination step via TS-4′, delivering INT-4′ with the product 2-phenyl acrylonitrile. On the other pathway, pre-COM undergoes the C−H activation of acrylonitrile through CMD mechanism via TS-3, generating INT-3 with two cis Pd−C bonds. The cross-coupling between the aryl and alkenyl groups is finished by the reductive elimination step via three-center transition state TS-4. TS-3′ is much more accessible than TS-3, because the total free-energy barrier of the C−H activation of acrylonitrile reaches at 26.8 kcal/mol and that of the migratory insertion step is merely 16.9 kcal/mol. Hence, the pathway involving two C−H activations could be ruled out. In the viewpoint of elementary steps, the C−H activation of acrylonitrile has the free-energy barrier of 18.8 kcal/mol (compare pre-COM to TS-3), while that of benzene with CAT is merely 13.2~13.4 kcal/mol (compare INT-1 with TS-2 in Fig. 3). These support that the palladium-catalyzed C−H activation would be hindered by a strongly σ-donating spectator ligand.
The electronic and steric environments of a transition metal could be tuned by their spectator ligands. This work aims at providing a quantitative measurement of the effects of diverse spectator ligands on palladium-catalyzed C−H activation. The main conclusions are summarized as follows:
(1) The univalent ligand generally influenced C−H activation more than the neutral ligand.
(2) The σ-donating ligand hindered the C−H activation in progress, and the retarding effect became more evident as the electron-donating ability of the ligand increased.
(3) The σ-donating ligand trans to C−H activation site exerted a larger effect than that cis to the C−H activation site.
(4) As σ-donating and π-accepting ligands, both cyanide and isonitrile hindered the C−H activation at their trans position, while they facilitated the C−H activation at their cis position.
(5) A chemical model was proposed to rationalize the observed ligand effects and was subsequently applied to mechanistic analyses on dehydrogenative Heck couplings.
Chen, X.; Engle, K. M.; Wang, D. H.; Yu, J. Q. Palladium(Ⅱ)-catalyzed C−H activation/C−C cross-coupling reactions: versatility and practicality. Angew. Chem. Int. Ed. 2009, 48, 5094−5115. doi: 10.1002/anie.200806273
Saini, G.; Kapur, M. Palladium-catalyzed functionalizations of acidic and non-acidic C(sp3)–H bonds – recent advances. Chem. Commun. 2021, 57, 1693−1714. doi: 10.1039/D0CC06892F
Chen, G.; Gong, W.; Zhuang, Z.; Andrä, M. S.; Chen, Y. Q.; Hong, X.; Yang, Y. F.; Liu, T.; Houk, K. N.; Yu, J. Q. Ligand-accelerated enantioselective methylene C(sp3)–H bond activation. Science 2016, 353, 1023–1027. doi: 10.1126/science.aaf4434
Wu, C.; McCollom, S. P.; Zheng, Z. P.; Zhang, J. D.; Sha, S. C.; Li, M. Y.; Walsh, P. J.; Tomson, N. C. Aryl fluoride activation through palladium-magnesium bimetallic cooperation: a mechanistic and computational study. ACS Catal. 2020, 10, 7934–7944. doi: 10.1021/acscatal.0c01301
Rocaboy, R.; Dailler, D.; Baudoin, O. A four-step synthesis of (±)-γ-lycorane via Pd0-catalyzed double C(sp2)–H/C(sp3)–H arylation. Org. Lett. 2018, 20, 772–775. doi: 10.1021/acs.orglett.7b03909
Zhang, Q.; Shi, B. F. From reactivity and regioselectivity to stereoselectivity: an odyssey of designing PIP amine and related directing groups for C−H activation. Chin. J. Chem. 2019, 37, 647–656. doi: 10.1002/cjoc.201900090
Shi, X. L.; Wang, Z. M.; Li, Y. X.; Li, X. W.; Li, X. Q.; Shi, D. Y. Palladium-catalyzed remote C−H phosphonylation of indoles at the C4 and C6 positions by a radical approach. Angew. Chem. Int. Ed. 2021, 60, 13871–13876. doi: 10.1002/anie.202103395
Li, H. T.; Huang, S. H.; Wang, Y. J.; Huo, C. D. Oxidative dehydrogenative [2+3]-cyclization of glycine esters with aziridines leading to imidazolidines. Org. Lett. 2018, 20, 92–95. doi: 10.1021/acs.orglett.7b03448
Wu, Y. B.; Xie, D.; Zang, Z. L.; Zhou, C. H.; Cai, G. X. Palladium-catalyzed aerobic regio- and stereo-selective olefination reactions of phenols and acrylates via direct dehydrogenative C(sp2)–O cross-coupling. Chem. Commun. 2018, 54, 4437–4440. doi: 10.1039/C8CC01226A
