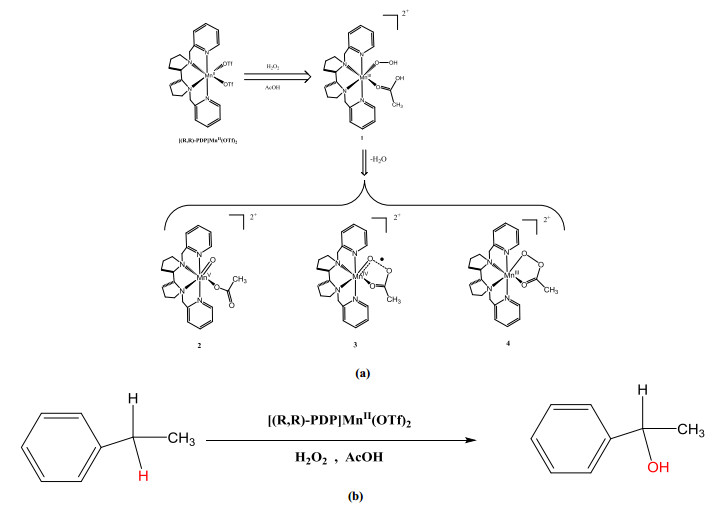
Scheme 1.
(a) Oxidation intermediates (2~4) obtained by the oxidation of [(R, R)-PDP]Mn(Ⅱ)(OTf)2 by H2O2 activation with carboxylic acid additives. (b) Selective catalytic hydroxylation of ethylbenzene by Mn(PDP) complexes

The activation of C–H bonds via hydroxylation is the focus of current research[1-5]. Many metalloenzymes, such as heme and non-heme iron enzymes, have remarkable activity toward the C–H bonds of substrates[6-9]. However, capturing the key intermediates in these catalytic reactions remains a difficult aspect of enzyme chemistry. The development of biomimetic chemistry has allowed the synthesis of metalloenzymes that can perform extremely challenging C–H bond activation reactions, such as the synthesis of iron porphyrins and similar compounds in cytochrome P450[10-13]. In recent years, a high-valent iron-oxo complex, [Fe(Ⅳ)(O)(Por)+·]+ (Cpd1), has been widely recognized as a key intermediate in the hydroxylation of alkanes. For example, Cpd1 can selectively abstract a hydrogen atom from a substrate to form [Fe(Ⅳ)(OH)(Por)]+ and substrate radicals[1]. According to the kinetic isotope effect experiments, H-abstraction is typically the key step in the entire reaction[14, 15]. After H-abstraction, the hydroxyl group in [Fe(Ⅳ)(OH)(Por)]+ quickly recombines with the substrate radical to give hydroxylation products, which is called the oxygen rebound mechanism[16, 17].
Recently, many Mn-oxo compounds have also been found to hydroxylate and oxidize C–H bonds selectively[18-20]. Similar to P450, manganese porphyrin compounds also activate the C–H bond of alkanes. By modifying manganese porphyrins, recombinant myoglobin can efficiently and selectively hydroxylate substrates such as ethylbenzene[21, 22]. Compounds containing chiral manganese complexes with carboxylic acid additives have been found to hydroxylate compounds like cyclohexane. Recently, Ottenbacher et al. have found that a [(PDP)Mn(Ⅱ)(OTf)2] complex (PDP = N,N΄-bis(pyridin-2-ylmethyl)-2,2΄-bipyrrolidine) is efficient for the selective hydroxylation of benzyl to form alcohols with high stereoselectivity[18]. However, the active intermediate has not yet been captured and the hydroxylation mechanism remains controversial.
Indirect experimental evidence has suggested that a Mn(Ⅴ)-oxo species, obtained by Mn(Ⅲ) activation with carboxylic acid and H2O2, is an active intermediate[23-25]. However, for other non-heme manganese complexes, an S = 3/2 signal has been observed using electron paramagnetic resonance spectroscopy, indicating that Mn(Ⅳ)-oxo species may also be reactive intermediates in catalytic reactions[26]. According to a study on the oxidation mechanism of non-heme iron, the electron transfer mechanism during H2O2 activation often determines the oxidation state of the metal center in a reactive intermediate[27]. To obtain a deeper understanding of the reaction process, we employed the density functional theory (DFT) method to explore the H2O2 activation of non-heme manganese complexes and the reaction mechanism of selective hydroxylation catalyzed by non-heme manganese oxidation intermediates. In particular, we studied the mechanism of a hydroxylation reaction catalyzed by a Mn-oxo species with a coordinated carboxylate (OAc). Similar to non-heme iron, we found three Mn(Ⅴ)-oxo species as possible oxidation intermediates, namely [(PDP)Mn(Ⅴ)(O)(OAc)]2+ (2), [(PDP)Mn(Ⅳ)(O···OC(O)CH3)2-·]2+ (3), and [(PDP)Mn(Ⅲ)(OOAc)-]2+ (4) (Scheme 1)[28]. For oxidation intermediate 3, AcOH played a "magic" role in the catalytic oxidation of non-heme manganese, with the carboxylic acid and Mn-oxo forming radicals with single-electron orbitals (σ*O–O) that assist the reaction. Finally, the formed oxidation intermediate, [Mn(Ⅳ)(O···OC(O)CH3)2-·]2+, acts as a two-electron oxidant to promote the single-step two-electron oxidation of a C–H bond[29].
