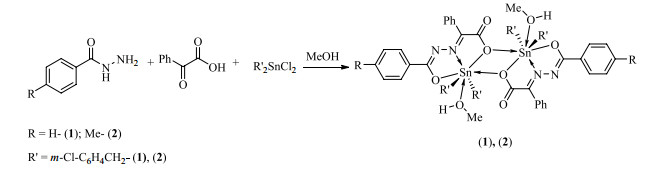
Figure 1.
Synthesis routes of the compounds
Syntheses, Crystal Structures and in vitro Anticancer Activities of Dibenzyltin Compounds Based on the N-(2-Phenylacetic acid)-aroyl Hydrazone
Wu-Jiu JIANG , Tian-Zi MO , Fu-Xing ZHANG , Dai-Zhi KUANG , Yu-Xing TAN

Cancer is a serious threat to human life and health as one of the important causes of human disease death. In the process of clinical use, cisplatin[1], a metal chemotherapy drug for early cancer treatment, has exposed some problems such as high toxicity and drug resistance. With further understanding of pharmacological action, many new metal compounds with anti-cancer activities have been synthesized[2-4]. Dialkyltin compounds have been concerned due to their similar structures to cisplatin and better anticancer activities in vitro than cisplatin and other platinum drugs[5, 6]. Researches show the biological activity of organotin compounds has been determined with the organic groups connected to tin atoms and the ligands coordinated with them[7, 8]. Therefore, we can obtain metal anticancer compounds with different activities with different organic groups or ligands.
Hydrazone is obtained by dehydration after nucleophilic addition reaction between hydrazine and aldehyde or ketone, and is a special class of Schiff base compounds with –CONHN=CH–. In addition, the groups are found such acyl, sub-amino and imide in the hydrazone compounds. The stability of these compounds is good because the unshared electron of nitrogen atom on sub-amino group can form p-π conjugation system with C=N groups of acyl group and imide. Furthermore, hydrazone compounds contain imino-nitrogen and carbonyl-oxygen atoms, as well as other coordination atoms with strong coordination ability, which can form stable compounds with transition metals, rare earth metals, major metals and nonmetals. Many experiments showed that the biological activity of acylhydrazone compounds was enhanced significantly compared with the corresponding ligands, and the compounds had extensive biological and drug activities[9-11].
Based on the previous researches, in the work, two new dibenzyltin compounds have been synthesized by the microwave "one pot" with benzoylhydrazine (or p-methyl benzhydrazine), phenylglyoxylic acid and di-m-chlorobenzyltin dichloride. The groups of aroyl hydrazone ligands to inhibit cancer cell activity have been investigated preliminarily, which lay a foundation for screening new organic-metallic compounds with high anticancer activity.
Italian MILESTONE microwave synthesizer was employed for the compounds. IR spectra were recorded using the Shimadzu Prestige 21 infrared spectrometer in the range of 4000~400 cm–1 (KBr pellets). 1H, 13C and 119Sn NMR were measured by Bruker AVANCE-500 NMR. Elemental analysis was performed by the PE-2400(Ⅱ) element analyzer. H RMS was determined by the Thermo Scientific LTQ Orbitrap XL (ESI source). Melting point was measured by Beijing Tektronix X-4 binocular photomicrograph (thermometer not corrected). Thermogravimetric analyses were executed on a NETZSCH TG 209 F3 thermogravimetric analyzer.
The di-m-chlorobenzyltin dichloride was obtained according the literature[12]. Carboplatin was purchased from Balinway technologies Co. LTD. NCI-H460, HepG2 and MCF7 cells were obtained from the U.S. tissue culture library (ATCC). RPMI 1640 medium with 10% fetal bovine serum was purchased from GIBICO.
The methanol solution (20.0 mL) of benzoylhydrazine (or p-methyl benzoylhydrazine, 1.0 mmol), phenylglyoxylic acid (1.0 mmol) and di-m-chlorobenzyltin dichloride (1.0 mmol) was added into 100.0 mL microwave reaction tank with microwave reaction for 30 min at 100 ℃, then the mixture was cooled to room temperature and filtered into a conical flask. The solvent was controlled for volatilezation into crystals 1, 2a and 2b.
Crystal 1: The product was yellow crystal. Yield: 68%. m. p.: 108~110 ℃. Anal. Calcd. (C30H26Cl2N2O4Sn): C, 53.93; H, 3.92; N, 4.19%. Found: C, 53.94; H, 3.92; N, 4.20%. FT-IR (KBr, cm–1): 3489, 3057, 2961, 2941, 1618, 1593, 1566, 1493, 1479, 1435, 1425, 1391, 1317, 1287, 1252, 1173, 1155, 1088, 1070, 1020, 999, 866, 804, 781, 731, 712, 687, 631, 594, 577, 498, 476, 432. 1H NMR (500 MHz, CDCl3, δ/ppm): 7.97 (d, J = 7.3 Hz, 2H), 7.52~7.55 (m, 6H), 7.40~7.43 (m, 2H), 7.09 (s, 2H), 6.88 (s, 6H), 3.39 (s, 4H). 13C NMR (126 MHz, CDCl3, δ/ppm): 176.06, 168.79, 147.38, 138.77, 134.35, 132.47, 132.31, 131.40, 130.98, 129.70, 129.19, 128.38, 128.31, 128.18, 127.47, 126.43, 125.72, 35.81. 119Sn NMR (Me4Sn, 187 MHz, CDCl3, δ/ppm): –638.95. H RMS (ESI) m/z calcd. for C29H23Cl2N2O3Sn+ [M-CH3OH+H]+ 637.01022, found 637.01093.
