Table 1.
Selected Bond Lengths (Å) and Bond Angles (°)
Citation:
WANG Qing, MA Cheng-Bing, CHEN Hui, HUANG De-Guang, CHEN Chang-Neng. Five-heterocyclic-biphosphinesubstituted Fe-only Hydrogenase Mimic: Synthesis, Characterization and Properties[J]. Chinese Journal of Structural Chemistry,
2016, 35(12): 1972-1979.
doi:
10.14102/j.cnki.0254-5861.2011-1211

Five-heterocyclic-biphosphinesubstituted Fe-only Hydrogenase Mimic: Synthesis, Characterization and Properties
English
Five-heterocyclic-biphosphinesubstituted Fe-only Hydrogenase Mimic: Synthesis, Characterization and Properties
Abstract:
A new five-heterocyclic-biphosphine-substituted Fe-only hydrogenase mimic,[(μ-pdt)Fe2(CO)5]2(PTP) (1), has been synthesized at room temperature. 1·H2O crystallizes in triclinic system, space group P1, with a=11.5897(4), b=13.6156(4), c=18.0333(6)Å, α=76.306(3), β=72.742(3), γ=68.939(3)°, V=2508.84(14)Å3, Dc=1.570 g/cm3, Z=2, Mr=1186.37, F(000)=1204, the final R=0.0748, and wR=0.2012. In the tetranuclear complex 1·H2O, each[2Fe2S] butterfly unit is attached to one P atom of the diphosphine bridge and exhibits a square-pyramidal geometry. Complex 1 was characterized by elemental analysis, IR spectra, UV-vis absorption spectra, 1H-NMR and 31P-NMR. The cyclic voltammetry behavior of compound 1 was investigated as well.
-
Key words:
- Fe-only hydrogenase mimic
- / spectral analysis
- / cyclic voltammetry
-
1. INTRODUCTION
Owing to the severe global energy crisis,hy-drogen,a clean and high efficient energy carrier,has come into the focus.Over the past decade,studies on structural and functional mimics of hydrogenases active site has made some progress[1-4]. Mimics of Fe-only hydrogenases have been more widely studied since the catalytic efficiency of Fe-only hydrogenases is about 100 times that of the Ni-Fe hydrogenases[5]. The active site of Fe-only hydro-genases is a [2Fe2S] unit,which consists of two Fe atoms coordinated by CO and CN-,as well as a bridging 1,3-dithiolato ligand[6-8]. Obviously,elec-tron-donating phosphine ligands instead of carbonyl ligands might prepare more stable diiron models and avoid polymerization of the diiron units[9-11]. Among the phosphine ligands,bisphosphine ligand s have morecoordination modes,which could make some contributions to the study of catalytic reaction mechanisms[12-15]. Up to now,flexible bisphosphine ligands were widelyused in these studies[16-18]. Moreover,a rigid and conjugate bisphosphine bri-dging to the diiron complexes makes the electro-chemical properties different from that with flexible bridges[19-22]. Therefore,we attempt to explore [2Fe2S] complexes using rigid bisphosphine ligands asbridges.
2,5-Dis(diphenylphosphino)thiophene (PTP) con-sists of S and P atom coordinationsites so that it was widely used in synthesizing metal complexes[23-26]. Its highlight part is that the basic sulfur atom may act as a proton relay and the diphosphine-thiophene mayr esult in different properties from other thio-phosphine ligands,which contain only one P atom[27, 28]. In this paper,PTP was selected as a bridge to link the [2Fe2S] centers. The electron-withdrawing inductive and conjugative effects of the S atoms may exhibit different properties in iron-sulphur com-plexes. Thus,a new diphosphine-t hiophene ligand was designed and synthesized. A tetranuclear iron-sulphur complex [(μ-PDT)Fe2(CO)5]2(PTP) (1,PDT= CH2CH2CH2) with the above ligand was prepared. The preparation,molecular structure and characteri-zations of complex 1 are presented in our work.