Rej, S.; Das, A.; Chatani, N. Strategic evolution in transition metal-catalyzed directed C−H bond activation and future directions. Coord. Chem. Rev. 2021, 431, 213683–37. doi: 10.1016/j.ccr.2020.213683
Li, R.; Xu, X.; Ye, M. Construction of medium rings via transition metal-catalyzed insertion of π-unsaturated compounds into C−H bonds. Chin. J. Org. Chem. 2020, 40, 3196−3202 doi: 10.6023/cjoc202005056
Carole, W. A.; Colacot, T. J. Understanding palladium acetate from a user perspective. Chem. Eur. J. 2016, 22, 7686−7695. doi: 10.1002/chem.201601450
Shao, Q.; Wu, K.; Zhuang, Z.; Qian, S.; Yu, J. Q. From Pd(OAc)2 to chiral catalysts: the discovery and development of bifunctional mono-N-protected amino acid ligands for diverse C−H functionalization reactions. Acc. Chem. Res. 2020, 53, 833−851. doi: 10.1021/acs.accounts.9b00621
Meng, G.; Lam, N. Y. S.; Lucas, E. L.; Saint-Denis, T. G.; Verma, P.; Chekshin, N.; Yu, J. Q. Achieving site-selectivity for C−H activation processes based on distance and geometry: a carpenter's approach. J. Am. Chem. Soc. 2020, 142, 10571−10591. doi: 10.1021/jacs.0c04074
Lin, T. Z.; Qian, P. C.; Wang, Y. E.; Ou, M. J.; Jiang, L.; Zhu, C.; Xu, Y. C.; Xiong, D.; Mao, J. Y. Palladium-catalyzed direct arylation of 2-pyridylmethyl silanes with aryl bromides. Org. Lett. 2021, 23, 3000−3003. doi: 10.1021/acs.orglett.1c00677
Halder, P.; Roy, T.; Das, P. Recent developments in selective N-arylation of azoles. Chem. Commun. 2021, 57, 5235−5249. doi: 10.1039/D1CC01265G
Thongpaen, J.; Manguin, R.; Baslé, O. Chiral N-heterocyclic carbene ligands enable asymmetric C−H bond functionalization. Angew. Chem. Int. Ed. 2020, 59, 10242–10251. doi: 10.1002/anie.201911898
Zhang, L.; Fang, D. C. An explicit interpretation of the directing group effect for the Pd(OAc)2-catalyzed aromatic C–H activations. J. Org. Chem. 2016, 81, 7400−7410. doi: 10.1021/acs.joc.6b00997
Zhou, J.; Zhou, Y.; Li, Y.; Zhang, J.; Zhang, L. DFT mechanistic study on palladium-catalyzed redox-neutral hydroarylation of unactivated alkenes with arylboronic acids. Asian J. Org. Chem. 2021, 10, 412−420. doi: 10.1002/ajoc.202000647
Davies, D. L.; Macgregor, S. A.; McMullin, C. L. Computational studies of carboxylate-assisted C–H activation and functionalization at group 8~10 transition metal centers. Chem. Rev. 2017, 117, 8649−8709. doi: 10.1021/acs.chemrev.6b00839
Ling, B.; Liu, Y.; Jiang, Y. Y.; Liu, P.; Bi, S. Mechanistic insights into the ruthenium-catalyzed [4+1] annulation of benzamides and propargyl alcohols by DFT studies. Organometallics 2019, 38, 1877−1886. doi: 10.1021/acs.organomet.8b00769
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A. Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision A. 02, Gaussian, Inc., Wallingford, CT 2009.
Becke, A. D. Density-functional thermochemistry. Ⅲ. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. doi: 10.1063/1.464913
Godbout, N.; Salahub, D. R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part Ⅰ. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560−571. doi: 10.1139/v92-079
Sosa, C.; Andzelm, J.; Elkin, B. C.; Wimmer, E.; Dobbs, K. D.; Dixon, D. A. A local density functional study of the structure and vibrational frequencies of molecular transition-metal compounds. J. Phys. Chem. 1992, 96, 6630−6636. doi: 10.1021/j100195a022
Scalmani, G.; Frisch, M. J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110−114124. doi: 10.1063/1.3359469
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787−1799. doi: 10.1002/jcc.20495
Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057−1065. doi: 10.1039/b515623h
Zhang, L.; Jiang, B.; Chen, Y.; Lv, J. F.; Feng, W. C. A computational study on the reaction mechanisms of nickel-catalyzed diarylation of alkenes. Eur. J. Org. Chem. 2019, 2019, 6217–6224. doi: 10.1002/ejoc.201900940
Zhou, Y.; Xue, R. C.; Feng, Y.; Zhang, L. How does HOTf/HFIP cooperative system catalyze the ring-opening reaction of cyclopropanes? A DFT study. Asian J. Org. Chem. 2020, 9, 311–316. doi: 10.1002/ajoc.202000031
Pearson, R. G. Antisymbiosis and the trans effect. Inorg. Chem. 1973, 12, 712–713. doi: 10.1021/ic50121a052