In this study, DFT calculations were performed using the B3LYP hybrid density functional[30] with two basis sets: (a) the Def2-SVP basis set, denoted as B1, was used for all atoms to optimize the minimal point and transition state and to analyze the frequency; and (b) the Def2-TZVP basis set[31], denoted as B2, was used for all atoms to perform single-point energy corrections. Frequency calculations to check the reactants, intermediates, and products showed only real frequencies, whereas those for transition states showed single imaginary frequencies. The dispersion corrections computed using Grimme's D3BJ method were added to all calculations[32]. The self-consistent reaction field (SCRF) was used for optimization and single-point energy calculations. Acetonitrile (ε = 35.688) was introduced as the solvent using the polarizable continuum model (PCM)[33]. For the reactants, other functionals (B3P86, PBE0, TPSSh, M06L, etc.) were also tested. To consider the DFT self-interaction error[34] caused by multiple positive charges (+1 or +2) in the system, the OTf (Triflate, CF3SO3-) group was added to obtain neutral species for calculation. Similar results were obtained for the calculations in the presence and absence of added OTf ions. Zero-point energy (ZPE) corrections were calculated for all species, and energies reported in this work are B2 values with corresponding ZPE corrections (at the B1 level). All DFT calculations were performed using the Gaussian 09 software package[35].
Although there is little information available on non-heme manganese oxidation intermediates, there have been many studies on non-heme iron oxidation intermediates. According to previous reports[28, 36, 37], three non-heme iron oxidation intermediates, [Fe(Ⅴ)(O)(OAc)], [Fe(Ⅳ)(O)(OAc·)], and [Fe(Ⅲ)–OOAc], are in dynamic equilibrium. Considering that the structure of Mn(PDP) complex is similar to that of non-heme iron complexes, we simulated three possible oxidation intermediates (2~4). The optimized structures and relative energies of these oxidation intermediates are presented in Fig. 1. Oxidation intermediates 2~4 have triplet (S = 1) ground states (Table S1). In 32, the O2ac atom of the carboxylate is far away from the Mn=O moiety, and the distance between Ooxo and O2ac is 4.754 Å. In 33, the O2ac atom of the carboxylate is close to the Mn=O moiety, and the distance between Ooxo and O2ac is 2.117 Å. The distances between Mn and Ooxo in 32 and 33 are 1.676 and 1.650 Å, respectively. Some studies on non-heme iron have also suggested cyclic isomers as potential oxidation intermediates[27, 28]. In our calculations, cyclic isomer 34 has a shorter Ooxo–O2ac distance of 1.461 Å. A comparison of the relative energies of the triplet ground state of 2~4 showed that 33 is the most stable structure, being 11.4 and 8.1 kcal/mol lower in energy than 32 and 34, respectively.
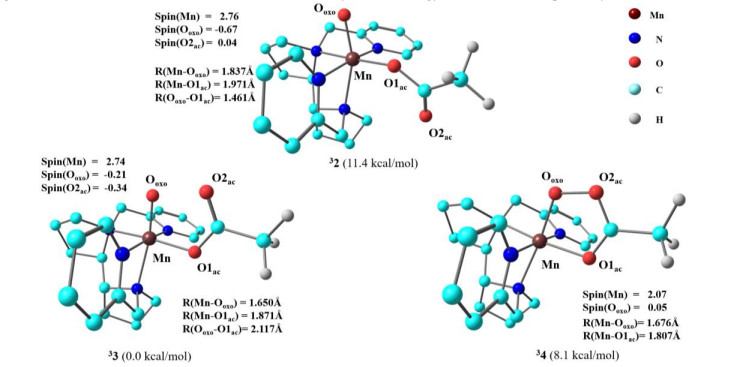
According to the spin density analysis of the oxidation intermediates in Fig. 1, the spin density of Mn in 33 is 2.74, whereas those of Ooxo and O2ac are –0.21 and –0.34, respectively. Interestingly, the spin density of Mn in 32 (2.74) is similar to that in 33, but that of O2ac is only 0.04 and that of Ooxo is –0.67. Furthermore, for 34, the spin density of Mn is only 2.07. To better understand the electronic structures of different oxidation intermediates, we examined their unrestricted corresponding orbitals (UCOs, Fig. 2).
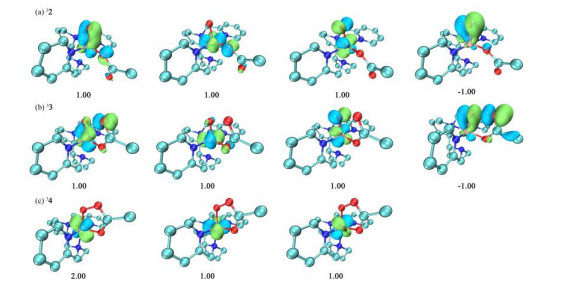
As shown in Fig. 2, 32 has three α-electron single-occupied orbitals corresponding to the Mn d-orbitals and a β-electron occupied orbital corresponding to the pOoxo orbital. By analogy to the electronic structure of many non-heme iron complexes[40, 41], the electronic structure of 32 corresponds to [Mn(Ⅴ)(O)(OAc)]2+. In contrast, 33 is shown to have three α-electron single-occupied orbitals corresponding to the Mn-d orbitals and a β-electron remaining in the σ*Ooxo+Oac orbital. Furthermore, the spin densities of O2ac and Ooxo (–0.34 and –0.21, respectively) indicate that the single β-electron is located on the orbital between O2ac and Ooxo. Similar conclusions have been reported in many non-heme iron calculation studies[27, 29, 42]. Therefore, it has been suggested that these structures contain OAc radicals[27], with an electronic structure of oxo-iron(Ⅳ)-AcO·. In contrast, based on CASSCF calculations of non-heme iron, Mondal et al.[29] proposed that the electronic structure is similar to [Fe(Ⅳ)(O…OC(O)CH3)2-·]2+. As the σ*O–O orbital in 33 is composed of Ooxo and O2ac instead of being localized on the carboxylate, we regard the electronic structure of 33 as [Mn(Ⅳ)(O…OC(O)CH3)2-·]2+. Cyclic isomer 34 has one filled d-orbital and two α-electron single-occupied orbitals, consistent with an electronic structure of [Mn(Ⅲ)-OOAc]2+.