Crystal 2a: The product was yellow crystal in the yield of 37%. m. p.: 144~146 ℃. Anal. Calcd. (C31H28Cl2N2O4Sn): C, 54.58; H, 4.14; N, 4.11%. Found: C, 54.60; H, 4.16; N, 4.10%. FT-IR (KBr, cm–1): 3462, 3049, 3011, 2938, 2828, 1624, 1609, 1595, 1584, 1570, 1476, 1427, 1393, 1306, 1281, 1250, 1177, 1155, 1090, 1078, 1063, 1018, 870, 806, 781, 745, 729, 691, 619, 594, 575, 471, 432. 1H NMR (500 MHz, CDCl3, δ/ppm): 7.86 (d, J = 7.9 Hz, 2H), 7.51~7.52 (m, 5H), 7.21 (d, J = 7.6 Hz, 2H), 7.09 (s, 2H), 6.84~6.89 (m, 6H), 3.40 (s, 4H), 2.43 (s, 3H). 13C NMR (126 MHz, CDCl3, δ/ppm): 176.20, 169.51, 146.39, 143.09, 138.96, 134.23, 131.33, 130.75, 129.62, 129.29, 128.91, 128.57, 128.37, 128.17, 127.40, 126.45, 125.59, 36.57, 21.82. 119Sn NMR (Me4Sn, 187 MHz, CDCl3, δ/ppm): –637.74. HRMS (ESI) m/z calcd. for C30H25Cl2N2O3Sn+ [M-CH3OH+H]+ 651.02587, found 651.02594.
Crystal 2b: The product was red-brown crystal in 35% yield. m.p.: 144~146 ℃. Anal. Calcd. (C31H28Cl2N2O4Sn): C, 54.58; H, 4.14; N, 4.11%. Found: C, 54.55; H, 4.13; N, 4.12%. FT-IR (KBr, cm–1): 3433, 3059, 3013, 2951, 1624, 1595, 1570, 1493, 1476, 1443, 1425, 1391, 1310, 1290, 1254, 1175, 1155, 1088, 1078, 1016, 866, 837, 804, 785, 746, 729, 692, 621, 594, 573, 488, 473, 434. 1H NMR (500 MHz, CDCl3, δ/ppm): 7.86 (d, J = 7.9 Hz, 2H), 7.50~7.54 (m, 5H), 7.21 (d, J = 7.6 Hz, 2H), 7.09 (s, 2H), 6.84~6.89 (m, 6H), 3.38 (s, 4H), 2.43 (s, 3H). H RMS (ESI) m/z calcd. for C30H25Cl2N2O3Sn+ [M-CH3OH+H]+ 651.02587, found 651.02661.
The suitable samples (1: 0.23 × 0.21 × 0.21 mm3, 2a: 0.22 × 0.21 × 0.21 mm3 and 2b: 0.22 × 0.21 × 0.21 mm3) were chosen for crystallographic study and then mounted at the Bruker SMART APEX Ⅱ CCD single crystal diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.071073 nm) with a φ-ω scan mode. All the data were corrected by Lp factors and empirical absorbance. The structure was solved by direct methods. All non-hydrogen atoms were determined in successive difference Fourier synthesis, and all hydrogen atoms were added according to theoretical models. All hydrogen and non-hydrogen atoms were refined by their isotropic and anisotropic thermal parameters through full-matrix least-squares techniques. All calculations were completed by the SHELXTL-97 program[13]. The selected bond lengths and bond angles for 1, 2a, 2b are listed in Tables 2, respectively.
Thermogravimetric analyses were executed on a NETZSCH TG 209 F3 thermogravimetric analyzer with the heating speed at 20 ℃·min–1 and gas flow rate of 20 mL·min–1 in the air.
The drug was dissolved in a small amount of DMSO. The mixture was diluted with water to the required concentration, and maintain the final concentration of DMSO < 0.1%. NCI-H460, HepG2 and MCF7 cells were cultured in vitro using RPMI 1640 (GIBICO) culture medium containing 10% fetal bovine serum in a 5% (volume fraction) CO2, 37 ℃ saturated humidity incubator. In vitro anticancer drug sensitivity test was determined by MTT assay. Graph Pad Prism version 7.0 program was used for data processing, and IC50 was obtained by fitting the nonlinear regression model with S-shaped dose response in the program.
The synthetic routes of compounds are depicted in Fig. 1. As we know, the conventional synthesis methods of organotin compound include the reflux method, solvent-thermal and interfacial diffusion one[14], while the first one is the most common. However, we adopted the microwave "one pot", which has the characteristics of short reaction time, simple reaction steps, easy purification and good reproducibility compared with traditional heating reflux method. Moreover, the yields of dibenzyltin compounds are around 70%, suggesting the yield of microwave methods is improved greatly[15].
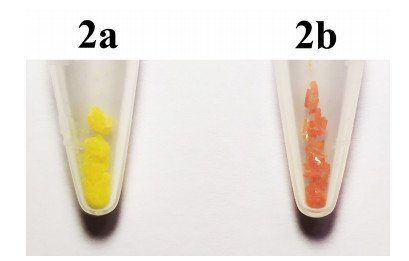
In addition, in the syntheses of 2, two kinds of crystals with different colors and shapes were precipitated at the same time. One is a yellow fine-block crystal (2a), and the other a red-brown rod-shaped crystal (2b), with their crystal morphology shown in Fig. 2. Two crystals were characterized by elemental analysis, IR, NMR, and X-ray single-crystal diffraction, showing their chemical compositions and molecular structures are the same, but the cell parameters were different, indicating 2a and 2b are homogeneous polycrystalline.