2. EXPERIMENTAL
2.1. Reagents and instructions
All operations were performed under dry oxygen-freenitrogen atmosphere with standard Schlenk techniques. Solvents were distilled from appropriate drying agents according to the standard methods.
Fe2(μ-PDT)(CO)6[29] and 2,5-bis(Diphenylphos-phino)thiophene(PTP)[18, 30] were preparedaccording to literature procedures and other chemicals and reagents were obtained from commercial sources and used as received. The elemental(C,H,O,S) analyses were carried out by Perkin-Elmer model 240C elemental analyzer. IR spectra (KBr discs,4000~400 cm-1) were recorded on a Perkin-Elmer Spectrum One FTIR spectrometer. NMR spectra were carried out on a Bruker Avance III (400 MHz) spectrometer. UV-Vis was performed on a Perkin-Elmer Lambda900 spectrometer.
2.2. Electrochemistry
The cyclic voltammograms (CV) were obtained using a CH instruments Model 630A Electrochemi-cal Workstation in CH2Cl2 solution containing 0.1 M (Bu4N)PF6 as the supporting electrolyte. All electro-chemical measurements were carried out at a scan rate of 100 mV·s-1 in the cathodic direction in a three-electrode cell under Ar atmosphere. The potential was measuredagainst a reference electrode Ag/AgCl,a 3 mm diameter glassy carbonworking electrodeand a platinum counter electrode.
2.3. X-ray crystallography
Single-crystal X-ray diffraction data of complex 1∙H2O were collected on a SuperNova diffractometer with a CuΚα radiation (λ = 1.54184 Å) at 293(2) K. A to tal of 17076 reflections were collected in the range of 3.52≤θ≤71.99° by using an ω-φ mode,of which 9670 were unique with Rint= 0.0476 and7644 were observed with I > 2σ(I). The structure was solved by direct methods using SHELXS-97[31] and refined by full-matrix least-squares on F2 techniques using SHELXL-97[32]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were deter-mined with difference Fourier maps or geometrical calculations riding on the related atoms,and their positions and thermal parameters were fixed during the structure refinement. The final R = 0.0748,wR =0.2012 (w= 1/[σ2(Fo2) + (0.1500P)2 + 1.8290P],P=(Fo2 + 2F 2) /3) ,S= 0.998,(Δ/σ) = 0.000,(Δρ)= 2.192 and (Δρ)min = -1.337 e/Å3. The crystallo-graphic data and structural refinements information for complex 1∙H2O are listed in Table 1.
Table 1

 DownLoad:
CSV
DownLoad:
CSV
Bond Dist. Bond Dist. Bond Dist. Fe(1) -C(7) 1.769(7) Fe(2) -S(1) 2.252(2) S(2) -C(1) 1.813(8) Fe(1) -P(1) 2.2347(16) P(1) -C(21) 1.822(6) S(3) -C(21) 1.719(6) Fe(1) -S(1) 2.2603(18) P(1) -C(9) 1.835(7) O(11) -H(11A) 0.8469 Fe(1) -Fe(2) 2.5114(13) P(1) -C(15) 1.845(7) Fe[a]-CCO,ba[b] 1.1300(26) Fe(2) -C(5) 1.799(8) S(1) -C(3) 1.853(9) Angle (°) Angle (°) Angle (°) C(7) -Fe(1) -C(8) 90.9(3) C(5) -Fe(2) -C(6) 89.4(4) C(21) -P(1) -Fe(1) 116.7(2) C(7) -Fe(1) -P(1) 98.1(2) C(4) -Fe(2) -C(6) 100.8(4) C(9) -P(1) -Fe(1) 116.4(2) S(1) -Fe(1) -S(2) 84.59(7) S(1) -Fe(2) -S(2) 