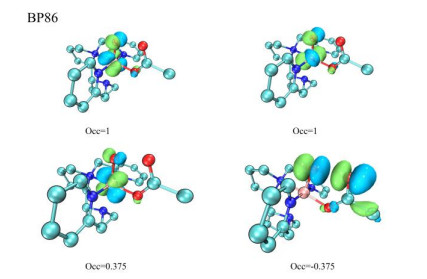
Since the electronic configuration of 33 has a special σ*Ooxo+Oac orbital, in order to consider whether this is only a special case of the B3LYP functional, we used different DFT functionals to clarify the electronic configuration of 33. Table 1 shows key data for the structure of 33 optimized by using different DFT functionals. All the optimized structures have similar geometries and electronic structures. For example, the Ooxo–Mn–O1ac–O2ac dihedral angle is usually less than 10°, and the distance between O2ac and Ooxo is approximately 2.1 Å. Furthermore, the spin natural orbitals (SNOs) obtained using different DFT functionals reveal an σ*O–O orbital (Table 1 and Fig. 3). Although the occupancy rates of the σ*O-O orbitals differ, all the DFT functionals assume that a σ*O–O orbital exists in 33. Therefore, we believe that the active intermediate is a Mn(Ⅴ)-oxo species but intermediate 33 has the characteristics of [Mn(Ⅳ)(O···OC(O)CH3)2-·]2+, where S(Mn) = 3/2 and a σ*O–O radical is present.
 DownLoad:
CSV
DownLoad:
CSV
| Spin (Mn) | Spin (Ooxo) | Spin (O2ac) | LOBA (Mn) | Ooxo–Mn–O1ac–O2ac | d (OoxoO2ac) | d (Mn–Ooxo) | Mayer (Mn–O) |
Occ (σ*O-O) | |
| B3LP | 2.74 | –0.21 | –0.34 | 4 | 5.880 | 2.090 | 1.650 | 1.667 | –0.754 |
| B3P86 | 2.74 | –0.19 | –0.34 | 4 | 5.880 | 2.090 | 1.639 | 1.681 | –0.741 |
| BLYP | 2.15 | 0.10 | –0.17 | 4 | 5.684 | 2.268 | 1.632 | 1.924 | –0.400 |
| BP86 | 2.13 | 0.11 | –0.16 | 4 | 5.471 | 2.230 | 1.621 | 1.928 | –0.376 |
| M06L | 2.73 | –0.20 | –0.30 | 4 | 10.402 | 2.179 | 1.648 | 1.654 | –0.679 |
| PBE0 | 2.90 | –0.30 | –0.35 | 4 | 9.282 | 2.092 | 1.648 | 1.612 | –0.798 |
| TPSS | 2.26 | 0.04 | –0.21 | 4 | 6.129 | 2.194 | 1.629 | 1.856 | –0.471 |
| TPSH | 2.60 | –0.13 | –0.31 | 4 | 5.803 | 2.127 | 1.641 | 1.721 | –0.677 |
| Occa: Occupied number of σ*O–O orbitals in SNO (spin natural orbital) | |||||||||
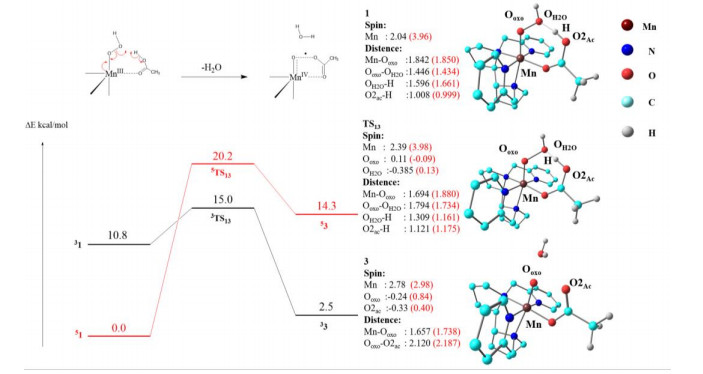
To further study the relationship between oxidation intermediate 3 and other oxidants, we investigated the formation mechanism of oxidation intermediate 3 from precursor 1. DFT calculations suggested that complex 1, [(PDP)Mn(Ⅲ)(OOH)(AcOH)], undergoes an O–OH bond homolytic cleavage process via the transfer of a hydrogen atom from AcOH, resulting in the generation of H2O and the formation of complex 3. Fig. 4 shows the formation mechanism results for the triplet (S = 1) and quintet (S = 2) states. According to previous theoretical results[43, 44], the ground state of complex 1 is a quintet state, and the calculated energy of the triplet state is 10.8 kcal/mol higher than that of the ground quintet state. After cleavage of the O–OH bond, oxidation intermediate 3, [(PDP)Mn(Ⅳ)(O···OC(O)CH3)2-·]2+, is formed. The lowest transition state is 3TS13 with an activation energy of 15.0 kcal/mol. The direct cleavage of the O–OH bond of complex 1 on the triplet state surface generates the most stable oxidation intermediate (3). We also examined the formation mechanism of oxidation intermediate 4, [(PDP)Mn(Ⅲ)(OOAc)-]2+. An energy barrier of 6.5 kcal/mol exists for bond formation between Ooxo and O2ac to obtain cyclic intermediate 4, and the relative energy of 4 is higher than that of 3 (Fig. S2). Therefore, the theoretical results clearly show that in the presence of acetic acid, 3 is the most likely oxidant in the non-heme manganese catalytic system.