In the infrared spectra, the absorption bands at 3489 (1), 3462 (2a) and 3433 (2b) cm–1 indicate the O–H stretching of coordinated methanol molecules. The peaks of 1566, 1570 and 1570 cm–1 can be attributed to the C=N–N=C of acyl hydrazone[16, 17]. The carboxylate group appeared at 1618 (1), 1624 (2a) and 1624 (2b) cm–1 for the antisymmetric stretching vibration and 1391 (1), 1393 (2a) and 1391 (2b) cm–1 for symmetric stretching vibration. Furthermore, the Δν values (Δν = νasym(COO–) – νsym(COO–)) are 227 (1), 231 (2a), 233 (2b) cm–1 (2b), respectively, suggesting all Sn atoms of compounds coordinated the O atoms of carboxylic with a monodentate chelating modes[18], which are consistent with their crystal structures. The characteristic peaks of Sn–O–Sn, Sn–O, Sn–N and Sn–C are located at 619~631, 573~577, 471~476 and 432~434 cm–1, respectively, which are consistent with the peak locations of similar compounds reported in the literature[19, 20], indicating preliminarily that 1, 2a and 2b have similar structures.
In 1H NMR spectrum, the ratio of the integral area of each group of the three crystals is consistent with the number of protons in each group of the expected structure[21]. Furthermore, it can be found the chemical displacements and peak shapes of all hydrogen protons in 2a and 2b remain the same, which further suggests 2a and 2b have the same asymmetric units. While the peak situation of hydrogen protons in 1 is similar to that of 2a and 2b, which speculated 1 has similar asymmetric unit structure to 2a and 2b.
In the 13C NMR spectrum, the peak positions of carboxyl carbon, hydrazine carbon and imino carbon in compounds 1 and 2a are consistent, and the peaks of the remaining groups are consistent with the number of carbon atoms in the theoretical structure[21]. Based on the 1H NMR and 13C NMR spectra, it can be further inferred that the structures of 1 and 2a are similar, which is consistent with the X-ray single-crystal diffraction results.
In 119Sn NMR spectrum, the single peak of tin atoms present at 638.95 and 637.74 ppm in 1 and 2a, respectively indicate 1 and 2a show only a class of tin, and their chemical shifts of the tin atoms are close. Therefore, tin atoms have similar chemical environments with similar structures.
In HRMS spectrum, the mass peaks at m/z 637.01093 (1), 651.02594 (2a) and 651.02661 (2b) are attributed to the absorption peaks of [M– CH3OH+H]+, indicating the weak Sn–O bonds will be broken in the solution state and present monomer structure in three crystals, which also further suggests 2a and 2b show similar structures in solution state.
As shown in Table 1, compound 2 has different crystal systems. 2a crystallizes in the triclinic P
 DownLoad:
CSV
DownLoad:
CSV
| Compounds | 1 | 2a | 2b | |||
| Empirical formula | C30H26Cl2N2O4Sn | C31H28Cl2N2O4Sn | C31H28Cl2N2O4Sn | |||
| Mr | 668.12 | 682.14 | 682.14 | |||
| Temperature/K | 296(2) | 296(2) | 296(2) | |||
| Crystal system | Triclinic | Triclinic | Monoclinic | |||
| Space group | P |
P |
C2/c | |||
| a/Å | 9.5731(15) | 8.0846(13) | 33.4153(13) | |||
| b/Å | 11.0889(18) | 13.129(2) | 11.7891(5) | |||
| c/Å | 15.431(3) | 15.013(2) | 16.6328(7) | |||
| α/° | 95.938(2) | 109.084(2) | 90 | |||
| β/° | 104.805(2) | 92.354(2) | 113.608(1) | |||
| γ/° | 110.500(2) | 94.379(2) | 90 | |||
| Volume/Å3 | 1449.4(4) | 1497.9(4) | 6003.9(4) | |||
| Z | 2 | 2 | 8 | |||
| Dc (mg/m3) | 1.531 | 1.512 | 1.509 | |||
| Absorption coefficient/mm–1 | 1.103 | 1.069 | 1.067 | |||
| F(000) | 672 | 688 | 2752 | |||
| Crystal size/mm | 0.23 × 0.21 × 0.21 | 0.22 × 0.21 × 0.21 | 0.22 × 0.21 × 0.21 | |||
| θ range/° | 2.36~25.10 | 2.52~ 25.09 | 1.85~25.10 | |||
| Limiting indices | –11≤h≤11, –13≤k≤13, –18≤l≤18 |
–9≤h≤9, –15≤k≤15, –17≤l≤17 |
–39≤h≤39, –14≤k≤≤9, –19≤l≤19 |
|||
| Reflections collected/unique | 14980/5141(Rint = 0.0191) | 15471/5319 (Rint = 0.0144) | 14916/5338 (Rint = 0.0140) | |||
| Completeness | 99.6% | 99.7% | 99.9% | |||
| Max/min. transmission | 0.8014, 0.7854 | 0.8066, 0.7988 | 0.8069, 0.7991 | |||
| Data/restraints/parameters | 5141/40/357 | 5319/0/367 | 5338/0/367 | |||
| Goodness-of-fit on F2 | 1.032 | 1.038 | 1.050 | |||
| Final R indices (I > 2σ(I)) |
R = 0.0343, wR = 0.1015 | R = 0.0260, wR = 0.0698 | R = 0.0207, wR = 0.0501 | |||
| R indices (all data) | R = 0.0375, wR = 0.1042 | R = 0.0278, wR = 0.0712 | R = 0.0252, wR = 0.0523 | |||
| (Δρ)max (e·Å–3) | 2.102 | 0.729 | 0.375 | |||
| (Δρ)min (e·Å–3) | –0.817 | –0.952 | –0.352 |
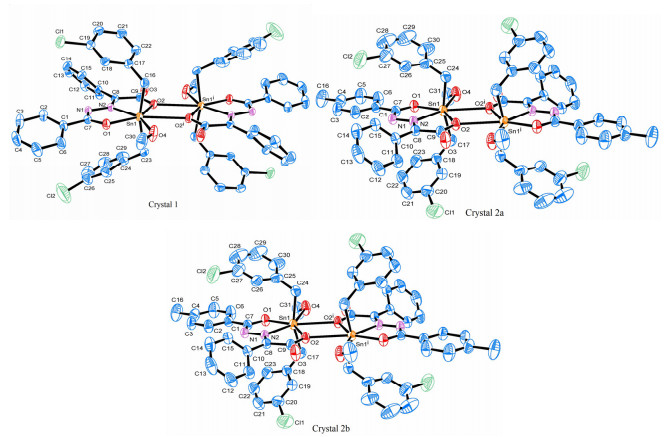
Compounds 1 and 2 have similar structural units, showing a distorted seven-coordinated pentagonal bipyramidal configuration with only little difference in bond parameters, which is consistent with the conclusion obtained by spectral characterization.