85.23(7) C(15) -P(1) -Fe(1) 117.2(2) P(1) -Fe(1) -S(2) 110.48(7) C(4) -Fe(2) -S(2) 97.7(3) C(5) -Fe(2) -Fe(1) 100.6(3) C(7) -Fe(1) -Fe(2) 100.4(2) C(6) -Fe(2) -Fe(1) 105.9(3) C(8) -Fe(1) -Fe(2) 96.7(2) S(1) -Fe(1) -Fe(2) 56.01(5) S(1) -Fe(2) -Fe(1) 56.34(5) H(11A)-O(11) -H(11B) 126.1 P(1) -Fe(1) -Fe(2) 155.51(6) C(4) -Fe(2) -Fe(1) 144.6(3) C(3) -S(1) -Fe(1) 114.0(3) Fe(2) -S(1) -Fe(1) 67.64(6) Fe(2) -S(2) -Fe(1) 67.46(6) C(1) -S(2) -Fe(1) 116.1(3) C(1) -S(2) -Fe(2) 109.7(3) C(3) -S(1) -Fe(2) 110.0(3) [a] The Fe atoms are the ones only in one [Fe2S2] unit. [b] Average of five Fe-CCO,ba bonds 2.4. Synthesis
Synthesis of PTP
Diphenylphosphine (Ph2PCl,4.0 mL,21.7 mmol) indry tetrahydrofuran (THF,30 mL) was added to the THF solution containing excess lithium (0.455 g,65.0 mmol) in an ice bath. Then the mixture was warmed to room temperature and refluxed for 3 h. After cooling,the red solution was added dropwise to the solution of 2,5-dibromothiophene (2.178 g,9 mmol) in THF (15 mL) at 0 ℃. The new black mixture was then stirred at room temperature until the reaction was completelyfinished. Saturated NH4Cl solution was added after the solvent was evaporated. The aqueous phase was extracted with dichloromethane (3*50 mL) and the organic phase was dried with anhydrous MgSO4. After the solvent was removed,the crude product was purified by chromatography on silica gel (100 ~ 200) with dichloromethane/petroleum (v/v = 1:5) and dichloro-methane as gradient eluent to give an off-white solid. Theyield was 1.86 g (46.1%). Anal. Calcd. for C28H22P2S: C,74.34; H,4.87; S,7.08%. Found: C,74.14; H,4.87; S,7.30%.1H NMR (400 MHz,CDCl3,ppm): 7.37~7.29 (m,20H),7.17 (dd,1H),7.01 (ddd,1H).31P NMR (162 MHz,CDCl3) : -19 ppm.
Synthesis of [(μ-PDT)Fe2(CO)5]2(PTP) (1)
A solution of Fe2(μ-PDT)(CO)6 (0.386 g,1 mmol) and Me3NO·2H2O (0.122 g,1.1 mmol) in 20 mL dry acetonitrile was stirred for 15 min at room tem-perature. Then the ligand PTP (0.276 g,0.5 mmol) was added and the reaction mixture was stirredfor 12 h at room temperature. The resulting solution was evaporated under reduced pressure.The residue was purified by chromatography on silica gel (100~200) with dichloromethane/petroleum (v/v = 1:5) as eluent to give a red solid. The yield was 0.411 g (67.8%). The crystals of 1∙H2O suitable for X-ray crystallography study were grown upon slow diffusion of hexane into a dichloromethanesolution containing 1 at 4 ℃ . Through high-temperature (150 ℃)vacuum drying,complex 1 could be got from 1∙H2O,then characterized by elemental analy-sis,IR spectra,UV-vis absorption spectra,1H-NMR and 31P-NMR. Anal. Calcd. for C44H34Fe4O10P2S5: C,45.21; H,2.91; O,13.70; S,13.70%. Found: C,45.53; H,3.09; O,13.13; S,14.24%. IR (KBr,cm-1): v(CO) 2044,1984,1953,1935.1H NMR (400 MHz,CDCl3,ppm): d 7.74~7.34 (m,20H),7.28 (s,2H),1.78 (t,8H),1.67~1.54 (m,4H).31P NMR (162 MHz,CDCl3) : d 54.39 (s).