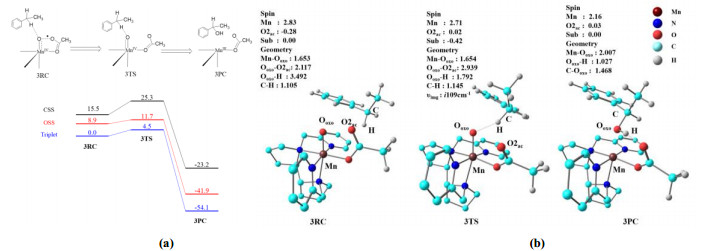
Subsequently, we studied the hydroxylation reaction with oxidation intermediate 3 to establish the C–H bond activation ability of this species. According to the above analysis of oxidation intermediate 3, the σ*O–O orbital with radical character spans the Ooxo and O2ac atoms. Therefore, it was speculated that either O2ac or Ooxo could abstract the H atom during the H-abstraction process. However, our calculation results showed that H-abstraction from the substrate by Ooxo is much more favorable, with an energy barrier of 4.5 kcal/mol on the triplet state surface (Fig. 5a). In contrast, H-abstraction from the substrate by O2ac is more difficult, with a higher activation energy of 14.5 kcal/mol (Fig. S5).
As shown in Fig. 5b, 33RC is composed of oxidation intermediate 33 and ethylbenzene. In 33RC, the Mn, Ooxo, and OAc groups (O2ac, O1ac, and C) are almost on the same plane, and the distance between Ooxo and O2ac is 2.117 Å. The substrate, ethylbenzene, is far away from oxidation intermediate 3, with a H–Ooxo distance of 3.492 Å. During the H-abstraction process, when the Ooxo–H distance is approximately 1.792 Å, transition state 33TS is formed. Fig. 5b shows the geometric structure of 33TS, which is a "reactant-like" transition state. The characteristics of this early transition state are consistent with a lower energy barrier, as the C–H bond is only slightly stretched (1.145 Å) and the Ooxo–H bond is long (1.792 Å). Notably, the Ooxo–O2ac distance increased from 2.117 Å in 33RC to 2.939 Å in 33TS. During the formation of the transition state, the substrate spin density changes from 0 in 33RC to –0.42 in 33TS, and the spin density of the OAc group changes from –0.28 in 33RC to –0.02 in 33TS. These observations indicate that C–H bond cleavage proceeds, with the electrons from σC–H in the substrate being transferred to the σ*O–O orbital, leading to a longer distance between Ooxo and O2ac. Upon completion of the H-abstractions process, the reaction proceeds directly to the hydroxylated product. According to the previous analysis of the H2O2 activation mechanism, the hydroxylated product is then replaced by a H2O2 molecule to re-form complex 1 and start a new reaction cycle.
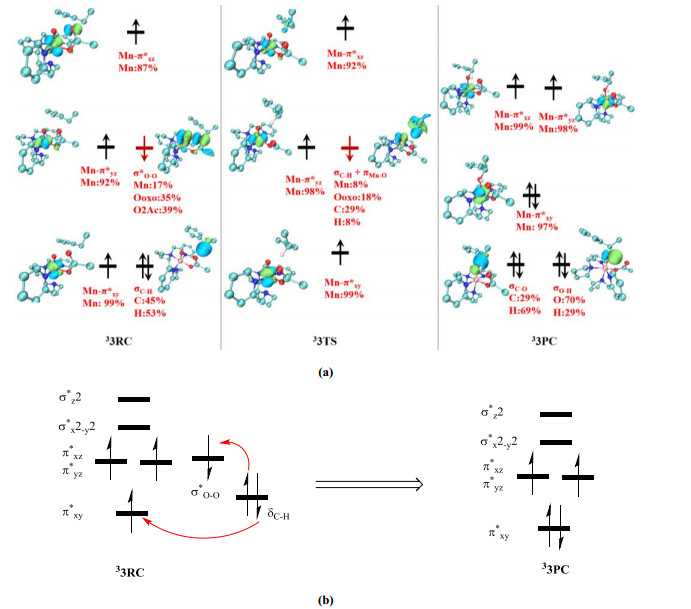
To understand the hydroxylation reactivity of 3, we carefully analyzed the evolution of its electronic structure during the hydroxylation process. For 33RC (Fig. 6), there are three α-electron single-occupied orbitals (π*xy, π*xz, and π*yz), which mainly correspond to the d-orbitals (dxy, dxz, and dyz) of Mn, and one β-electron single-occupied orbital corresponding to σ*O–O (35% Ooxo + 39% O2ac). In 33TS, the C–H bond approaches the Ooxo atom at a Mn–Ooxo–H angle of 113°. The calculated valence orbitals of 33TS reveal that the three single-occupied orbitals on the Mn atom do not change, but the β-electron single-occupied orbital changes from the σ*O–O orbital in 33RC to a newly mixed orbital of σC–H and πMn–O (Mn: 8%, Ooxo: 18%, C: 29%, H: 8%) in 33TS (Fig. 6a). This obvious change in the valence orbital indicates that the α-electron of σC–H is transferred to the σ*O–O orbital during the formation of 33TS. However, as the newly formed mixed orbital in 33TS mainly consists of the σC–H orbital with a small contribution from the Mn-d orbital, subsequent β-electron transfer from the mixed orbital to the α-electron single-occupied π*xy orbital could be favorable, resulting in the formation of 33PC. These results show that oxidation intermediate 3 has two single electron-accepting orbitals (α Mn-dxy and β σ*O–O), which can accept two electrons in a single step. Fig. 6b shows a schematic diagram of the electron transfer processes during C–H bond activation. The two electrons of the σC–H orbital in the reactant could be transferred in a single step to obtain the hydroxylated product quickly. This electronic structure analysis verifies that oxidation intermediate 3 can act as a two-electron oxidant for C–H bond activation[29].