Thermal analyses were performed for the synthesized compounds under an air atmosphere, as shown in Fig. 4. Three crystals exhibit similar weight loss processes with three stages. At the initial stage of 40~180 ℃, the weight loss of 1 was 5.1% (calcd. 4.8%), 2a was 4.9% (calcd. 4.7%) and 2b was 4.9% (calcd. 4.7%), which are attributed to the removal of methanol molecules. And further weight loss begins at 180 ℃, corresponding to the elimination of organic molecules by assuming a SnO2 21.0% (1, calcd. 22.4%), 21.1% (2a, calcd. 21.9%) and 21.8% (2b, calcd. 21.9%) phase as the final residue. As a result, three crystals are stable until 100 ℃, and 2a and 2b have the same weight loss processes and ratios, indicating they are homogeneous polycrystalline.
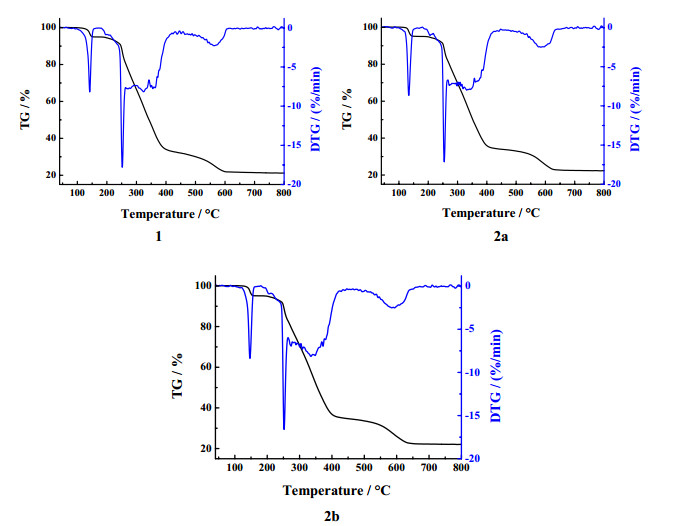
Table 3 lists the inhibitory activities of 1, 2a and 2b and carboplatin on the cultured cancer cells NCI-H460, HepG2 and MCF7 in vitro. It can be seen that the approximation IC50 of 2a and 2b to three cancer cells indicates that different crystal systems have little influence on the activity of compounds. In addition, compared with IC50, a conclusion can be drawn that the inhibitory activities of 1, 2a and 2b on NCI-H460 are similar with no significant difference, and are weaker than one of carboplatin. However, for HepG2 and MCF-7, the inhibitory activities of 2a and 2b were significantly better than that of 1, and even the one on MCF7 was better than that of carboplatin. Therefore, three crystals had certain inhibitory effects on the three cancer cells, and 2a and 2b are stronger than 1. Furthermore, combined with the structures of 1, 2a and 2b, they have similar structures, but only the aromatic ring of the hydrazone ligand is different. The aromatic ring of 1 is phenyl, while the aromatic ring of 2a and 2b is p-methyl phenyl, which indicates the different aromatic ring of ligand has also certain influence on the anticancer activity of the compound.