Table 2
Table 2. Hydrogen Bond Lengths (Å) and Bond Angles (°)DownLoad:
CSV
D-H···A d(D-H) d(H···A) d(D···A) ∠DHA O(11) -H(11A)···O(11) a 0.8462(5) 1.9549(5) 2.8013(7) 176.2(3) O(11) -H(11B)···O(4) 0.8435(6) 2.5494(6) 3.3919(8) 176.6(1) Symmetry code: (a) -x,-y+2,-z+1 Table 3
Table 3. Characteristic IR Absorption Peaks of v(CO) BandsDownLoad:
CSV
Complexes v(CO) KBr/cm-1 ve1st(CO)KBr/cm-1 Fe2(μ-PDT)(CO)6 2074,2029,1988 0 1 2044,1984,1953,1935 30 [(μ-pdt)Fe2(CO)5]2(PNP)d 2041,1977,1985,1933 33 [(μ-pdt)Fe2(CO)5]2(PNNP)d 2044,1983,1957,1935 30 d reported by our group[18]. e v1st(CO) = ν1st(CO)1 -ν1st(CO)mono-substituted Figure 1
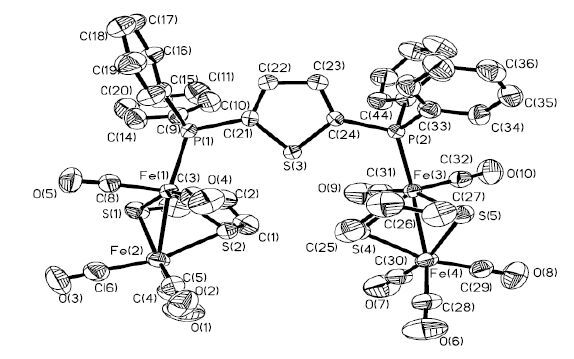
 Figure 1. ORTEP drawing ofcomplex 1 with thermal ellipsoids at 30% probability. Hydrogen atoms are omitted for clarity
Figure 1. ORTEP drawing ofcomplex 1 with thermal ellipsoids at 30% probability. Hydrogen atoms are omitted for clarityFigure 2
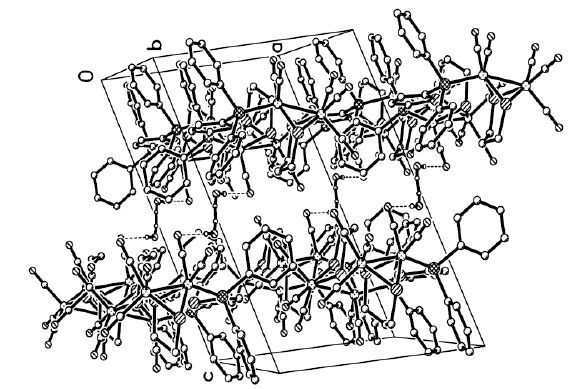
 Figure 2. Packing diagram of complex 1,showing the formation of the three-dimensional supramolecular framework
Figure 2. Packing diagram of complex 1,showing the formation of the three-dimensional supramolecular frameworkFigure 3
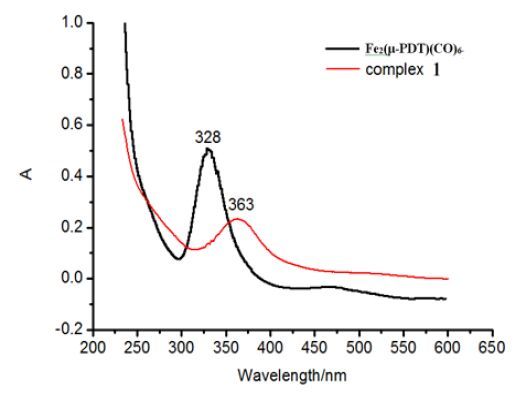
 Figure 3. UV-visabsorption spectra of Fe2(μ-PDT)(CO)6 and 1 recorded in theCH2Cl2 solution
Figure 3. UV-visabsorption spectra of Fe2(μ-PDT)(CO)6 and 1 recorded in theCH2Cl2 solutionFigure 4
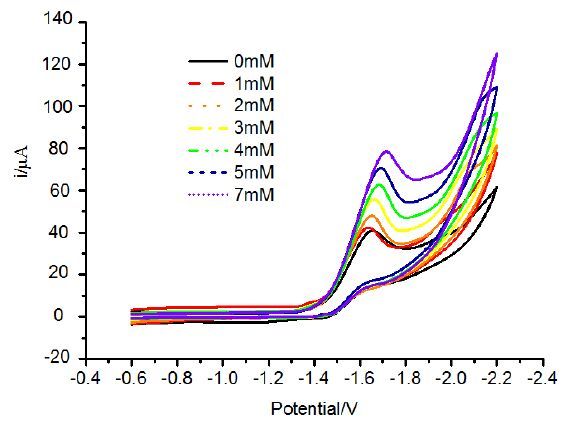
 Figure 4. Cyclic voltammogram of 1 (1.0 mM) in the presence of CH3COOH at a scan rateof 100 mV s-1 in 0.1 M n-Bu4NPF6 CH2Cl2 solution
Figure 4. Cyclic voltammogram of 1 (1.0 mM) in the presence of CH3COOH at a scan rateof 100 mV s-1 in 0.1 M n-Bu4NPF6 CH2Cl2 solution3. RESULTS AND DISCUSSION
3.1. Structure description