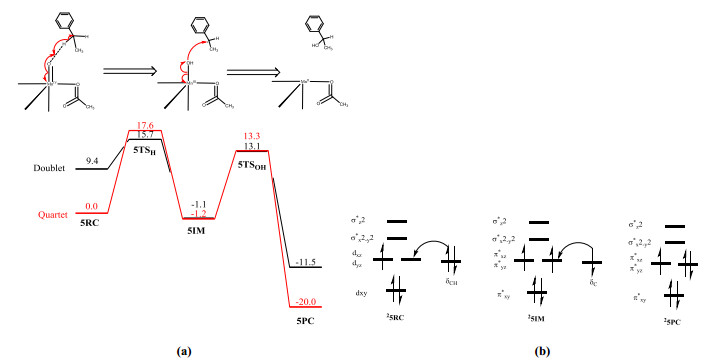
To confirm the advantages of the [(L)Mn(Ⅳ)(O···OC(O)CH3)2-·]2+ species (3) for the hydroxylation reaction, we also studied the hydroxylation reaction mechanism of the Mn(Ⅳ)-oxo species (5). Similar to many other non-heme species[45], the ground state of the Mn(Ⅳ)-oxo species is usually a quartet state, with the energy of the doublet being 9.4 kcal/mol higher than that of the quartet (Fig. 7a). The C–H bond hydroxylation reaction with the Mn(Ⅳ)-oxo species requires two steps. First, a stable intermediate configuration is obtained by a H-abstraction process, and then the hydroxylated product is obtained by oxygen rebound. Both the doublet and quartet states have high activation energies (15.7 and 17.6 kcal/mol, respectively) for the H-abstraction process. A high activation energy is also required for oxygen rebound from the stable intermediate. As shown in Fig. 7b, for the double state (S = 1/2), in the first step, one α-electron of σC–H is transferred to πyz* with one β-electron remaining on ethylbenzene. In the second step, during oxygen rebound, the electron on the ethylbenzene radical is transferred to π*yz. Unlike the single-step electron transfer process with oxidation intermediate 3, a two-step electron transfer process is required with the Mn(Ⅳ)-oxo species and the theoretical reaction rate is very low. Therefore, the reaction of ethylbenzene with the Mn(Ⅳ)-oxo species is much less favorable than that with oxidation intermediate 3.
In summary, we performed DFT calculations to explore the activation and hydroxylation mechanisms of a non-heme manganese complex. The calculation results showed that after H2O2 activation, the non-heme manganese complex can form Mn(Ⅴ)-oxo intermediates, with oxidation intermediate 3 being the main species involved in the hydroxylation reaction. This oxidation intermediate corresponds to [(PDP)Mn(Ⅳ)(O…OC(O)CH3)2-·]2+ with a S(Mn) = 3/2 center and a σ*O–O radical, where the O–O bond is not completely broken. With oxidation intermediate 3, C–H bond hydroxylation was found to proceed via a single step to produce a hydroxylated product. Our analysis of the changes to the electronic structure of oxidation intermediate 3 during C–H bond hydroxylation revealed the presence of two electron-accepting orbitals (α Mn-dxy and β σ*O–O), which could simultaneously interact with the doubly occupied electron-donating orbital (σC–H) of the substrate. Therefore, oxidation intermediate 3 can act as a two-electron oxidant for C–H bond activation. In comparison, the hydroxylation reaction with [(PDP)Mn(Ⅳ)(O)(OAc)]+ required a higher activation energy. Therefore, the σ*O–O orbital formed by O2ac and Ooxo is beneficial to the reaction system and explains the experimental observation of improved reactivity in the presence of carboxylates. These findings provide new insights into the catalytic effects of additives such as acetic acid on the non-heme Mn/H2O2 system, which may aid experimentalists in designing improved catalysts with higher efficiencies.
Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 enzymes: their structure, reactivity, and selectivity-modeled by QM/MM calculations. Chem. Rev. 2010, 110, 949−1017. doi: 10.1021/cr900121s
Lu, Q.; Song, J.; Wu, P.; Li, C.; Thiel, W. Mechanistic insights into the directing effect of Thr303 in ethanol oxidation by cytochrome P450 2E1. ACS Catal. 2019, 9, 4892−4901. doi: 10.1021/acscatal.9b00907
Lu, J.; Wang, B.; Shaik, S.; Lai, W. QM/MM calculations reveal the important role of α-heteroatom substituents in controlling selectivity of mononuclear nonheme HppE-catalyzed reactions. ACS Catal. 2020, 10, 9521−9532. doi: 10.1021/acscatal.0c01803
Yan, X.; Song, J.; Lu, Q.; Li, C. Mechanistic insights into the crucial roles of Glu76 residue in nickel-dependent quercetin 2, 4-dioxygenase for quercetin oxidative degradation. J. Catal. 2020, 387, 73−83. doi: 10.1016/j.jcat.2020.04.016
Kalita, S.; Shaik, S.; Kisan, H. K.; Dubey, K. D. A paradigm shift in the catalytic cycle of P450 – the preparatory choreography during O2 binding, and origins of the necessity for two protonation pathways. ACS Catal. 2020, 10, 11481−11492. doi: 10.1021/acscatal.0c02775
Wang, J. B.; Huang, Q.; Peng, W.; Wu, P.; Yu, D.; Chen, B.; Wang, B.; Reetz, M. T. P450-BM3-catalyzed sulfoxidation versus hydroxylation: a common or two different catalytically active species? J. Am. Chem. Soc. 2020, 142, 2068−2073. doi: 10.1021/jacs.9b13061
Wang, B.; Cao, Z.; Rovira, C.; Song, J.; Shaik, S. Fenton-derived OH radicals enable the MPnS enzyme to convert 2-hydroxyethylphosphonate to methylphosphonate: insights from ab initio QM/MM MD simulations. J. Am. Chem. Soc. 2019, 141, 9284−9291. doi: 10.1021/jacs.9b02659
Li, H.; Liu, Y. Mechanistic investigation of isonitrile formation catalyzed by the nonheme iron/α-KG-D ependent decarboxylase (ScoE). ACS Catal. 2020, 10, 2942−2957. doi: 10.1021/acscatal.9b05411
Su, H.; Wang, B.; Shaik, S. Quantum-mechanical/molecular-mechanical studies of CYP11A1-catalyzed biosynthesis of pregnenolone from cholesterol reveal a C−C bond cleavage reaction that occurs by a compound Ⅰ-mediated electron transfer. J. Am. Chem. Soc. 2019, 141, 20079−20088. doi: 10.1021/jacs.9b08561
Li, G.; Kates, P. A.; Dilger, A. K.; Cheng, P. T.; Ewing, W. R.; Groves, J. T. Manganese-catalyzed desaturation of N-acyl amines and ethers. ACS Catal. 2019, 9, 9513−9517. doi: 10.1021/acscatal.9b03457
Rebelo, S. L. H.; Pires, S. M. G.; Simões, M. M. Q.; de Castro, B.; Neves, M. G. P. M. S.; Medforth, C. J. Biomimetic oxidation of benzofurans with hydrogen peroxide catalyzed by Mn(Ⅲ) porphyrins. Catalysts 2020, 10−14.
Liu, W.; Groves, J. T. Manganese catalyzed C−H halogenation. Acc. Chem. Res. 2015, 48, 1727−35. doi: 10.1021/acs.accounts.5b00062
Li, G.; Dilger, A. K.; Cheng, P. T.; Ewing, W. R.; Groves, J. T. Selective C−H halogenation with a highly fluorinated manganese porphyrin. Angew. Chem. Int. Ed. 2018, 57, 1251−1255. doi: 10.1002/anie.201710676
Mandal, D.; Mallick, D.; Shaik, S. Kinetic isotope effect determination probes the spin of the transition state, its stereochemistry, and its ligand sphere in hydrogen abstraction reactions of oxoiron(Ⅳ) complexes. Acc. Chem. Res. 2018, 51, 107−117. doi: 10.1021/acs.accounts.7b00442
Li, C. S.; Wu, W.; Cho, K. B.; Shaik, S. Oxidation of tertiary amines by cytochrome P450-kinetic isotope effect as a spin-state reactivity probe. J. Am. Chem. Soc. 2009, 15, 8492−8503.
Kamachi, T.; Yoshizawa, K. A theoretical study on the mechanism of camphor hydroxylation by compound Ⅰ of cytochrome P450. J. Am. Chem. Soc. 2003, 125, 4652−4661. doi: 10.1021/ja0208862
Cho, K. B.; Hirao, H.; Shaik, S.; Nam, W. To rebound or dissociate? This is the mechanistic question in C–H hydroxylation by heme and nonheme metal-oxo complexes. Chem. Soc. Rev. 2016, 45, 1197−1210. doi: 10.1039/C5CS00566C
Ottenbacher, R. V.; Talsi, E. P.; Bryliakov, K. P. Highly enantioselective undirected catalytic hydroxylation of benzylic CH2 groups with H2O2. J. Catal. 2020, 390, 170−177. doi: 10.1016/j.jcat.2020.08.005
Srour, H.; Le Maux, P.; Simonneaux, G. Enantioselective manganese-porphyrin-catalyzed epoxidation and C–H hydroxylation with hydrogen peroxide in water/methanol solutions. Inorg. Chem. 2012, 51, 5850−6. doi: 10.1021/ic300457z
Song, W. J.; Seo, M. S.; George, S. D.; Ohta, T.; Song, R.; Kang, M. J.; Tosha, T.; Kitagawa, T.; Solomon, E. I.; Nam, W. Synthesis, characterization, and reactivities of manganese(Ⅴ)-oxo porphyrin complexes. J. Am. Chem. Soc. 2007, 129, 1268−1277. doi: 10.1021/ja066460v
Oohora, K.; Kihira, Y.; Mizohata, E.; Inoue, T.; Hayashi, T. C(sp3)–H bond hydroxylation catalyzed by myoglobin reconstituted with manganese porphycene. J. Am. Chem. Soc. 2013, 135, 17282−17285. doi: 10.1021/ja409404k
Ohora, K.; Meichin, H.; Kihira, Y.; Sugimoto, H.; Shiro, Y.; Hayashi, T. Manganese(Ⅴ) porphycene complex responsible for inert C–H bnd hydroxylation in a myoglobin matrix. J. Am. Chem. Soc. 2017, 139, 18460−18463. doi: 10.1021/jacs.7b11288
Balcells, D.; Raynaud, C.; Crabtree, R. H.; Eisenstein, O. C−H oxidation by hydroxo manganese(Ⅴ) porphyrins: a DFT study. Chem. Commun. 2009, 13, 1772−4.