DownLoad:
CSV
| 1 | |||||||
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| Sn(1)–C(16) | 2.134(4) | Sn(1)–O(1) | 2.136(2) | Sn(1)–C(23) | 2.150(4) | ||
| Sn(1)–N(2) | 2.222(3) | Sn(1)–O(2) | 2.277(2) | Sn(1)–O(4) | 2.433(3) | ||
| Sn(1)–O(2)ⅰ | 2.837(2) | ||||||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| C(16)–Sn(1)–O(1) | 94.02(12) | C(16)–Sn(1)–C(23) | 161.91(15) | O(1)–Sn(1)–C(23) | 94.74(13) | ||
| C(16)–Sn(1)–N(2) | 98.67(11) | O(1)–Sn(1)–N(2) | 71.52(9) | C(23)–Sn(1)–N(2) | 99.11(14) | ||
| C(16)–Sn(1)–O(2) | 92.92(12) | O(1)–Sn(1)–O(2) | 142.18(9) | C(23)–Sn(1)–O(2) | 89.83(13) | ||
| N(2)–Sn(1)–O(2) | 70.69(9) | C(16)–Sn(1)–O(4) | 82.75(15) | O(1)–Sn(1)–O(4) | 77.41(10) | ||
| C(23)–Sn(1)–O(4) | 83.76(17) | N(2)–Sn(1)–O(4) | 148.92(10) | O(2)–Sn(1)–O(4) | 140.39(9) | ||
| O(2)–Sn(1)–O(2)ⅰ | 66.27(2) | O(2)ⅰ–Sn(1)-C(16) | 84.81(9) | O(2)ⅰ–Sn(1)–C(23) | 79.95(11) | ||
| O(2)ⅰ–Sn(1)–O(4) | 74.1 (1) | ||||||
| Symmetry code: ⅰ –x, –y, –z | |||||||
| 2a | |||||||
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| Sn(1)–O(1) | 2.137(2) | Sn(1)–C(17) | 2.138(3) | Sn(1)–C(24) | 2.141(3) | ||
| Sn(1)–N(2) | 2.234(1) | Sn(1)–O(2) | 2.280(1) | Sn(1)–O(4) | 2.430(2) | ||
| Sn(1)–O(2)ⅰ | 2.818(1) | ||||||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| O(1)–Sn(1)–C(17) | 93.96(9) | O(1)–Sn(1)–C(24) | 97.56(9) | C(17)–Sn(1)–C(24) | 160.82(11) | ||
| O(1)–Sn(1)–N(2) | 71.04(6) | C(17)–Sn(1)–N(2) | 99.80(8) | C(24)–Sn(1)–N(2) | 98.43(9) | ||
| O(1)–Sn(1)–O(2) | 141.48(6) | C(17)–Sn(1)–O(2) | 89.75(9) | C(24)–Sn(1)–O(2) | 90.72(9) | ||
| N(2)–Sn(1)–O(2) | 70.55(6) | O(1)–Sn(1)–O(4) | 76.95(7) | C(17)–Sn(1)–O(4) | 85.47(9) | ||
| C(24)–Sn(1)–O(4) | 82.27(10) | N(2)–Sn(1)–O(4) | 147.81(7) | O(2)–Sn(1)–O(4) | 141.57(6) | ||
| O(2)–Sn(1)–O(2)ⅰ | 65.071(51) | O(2)ⅰ–Sn(1)–C(17) | 82.52(8) | O(2)ⅰ–Sn(1)–C(24) | 80.31(8) | ||
| O(2)ⅰ–Sn(1)–O(4) | 76.495(63) | ||||||
| Symmetry code: ⅰ 1–x, –y, –z | |||||||
| 2b | |||||||
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| Sn(1)–C(17) | 2.137(2) | Sn(1)–O(1) | 2.140(1) | Sn(1)–C(24) | 2.144(2) | ||
| Sn(1)–N(2) | 2.2370(1) | Sn(1)–O(2) | 2.2980(1) | Sn(1)–O(4) | 2.461(2) | ||
| Sn(1)–O(2)ⅰ | 2.763(1) | ||||||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| C(17)–Sn(1)–O(1) | 94.92(7) | C(17)–Sn(1)–C(24) | 161.57(8) | O(1)–Sn(1)–C(24) | 95.51(7) | ||
| C(17)–Sn(1)–N(2) | 100.11(7) | O(1)–Sn(1)–N(2) | 71.14(5) | C(24)–Sn(1)–N(2) | 97.63(7) | ||
| C(17)–Sn(1)–O(2) | 89.07(6) | O(1)–Sn(1)–O(2) | 141.54(5) | C(24)–Sn(1)–O(2) | 92.16(7) | ||
| N(2)–Sn(1)–O(2) | 70.50(5) | C(17)–Sn(1)–O(4) | 84.97(7) | O(1)–Sn(1)–O(4) | 76.05(5) | ||
| C(24)–Sn(1)–O(4) | 82.86(8) | N(2)–Sn(1)–O(4) | 147.09(6) | O(2)–Sn(1)–O(4) | 142.38(5) | ||
| O(2)–Sn(1)–O(2)ⅰ | 64.59(5) | O(2)ⅰ–Sn(1)–C(17) | 82.40(6) | O(2)ⅰ–Sn(1)–C(24) | 81.54 (7) | ||
| O(2)ⅰ–Sn(1)–O(4) | 77.81(6) | ||||||
| Symmetry code: ⅰ 1–x, 1–y, 1–z | |||||||
DownLoad:
CSV
| IC50 / μM | |||
| NCI-H460 | HepG2 | MCF-7 | |
| 1 | 8.67 ± 0.10 | 10.20 ± 0.18 | 9.41 ± 0.22 |
| 2a | 9.19 ± 0.17 | 8.40 ± 0.20 | 6.82 ± 0.15 |
| 2b | 9.33 ± 0.10 | 8.28 ± 0.15 | 6.87 ± 0.08 |
| Carboplatin | 7.26 ± 0.32 | 7.70 ± 0.25 | 8.22 ± 0.41 |
Two dibenzyltin compounds have been synthesized by the microwave "one pot" method. Compound 2 has two crystals of 2a and 2b with different colors and shapes. Three crystals contain four-membered ring Sn2O2 plane as the center of symmetry to construct a distorted pentagonal bipyramidal configuration. Three crystals can exist stably until 100 ℃. The in vitro inhibitory activity on cancer cells NCI-H460, HepG2 and MCF7 showed the inhibitory activity of 2a and 2b is the same, and three crystals have obvious inhibitory effect on the three cancer cells, and the inhibitory effects of 2a and 2b are better than that of 1.
Rosenberg, B.; Vancamp, L.; Trosko, J. E.; Mansour, V. H. Platinum compounds: a new class of potent antitumour agents. Nature 1969, 222, 385–386. doi: 10.1038/222385a0
Yan, Y. K.; Melchart, M.; Habtemariam, A.; Sadler, P. J. Organometallic chemistry, biology and medicine: ruthenium arene anticancer complexes. Chem. Commun. 2005, 4764–4776.