Figure 1 Two types of ligands affecting C−H bond cleavage. (a) MAPP serving as a reacting ligand; (b) group G serving as a spectator ligand

Figure 3 Mechanism and free energies (in kcal/mol) for C−H activation of CAT and benzene. Only the structures on the O2 pathway are given

Figure 5 Designed palladium catalysts with different univalent ligands (bond length in Å)

Figure 6 Three-dimensional geometries of the proton-transfer transition states (bond length in Å)

Figure 9 Designed palladium catalysts with different spectator ligands (bond length in Å)

Figure 10 Reaction pathways for dehydrogenative Heck couplings: the conventional mechanism (left) and a new mechanism (right)

Figure 11 Structures and free-energy variations (in kcal/mol) for the coupling between benzene and acrylonitrile through two different pathways
Table 1. Computed Free-energy Changes (∆G) and Barriers (∆G‡) for C−H Activation Reactions Mediated by Different Catalysts (in kcal/mol)
| Catalysts | Pathway | ∆G | ∆G⧧ |
| CAT | O1 | 4.9 | 18.7 |
| O2 | 10.6 | 21.4 | |
| CAT−OCH3 | O1 | 8.4 | 22.1 |
| O2 | 19.2 | 25.3 | |
| CAT−NH2 | O1 | 6.7 | 20.7 |
| O2 | 25.3 | 27.3 | |
| CAT−CH3 | O1 | 11.5 | 25.1 |
| O2 | 35.6 | 35.8 | |
| CAT−Ph | O1 | 9.8 | 23.7 |
| O2 | 33.7 | 32.8 | |
| CAT−CN | O1 | 0.5 | 18.3 |
| O2 | 18.6 | 25.2 | |
| CAT−F | O1 | 5.0 | 19.5 |
| O2 | 10.7 | 21.2 | |
| CAT−Cl | O1 | 2.0 | 19.1 |
| O2 | 10.5 | 21.1 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Computed Free-energy Changes (∆G) and Barriers (∆G‡) for C−H Activation Reactions Mediated by Different Catalysts (in kcal/mol)
| Catalysts | Pathway | ∆G | ∆G⧧ |
| CAT | O1 | 4.9 | 18.7 |
| O2 | 10.6 | 21.4 | |
| CAT−NMe3 | O1 | 14.0 | 23.6 |
| O2 | 14.6 | 28.0 | |
| CAT−PMe3 | O1 | 24.2 | 28.4 |
| O2 | 9.6 | 24.7 | |
| CAT−SMe2 | O1 | 14.1 | 23.7 |
| O2 | 9.8 | 23.6 | |
| CAT−C2H4 | O1 | 11.0 | 22.1 |
| O2 | 7.8 | 23.2 | |
| CAT−C2H2 | O1 | 9.1 | 22.0 |
| O2 | 7.9 | 22.8 | |
| CAT−NHC | O1 | 24.4 | 29.3 |
| O2 | 12.2 | 23.4 | |
| CAT−CNR | O1 | 15.0 | 25.5 |
| O2 | 7.1 | 20.5 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