In the tetranuclear complex 1∙H2O,each [2Fe2S] butterfly unit is attached to one P atom of the diphosphine bridge and exhibits a square-pyramidal geometry. Different from the behaviors of reported mono-or di-phosphine ligands[33-36],each P atom of the diphosphine in 1∙H2O selects the apical position andowns space group P1 . Besides,both the tilts of the propanedithiolate rings in 1∙H2O are away from the ligand PTP,which is similar to the structuresof [(μ-PDT)Fe2(CO)5]2(PNP) and [(μ-PDT)Fe2 (CO)5]2-(PNNP)[18]. The Fe-Fe distance is 2.5114(13) Å,consistent with those found in tertiary pho-sphane-substituted diiron analogues reported by our group and close to the starting material Fe2(μ-PDT)(CO)6(2.5109(6) Å)[37]. The Fe-P distance (2.2347(16) Å) is longer by 0.02 Å than that of [(μ-PDT)Fe2(CO)5]2(PNNP) (2.2155(7) Å)[18] and is almost in close proximity to that of [(μ-PDT)Fe2(CO)5]2(PNP) (2.2315(11) Å)[18],which is in good agreement with the poor donor ability of PTPligand. The angles C(1) -S(2) -Fe(2) (109.7(3) °) and C(3) -S(1) -Fe(2) (110.0(3) °) are slightly less than C(3) -S(1) -Fe(1) (114.0(3) °) and C(1) -S(2) -Fe(1) (116.1(3) °) because bulk ligand PTP causes the propanedithiolate ring to lean towards the Fe(CO)3 unit. Moreover,the different anglesof C(3) -S(1) -Fe(1) and C(1) -S(2) -Fe(1) for 1∙H2O are significantly larger than that of [(μ-PDT)Fe2 (CO)5]2-(PNP) (110.2(9) °,113.0(4) °),which indicates that the steric hindrance plays an important role in the tilts of the propanedithiolate rings. The 4.6° difference between P(1) -Fe(1) -S(2) (110.48(7) °) and C(6) -Fe(2) -Fe(1) (105.9(3) °) demonstrates that the introduction of π-acceptor phosphane ligand leads to the decreaseof electron donation of the iron centers and weakens the π-back-bonding from the ironatoms to the carbonyl atoms.
Strong hydrogen bond is observedin the complex 1∙H2O: the H atoms from water in the compound are combined to both the O atoms in another water and O(4) in the same molecule. The bond lengths of H(11A)…O(11) a and H(11B)…O(4) are 1.9549(5) and 2.5494(6) Å. In addition,the bond angles of O(11) -H(11A)…O(11) aand O(11) -H(11B)…O(4) are 176.2(3) ° and 176.6(1) °,respectively. The hydrogen bond is beneficial to the stability of the three-dimensional supramolecular framework.