Cao, W.; Qi, J.; Qian, K.; Tian, L.; Cheng, Z.; Wang, Y. Structure-activity relationships of 2-quinolinecarboxaldehyde thiosemicarbazone gallium(Ⅲ) complexes with potent and selective anticancer activity. J. Inorg. Biochem. 2019, 191, 174–182. doi: 10.1016/j.jinorgbio.2018.11.017
Díaz-García, D.; Cenariu, D.; Pérez, Y.; Cruz, P.; del Hierro, I.; Prashar, S.; Fischer-Fodor, E.; Gómez-Ruiz, S. Modulation of the mechanism of apoptosis in cancer cell lines by treatment with silica-based nanostructured materials functionalized with different metallodrugs. Dalton Trans. 2018, 47, 12284–12299. doi: 10.1039/C8DT01677A
Attanzio, A.; Ippolito, M.; Girasolo, M. A.; Saiano, F.; Rotondo, A.; Rubino, S.; Mondello, L.; Capobianco, M. L.; Sabatino, P.; Tesoriere, L.; Casella, G. Anti-cancer activity of di- and tri-organotin(Ⅳ) compounds with D-(+)-Galacturonic acid on human tumor cells. J. Inorg. Biochem. 2018, 188, 102–112. doi: 10.1016/j.jinorgbio.2018.04.006
Shang, X.; Meng, X.; Alegria, E. C. B. A.; Li, Q.; Guedes da Silva, M. F. C.; Kuznetsov, M. L.; Pombeiro, A. J. L. Syntheses, molecular structures, electrochemical behavior, theoretical study, and antitumor activities of organotin(ⅳ) complexes containing 1-(4-chlorophenyl)-1-cyclopentanecarboxylato ligands. Inorg. Chem. 2011, 50, 8158–8167. doi: 10.1021/ic200635g
Hong, M.; Yang, Y.; Li, C.; Xu, L.; Li, D.; Li, C. Z. Study of the effect of molecular structure and alkyl groups bound with tin(ⅳ) on their cytotoxicity of organotin(ⅳ) 2-phenyl-4-selenazole carboxylates. RSC Adv. 2015, 5, 102885–102894. doi: 10.1039/C5RA18445B
Basu Baul, T. S.; Dutta, D.; Duthie, A.; Guchhait, N.; Rocha, B. G. M.; Guedes da Silva, M. F. C.; Mokhamatam, R. B.; Raviprakash, N.; Manna, S. K. New dibutyltin(Ⅳ) ladders: syntheses, structures and, optimization and evaluation of cytotoxic potential employing A375 (melanoma) and HCT116 (colon carcinoma) cell lines in vitro. J. Inorg. Biochem. 2017, 166, 34–48. doi: 10.1016/j.jinorgbio.2016.10.008
Zong, X. J.; Liu, X. R.; Zhao, S. S.; Yang, Z. W. Preparation, thermal analyses and biological activities of Co(Ⅱ) and Cr(Ⅲ) complexes with 2-acetylpyridine-6-bromo-2-naphthoyl acylhydrazone. Polyhedron 2019, 170, 303–311. doi: 10.1016/j.poly.2019.05.059
Zhou, Y.; Wei, W.; Zhu, L.; Li, Y.; Li, Z. Synthesis and insecticidal activity study of novel anthranilic diamides analogs containing a diacylhydrazine bridge as effective Ca2+ modulators. Chem. Biol. Drug Des. 2018, 92, 1914–1919. doi: 10.1111/cbdd.13349
Wang, H.; Fu, Y.; Fan, Z.; Song, H.; Wu, Q.; Zhang, Y.; Belskaya, N. P.; Bakulev V. A. Synthesis, crystal structure and biological activity of N-tert-butyl-N-(4-methyl-1,2,3-thiadiazole)-5-yl-N-(4-methyl-1,2,3-thiadiazole)-5-formyl- N-3,5-dichloropyridin-2-yl-diacylhydrazine. Chin. J. Struct. Chem. 2011, 30, 412–416.
Sisido, K.; Takeda, Y.; Kinugawa, Z. Direct synthesis of organotin compounds. I. di- and tribenzyltin chlorides. J. Am. Chem. Soc. 1961, 83, 538–541. doi: 10.1021/ja01464a008
Sheldrick, G. M. SHELXL-97, A Program for Crystal Structure Refinement. University of Göttingen, Germany 1997.
Xu, R. R.; Pang, W. Q.; Huo, Q. S. Inorganic Synthesis and Preparative Chemistry. Higher Education Press, Beijing 2009.
Yin, H. D.; Chen, S. W.; Li, L. W.; Wang, D. Q. Synthesis, characterization and crystal structures of the organotin(Ⅳ) compounds with the Schiff base ligands of pyruvic acid thiophene-2-carboxylic hydrazone and salicylaldehyde thiophene-2-carboxylic hydrazone. Inorg. Chim. Acta 2007, 360, 2215–2223. doi: 10.1016/j.ica.2006.10.038
Liu, K.; Yan, H.; Chang, G.; Li, Z.; Niu, M.; Hong, M. Organotin(Ⅳ) complexes derived from hydrazone Schiff base: synthesis, crystal structure, in vitro cytotoxicity and DNA/BSA interactions. Inorg. Chim. Acta 2017, 464, 137–146. doi: 10.1016/j.ica.2017.05.017
Tan, Y.; Zhang, Z.; Tan, Y.; Kuang, D.; Yu, J.; Zhu, X.; Jiang, W. Syntheses, crystal structures and biological activity of the dialkytin complexes based on 2-oxo-3-phenylpropionic acid salicyloylhydrazone. J. Coord. Chem. 2017, 70, 2606–2624. doi: 10.1080/00958972.2017.1355460
Deacon, G. B.; Phillips, R. J. Relationships between the carbon-oxygen stretching frequencies of carboxylato complexes and the type of carboxylate coordination. Coordin. Chem. Rev. 1980, 33, 227−250. doi: 10.1016/S0010-8545(00)80455-5
Salam, M. A.; Hussein, M. A.; Ramli, I.; Islam, M. S. Synthesis, structural characterization, and evaluation of biological activity of organotin(Ⅳ) complexes with 2-hydroxy-5-methoxybenzaldehyde-N(4)-methylthiosemicarbazone. J. Organomet. Chem. 2016, 813, 71–77. doi: 10.1016/j.jorganchem.2016.04.007
Hong, M.; Yin, H.; Zhang, X.; Li, C.; Yue, C.; Cheng, S. Di- and tri-organotin(Ⅳ) complexes with 2-hydroxy-1-naphthaldehyde 5-chloro-2-hydroxybenzoylhydrazone: synthesis, characterization and in vitro antitumor activities. J. Organomet. Chem. 2013, 724, 23–31. doi: 10.1016/j.jorganchem.2012.10.031
Pretsch, E.; Bühlmann, P.; Badertscher, M. Structure Determination of Organic Compounds, Tables of Spectral Data. Fourth. Springer-Verlag, Berlin Heidelberg 2009.