Nuhant, P.; Oderinde, M. S.; Genovino, J.; Juneau, A.; Gagne, Y.; Allais, C.; Chinigo, G. M.; Choi, C.; Sach, N. W.; Bernier, L.; Fobian, Y. M.; Bundesmann, M. W.; Khunte, B.; Frenette, M.; Fadeyi, O. O. Visible-light-initiated manganese catalysis for C–H alkylation of heteroarenes: applications and mechanistic studies. Angew. Chem. Int. Ed. 2017, 56, 15309−15313. doi: 10.1002/anie.201707958
Li, X. X.; Guo, M.; Qiu, B.; Cho, K. B.; Sun, W.; Nam, W. High-spin Mn(Ⅴ)-oxo intermediate in nonheme manganese complex-catalyzed alkane hydroxylation reaction: experimental and theoretical approach. Inorg. Chem. 2019, 58, 14842−14852. doi: 10.1021/acs.inorgchem.9b02543
Biswas, S.; Mitra, A.; Banerjee, S.; Singh, R.; Das, A.; Paine, T. K.; Bandyopadhyay, P.; Paul, S.; Biswas, A. N. A high spin Mn(Ⅳ)-oxo complex generated via stepwise proton and electron transfer from Mn(Ⅲ)-hydroxo precursor: characterization and C–H bond cleavage reactivity. Inorg. Chem. 2019, 58, 9713−9722. doi: 10.1021/acs.inorgchem.9b00579
Wang, Y.; Janardanan, D.; Usharani, D.; Han, K.; Que, L.; Shaik, S. Nonheme iron oxidant formed in the presence of H2O2 and acetic acid is the cyclic ferric peracetate complex, not a perferryloxo complex. ACS Catal. 2013, 3, 1334−1341. doi: 10.1021/cs400134g
Oloo, W. N.; Banerjee, R.; Lipscomb, J. D.; Que, L. Jr. Equilibrating (L)Fe(Ⅲ)−OOAc and (L)Fe(Ⅴ)(O) species in hydrocarbon oxidations by bio-inspired nonheme iron catalysts using H2O2 and AcOH. J. Am. Chem. Soc. 2017, 139, 17313−17326. doi: 10.1021/jacs.7b06246
Mondal, B.; Neese, F.; Bill, E.; Ye, S. Electronic structure contributions of non-heme oxo-iron(Ⅴ) complexes to the reactivity. J. Am. Chem. Soc. 2018, 140, 9531−9544. doi: 10.1021/jacs.8b04275
Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. Ab-initio calculation of vibrational absorption and circular-dichroism spectra using density-functional force-fields. J. Phys. Chem. 1994, 98, 11623−11627. doi: 10.1021/j100096a001
Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297−3305. doi: 10.1039/b508541a
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H~Pu. J. Chem. Phys. 2010, 132, 154104−20. doi: 10.1063/1.3382344
Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999−3093. doi: 10.1021/cr9904009
Johansson, A. J.; Blomberg, M. R. A.; Siegbahn, P. E. M. Quantum chemical modeling of the oxidation of dihydroanthracene by the biomimetic nonheme iron catalyst (TMC)Fe-Ⅳ(O)2+. J. Phys. Chem. C 2007, 111, 12397−12406. doi: 10.1021/jp0730444
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A. Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision D. 01, Gaussian, Inc. : Wallingford, CT 2013.
Dantignana, V.; Serrano-Plana, J.; Draksharapu, A.; Magallon, C.; Banerjee, S.; Fan, R.; Gamba, I.; Guo, Y.; Que, L, Jr.; Costas, M.; Company, A. Spectroscopic and reactivity comparisons between nonheme oxoiron(Ⅳ) and oxoiron(Ⅴ) species bearing the same ancillary ligand. J. Am. Chem. Soc. 2019, 141, 15078−15091. doi: 10.1021/jacs.9b05758
Fan, R.; Serrano-Plana, J.; Oloo, W. N.; Draksharapu, A.; Delgado-Pinar, E.; Company, A.; Martin-Diaconescu, V.; Borrell, M.; Lloret-Fillol, J.; Garcia-Espana, E.; Guo, Y.; Bominaar, E. L.; Que, L. Jr.; Costas, M.; Munck, E. Spectroscopic and DFT characterization of a highly reactive nonheme Fe(Ⅴ)-oxo intermediate. J. Am. Chem. Soc. 2018, 140, 3916−3928. doi: 10.1021/jacs.7b11400
Lu, T.; Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580−592. doi: 10.1002/jcc.22885
Humphrey, W.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 1996, 14, 33−38. doi: 10.1016/0263-7855(96)00018-5
Chen, H.; Lai, W.; Yao, J.; Shaik, S. Perferryl Fe-v-oxo nonheme complexes: do they have high-spin or low-spin ground states? J. Chem. Theory Comput. 2011, 7, 3049−3053. doi: 10.1021/ct200614g
Bassan, A.; Blomberg, M. R. A.; Siegbahn, P. E. M.; Que, L. A density functional study of O–O bond cleavage for a biomimetic non-heme iron complex demonstrating an Fe-Ⅴ-intermediate. J. Am. Chem. Soc. 2002, 124, 11056−11063. doi: 10.1021/ja026488g