3.2. Optical spectroscopy
The IR spectra of 1 display four characteristic peaks in the range of 1935~2044 cm-1,belonging to the carbonyl stretching pattern for terminally bonded CO groups. The IR data of ν(CO) of 1,[(μ-PDT)Fe2(CO)5]2(PNP) and [(μ-PDT)Fe2(CO)5]2-(PNNP),together with that of compound (μ-PDT)Fe2(CO)6,are listed in Table 3 for clear comparison. Compared with CO bands in the IR spectra of (μ-PDT)Fe2(CO)6 shifts,the shiftsof (μ-PDT)Fe2(CO)6,[(μ-PDT)Fe2(CO)5]2(PNP) and [(μ-PDT)Fe2(CO)5]2(PNNP) move to lower energy due to the electron-donating effects of phosphine ligands PTP and PNP,PNNP. The S atom in thiophene ring and two N atoms in the pyridine ring cause a 3 cm-1 blue-shift of the first (CO) band of 1 and [(μ-PDT)Fe2(CO)5]2(PNNP) compared with that of [(μ-PDT)Fe2(CO)5]2(PNP). That is to say,the S atom in the thiophene ring and two N atoms in the pyridine ring nearly have the same effects on the shifts of the first (CO) band and the introduction of another N atom in the pyridine ring makes a differencein the shifts of the first(CO) band.
At room temperature,the UV-visibleabsorption spectrum of complexes (μ-PDT)Fe2(CO)6 and 1 in CH2Cl2 solution are shown in Fig. 3. Strong absorp-tion peaks near 230~300 nm are attributed to π-π* ligand charge transferto the center metal (LC),based on spectroscopic data center iron absorption in the visible region. Meanwhile,the moderate intensity weaker absorption peaks in the range of 300 ~ 450 nm are attributed to the transition metal-to-ligand charge (MLCT). For complex 1,the MLCT absorption is located at 363 nm,which is the same with [(μ-PDT)Fe2(CO)5]2(PNP) and has a red shift of 10 nm than [(μ-PDT)Fe2(CO)5]2(PNNP)[18]. That is to say,the PTP and PNP ligands have similar effects on the UV-vis absorption. In addition,the PTPligand has larger impact on the charge transfer than the PNNP ligand and indicates a large red shift than that in complex(μ-PDT)Fe2(CO)6[18].
3.3. Electrochemical properties
PTP-substituted complex 1 displays an irrever-sible reduction peak at -1.64 V in the CH2Cl2 solu-tion. As the acetic acid (CH3COOH) was added to the CH2Cl2 solution,the peaks moved from -1.64 V without CH3COOH to -1.72 V with 7 mM CH3COOH,which indicated that catalytic reaction occurred in the system. This is different from the result of PNP-substituted tetranuclear complex [(μ-PDT)Fe2(CO)5]2(PNNP),in which there are two reduction events.But it is similar to the results of PNP-substituted tetranuclear complex [(μ-PDT)-Fe2(CO)5]2(PNP),in which only the [2Fe2S]centres are reducedby one step at -1.60 V with consumptions of two electrons[18]. The only reduc-tion potential of 1 is comparable to those of dppe-substituted tetranuclear complexes,which are ascribed to the reduction processes of [FeIFeI] to [FeIFe0][38]. Anyway,the cyclic voltammograms show that complex 1 could catalyze hydrogen production (Fig. 4) .
4. CONCLUSION
In summary,a new five-heterocyclic-biphos-phine-substituted Fe-only hydrogenase mimic (complex 1) in a square-pyramidal geometry with P1 space group was synthesized at room tempera-ture. The rigid and conjugate bisphosphine (PTP) bridged to the diiron complex 1 can decreasethe electron donation of the iron centers and weaken the π-back-bonding from the iron atoms to the carbonyl atoms. Moreover,complex 1 can catalyze hydrogen production with a favorable reduction potential at -1.64 V.