Zhang, Z. Z.; Jiang, W. J.; Liu, Y.; Kuang, D. Z.; Yu, J. X.; Zhu, X. M.; Tan, Y. X. Syntheses, crystal structures and biological activity of N-(2-propionic acid)-aroyl hydrazone di-p-methylbenzytin complexes. Chin. J. Inorg. Chem. 2017, 33, 1603–1610.
Tan, Y. X.; Zhang, Z. Z.; Feng, Y. L.; Yu, J. X.; Zhu, X. M.; Kuang, D. Z.; Jiang, W. J. Syntheses, crystal structures and biological activities of the 2-oxo-butyric acid salicylacylhydrazone dibenzyltin complexes. Chin. J. Struct. Chem. 2017, 36, 925–936.

Figure 3 Molecular structures of compounds (Symmetry codes: 1, ⅰ –x, –y, –z; 2a, ⅰ 1–x, –y, –z; 2b, ⅰ 1–x, 1–y, 1–z)
Table 1. Crystallographic Data of the Compounds
| Compounds | 1 | 2a | 2b | |||
| Empirical formula | C30H26Cl2N2O4Sn | C31H28Cl2N2O4Sn | C31H28Cl2N2O4Sn | |||
| Mr | 668.12 | 682.14 | 682.14 | |||
| Temperature/K | 296(2) | 296(2) | 296(2) | |||
| Crystal system | Triclinic | Triclinic | Monoclinic | |||
| Space group | P |
P |
C2/c | |||
| a/Å | 9.5731(15) | 8.0846(13) | 33.4153(13) | |||
| b/Å | 11.0889(18) | 13.129(2) | 11.7891(5) | |||
| c/Å | 15.431(3) | 15.013(2) | 16.6328(7) | |||
| α/° | 95.938(2) | 109.084(2) | 90 | |||
| β/° | 104.805(2) | 92.354(2) | 113.608(1) | |||
| γ/° | 110.500(2) | 94.379(2) | 90 | |||
| Volume/Å3 | 1449.4(4) | 1497.9(4) | 6003.9(4) | |||
| Z | 2 | 2 | 8 | |||
| Dc (mg/m3) | 1.531 | 1.512 | 1.509 | |||
| Absorption coefficient/mm–1 | 1.103 | 1.069 | 1.067 | |||
| F(000) | 672 | 688 | 2752 | |||
| Crystal size/mm | 0.23 × 0.21 × 0.21 | 0.22 × 0.21 × 0.21 | 0.22 × 0.21 × 0.21 | |||
| θ range/° | 2.36~25.10 | 2.52~ 25.09 | 1.85~25.10 | |||
| Limiting indices | –11≤h≤11, –13≤k≤13, –18≤l≤18 |
–9≤h≤9, –15≤k≤15, –17≤l≤17 |
–39≤h≤39, –14≤k≤≤9, –19≤l≤19 |
|||
| Reflections collected/unique | 14980/5141(Rint = 0.0191) | 15471/5319 (Rint = 0.0144) | 14916/5338 (Rint = 0.0140) | |||
| Completeness | 99.6% | 99.7% | 99.9% | |||
| Max/min. transmission | 0.8014, 0.7854 | 0.8066, 0.7988 | 0.8069, 0.7991 | |||
| Data/restraints/parameters | 5141/40/357 | 5319/0/367 | 5338/0/367 | |||
| Goodness-of-fit on F2 | 1.032 | 1.038 | 1.050 | |||
| Final R indices (I > 2σ(I)) |
R = 0.0343, wR = 0.1015 | R = 0.0260, wR = 0.0698 | R = 0.0207, wR = 0.0501 | |||
| R indices (all data) | R = 0.0375, wR = 0.1042 | R = 0.0278, wR = 0.0712 | R = 0.0252, wR = 0.0523 | |||
| (Δρ)max (e·Å–3) | 2.102 | 0.729 | 0.375 | |||
| (Δρ)min (e·Å–3) | –0.817 | –0.952 | –0.352 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Parts of the Bond Lengths (Å) and Bond Angles (°) of (1), (2a) and (2b)
| 1 | |||||||
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| Sn(1)–C(16) | 2.134(4) | Sn(1)–O(1) | 2.136(2) | Sn(1)–C(23) | 2.150(4) | ||
| Sn(1)–N(2) | 2.222(3) | Sn(1)–O(2) | 2.277(2) | Sn(1)–O(4) | 2.433(3) | ||
| Sn(1)–O(2)ⅰ | 2.837(2) | ||||||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| C(16)–Sn(1)–O(1) | 94.02(12) | C(16)–Sn(1)–C(23) | 161.91(15) | O(1)–Sn(1)–C(23) | 94.74(13) | ||