Zima, A. M.; Lyakin, O. Y.; Bryliakov, K. P.; Talsi, E. P. High-spin and low‐spin perferryl intermediates in Fe(PDP)-catalyzed epoxidations. ChemCatChem. 2019, 11, 5345−5352. doi: 10.1002/cctc.201900842
Dai, W.; Shang, S.; Lv, Y.; Li, G.; Li, C.; Gao, S. Highly chemoselective and enantioselective catalytic oxidation of heteroaromatic sulfides via high-valent manganese(Ⅳ)-oxo cation radical oxidizing intermediates. ACS Catal. 2017, 7, 4890−4895. doi: 10.1021/acscatal.7b00968
Miao, C.; Wang, B.; Wang, Y.; Xia, C.; Lee, Y. M.; Nam, W.; Sun, W. Proton-promoted and anion-enhanced epoxidation of olefins by hydrogen peroxide in the presence of nonheme manganese catalysts. J. Am. Chem. Soc. 2016, 138, 936−943. doi: 10.1021/jacs.5b11579
Rice, D. B.; Massie, A. A.; Jackson, T. A. Experimental and multireference ab initio investigations of hydrogen-atom-transfer reactivity of a mononuclear Mn(Ⅳ)-oxo complex. Inorg. Chem. 2019, 58, 13902−13916. doi: 10.1021/acs.inorgchem.9b01761

Scheme 1 (a) Oxidation intermediates (2~4) obtained by the oxidation of [(R, R)-PDP]Mn(Ⅱ)(OTf)2 by H2O2 activation with carboxylic acid additives. (b) Selective catalytic hydroxylation of ethylbenzene by Mn(PDP) complexes

Figure 1 Optimized structures of oxidation intermediates 2~4 and spin densities for the lowest spin state. Bond lengths are in Å unit. The relative energies (based on oxidation intermediate 33), including zero-point energy (ZPE) and solvent corrections, were calculated at the UB3LYP/B2 level. The hydrogen atoms on the ligand skeleton are omitted for clarity

Figure 2 Unrestricted corresponding orbitals (UCOs) and occupation numbers for oxidation intermediates (a) 2, (b) 3, and (c) 4. A positive occupation number indicates α occupation and a negative occupation number indicates β occupation. Only the double-occupied d orbitals of the Mn atoms are shown. All orbital graphics were created using Multiwfn[38] and VMD software[39]

Figure 3 Spin natural orbitals (SNOs) and corresponding occupation numbers for 33 at the BP86/B1 level. A negative occupation number corresponding to a β spin

Figure 4 Energy profiles for the conversion of 1 to 3 with H2O2 (in kcal/mol). The energies, including zero-point energy (ZPE) and solvent corrections, were calculated at the UB3LYP/B2 level

Figure 5 (a) Energy profiles for the hydroxylation of oxidation intermediate 3 with ethylbenzene (in kcal/mol). The energies, including zero-point energy (ZPE) and solvent corrections were calculated at the UB3LYP/B2 level (OSS: open-shell singlet, CSS: closed-shell singlet); (b) Structures of oxidation intermediate3 3 during hydroxylation. Distances (in Å), spin densities, and imaginary frequencies are shown

Figure 6 (a) Schematic UCO diagrams for the H-abstraction reactant (33RC), transition state (33TS), and product (33PC). (b) Diagram of orbital occupancy and electron transfer during hydroxylation

Figure 7 Hydroxylation mechanism of ethylbenzene with Mn(Ⅳ)-oxo species 5. (a) Energy profiles for the hydroxylation of ethylbenzene with 5 (in kcal/mol). The energies, including zero-point energy (ZPE) and solvent corrections, were calculated at the UB3LYP/B2 level. (b) Diagram of orbital occupancy and electron transfer during hydroxylation
Table 1. Optimization of 33 Using Different DFT Functionals
| Spin (Mn) | Spin (Ooxo) | Spin (O2ac) | LOBA (Mn) | Ooxo–Mn–O1ac–O2ac | d (OoxoO2ac) | d (Mn–Ooxo) | Mayer (Mn–O) |
Occ (σ*O-O) | |
| B3LP | 2.74 | –0.21 | –0.34 | 4 | 5.880 | 2.090 | 1.650 | 1.667 | –0.754 |
| B3P86 | 2.74 | –0.19 | –0.34 | 4 | 5.880 | 2.090 | 1.639 | 1.681 | –0.741 |
| BLYP | 2.15 | 0.10 | –0.17 | 4 | 5.684 | 2.268 | 1.632 | 1.924 | –0.400 |
| BP86 | 2.13 | 0.11 | –0.16 | 4 | 5.471 | 2.230 | 1.621 | 1.928 | –0.376 |
| M06L | 2.73 | –0.20 | –0.30 | 4 | 10.402 | 2.179 | 1.648 | 1.654 | –0.679 |
| PBE0 | 2.90 | –0.30 | –0.35 | 4 | 9.282 | 2.092 | 1.648 | 1.612 | –0.798 |
| TPSS | 2.26 | 0.04 | –0.21 | 4 | 6.129 | 2.194 | 1.629 | 1.856 | –0.471 |
| TPSH | 2.60 | –0.13 | –0.31 | 4 | 5.803 | 2.127 | 1.641 | 1.721 | –0.677 |
| Occa: Occupied number of σ*O–O orbitals in SNO (spin natural orbital) | |||||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们