| C(16)–Sn(1)–N(2) | 98.67(11) | O(1)–Sn(1)–N(2) | 71.52(9) | C(23)–Sn(1)–N(2) | 99.11(14) | ||
| C(16)–Sn(1)–O(2) | 92.92(12) | O(1)–Sn(1)–O(2) | 142.18(9) | C(23)–Sn(1)–O(2) | 89.83(13) | ||
| N(2)–Sn(1)–O(2) | 70.69(9) | C(16)–Sn(1)–O(4) | 82.75(15) | O(1)–Sn(1)–O(4) | 77.41(10) | ||
| C(23)–Sn(1)–O(4) | 83.76(17) | N(2)–Sn(1)–O(4) | 148.92(10) | O(2)–Sn(1)–O(4) | 140.39(9) | ||
| O(2)–Sn(1)–O(2)ⅰ | 66.27(2) | O(2)ⅰ–Sn(1)-C(16) | 84.81(9) | O(2)ⅰ–Sn(1)–C(23) | 79.95(11) | ||
| O(2)ⅰ–Sn(1)–O(4) | 74.1 (1) | ||||||
| Symmetry code: ⅰ –x, –y, –z | |||||||
| 2a | |||||||
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| Sn(1)–O(1) | 2.137(2) | Sn(1)–C(17) | 2.138(3) | Sn(1)–C(24) | 2.141(3) | ||
| Sn(1)–N(2) | 2.234(1) | Sn(1)–O(2) | 2.280(1) | Sn(1)–O(4) | 2.430(2) | ||
| Sn(1)–O(2)ⅰ | 2.818(1) | ||||||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| O(1)–Sn(1)–C(17) | 93.96(9) | O(1)–Sn(1)–C(24) | 97.56(9) | C(17)–Sn(1)–C(24) | 160.82(11) | ||
| O(1)–Sn(1)–N(2) | 71.04(6) | C(17)–Sn(1)–N(2) | 99.80(8) | C(24)–Sn(1)–N(2) | 98.43(9) | ||
| O(1)–Sn(1)–O(2) | 141.48(6) | C(17)–Sn(1)–O(2) | 89.75(9) | C(24)–Sn(1)–O(2) | 90.72(9) | ||
| N(2)–Sn(1)–O(2) | 70.55(6) | O(1)–Sn(1)–O(4) | 76.95(7) | C(17)–Sn(1)–O(4) | 85.47(9) | ||
| C(24)–Sn(1)–O(4) | 82.27(10) | N(2)–Sn(1)–O(4) | 147.81(7) | O(2)–Sn(1)–O(4) | 141.57(6) | ||
| O(2)–Sn(1)–O(2)ⅰ | 65.071(51) | O(2)ⅰ–Sn(1)–C(17) | 82.52(8) | O(2)ⅰ–Sn(1)–C(24) | 80.31(8) | ||
| O(2)ⅰ–Sn(1)–O(4) | 76.495(63) | ||||||
| Symmetry code: ⅰ 1–x, –y, –z | |||||||
| 2b | |||||||
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| Sn(1)–C(17) | 2.137(2) | Sn(1)–O(1) | 2.140(1) | Sn(1)–C(24) | 2.144(2) | ||
| Sn(1)–N(2) | 2.2370(1) | Sn(1)–O(2) | 2.2980(1) | Sn(1)–O(4) | 2.461(2) | ||
| Sn(1)–O(2)ⅰ | 2.763(1) | ||||||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| C(17)–Sn(1)–O(1) | 94.92(7) | C(17)–Sn(1)–C(24) | 161.57(8) | O(1)–Sn(1)–C(24) | 95.51(7) | ||
| C(17)–Sn(1)–N(2) | 100.11(7) | O(1)–Sn(1)–N(2) | 71.14(5) | C(24)–Sn(1)–N(2) | 97.63(7) | ||
| C(17)–Sn(1)–O(2) | 89.07(6) | O(1)–Sn(1)–O(2) | 141.54(5) | C(24)–Sn(1)–O(2) | 92.16(7) | ||
| N(2)–Sn(1)–O(2) | 70.50(5) | C(17)–Sn(1)–O(4) | 84.97(7) | O(1)–Sn(1)–O(4) | 76.05(5) | ||
| C(24)–Sn(1)–O(4) | 82.86(8) | N(2)–Sn(1)–O(4) | 147.09(6) | O(2)–Sn(1)–O(4) | 142.38(5) | ||
| O(2)–Sn(1)–O(2)ⅰ | 64.59(5) | O(2)ⅰ–Sn(1)–C(17) | 82.40(6) | O(2)ⅰ–Sn(1)–C(24) | 81.54 (7) | ||
| O(2)ⅰ–Sn(1)–O(4) | 77.81(6) | ||||||
| Symmetry code: ⅰ 1–x, 1–y, 1–z | |||||||
下载: 导出CSV
Table 3. Inhibition Action of Compounds to Cancer Cell in Vitro
| IC50 / μM | |||
| NCI-H460 | HepG2 | MCF-7 | |
| 1 | 8.67 ± 0.10 | 10.20 ± 0.18 | 9.41 ± 0.22 |
| 2a | 9.19 ± 0.17 | 8.40 ± 0.20 | 6.82 ± 0.15 |
| 2b | 9.33 ± 0.10 | 8.28 ± 0.15 | 6.87 ± 0.08 |
| Carboplatin | 7.26 ± 0.32 | 7.70 ± 0.25 | 8.22 ± 0.41 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