-
-
[1]
Lawrence J. D, Li H, Rauchfuss T. B, Benard M, Rohmer M. M. Diiron azadithiolates as models for the iron-only hydrogenase active site: synthesis, structure, and stereoelectronics[J]. Angew. Chem., Int. Ed., 2001, 40: 1768-1771. doi: 10.1002/(ISSN)1521-3773
-
[2]
Ott S, Kritikos M, Akermark B, Sun L C, Lomoth R. A biomimetic pathway for hydrogen evolution from a model of the iron hydrogenase active site[J]. Angew. Chem., Int. Ed., 2004, 43: 1006-1009. doi: 10.1002/(ISSN)1521-3773
-
[3]
Felton G. A. N, Mebi C. A, Petro B. J, Vannucci A. K, Evans D. H, Glass R. S, Lichtenberger D. L. Review of electrochemical studies of complexes containing the Fe2S2 core characteristic of[J]. J. Organomet. Chem., 2009, 694: 2681-2699. doi: 10.1016/j.jorganchem.2009.03.017
-
[4]
Baltazar C. S. A, Marques M. C, Soares C. M, DeLacey A. M, Pereira I. A. C, Matias P. M. Nickel-iron-selenium hydrogenases-an overview[J]. Eur. J. Inorg. Chem., 2011, 7: 948-962.
-
[5]
Frey, M. Hydrogenases: hydrogen-activating enzymes. ChemBioChem. 2002, 2-3, 153-160.
-
[6]
Peters J. W, Lanzilotta W. N, Lemon B. J, Seefeldt L. C. X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution[J]. Science, 1998, 282: 1853-1858. doi: 10.1126/science.282.5395.1853
-
[7]
Peters J. W. Structure and mechanism of iron-only hydrogenases[J]. Curr. Opin. Struc. Biol., 1999, 9: 670-676. doi: 10.1016/S0959-440X(99)00028-7
-
[8]
Huo F. W, Hou J, Chen G. C, Guo D. M, Peng X. J. [FeFe]-Hydrogen models: overpotential control for electrocatalytic H2 production by tuning of the ligand π-acceptor ability. Eur. J[J]. Inorg. Chem., 2010, 25: 3942-3951.
-
[9]
Chong D, Georgakaki I. P, Mejia-Rodriguez R, Sanabria-Chinchilla J, Soriaga M. P, Darensbourg M. Y. Electrocatalysis of hydrogen production by active site analogues of the iron hydrogenase enzyme: structure/function relationships[J]. Dalton Trans., 2003, 21: 4158-4163.
-
[10]
Schwartz L, Eilers G, Eriksson L, Gogoll A, Lomoth R, Ott S. Iron hydrogenase active site mimic holding a proton and a hydride[J]. Chem. Commun., 2006, 5: 520-522.
-
[11]
Si G, Wang W. G, Wang H. Y, Tung C. H, Wu L. Z. Facile synthesis and functionality-dependent electrochemistry of Fe-only hydrogenase mimics[J]. Inorg. Chem., 2008, 47: 8101-8111. doi: 10.1021/ic800676y
-
[12]
Gloaguen F, Lawrence J. D, Rauchfuss T. B, Benard M, Rohmer M. M. Bimetallic carbonyl thiolates as functional models for Fe-only hydrogenases[J]. Inorg. Chem., 2002, 41: 6573-6582. doi: 10.1021/ic025838x
-
[13]
Gloaguen F, Lawrence J. D, Rauchfuss T. B. Biomimetic hydrogen evolution catalyzed by an iron carbonyl thiolate[J]. J. Am. Chem. Soc., 2001, 123: 9476-9477. doi: 10.1021/ja016516f
-
[14]
Chong D. S, Georgakaki I. P, Mejia-Rodriguez R, Samabria-Chinchilla J, Soriaga M. P, Darensbourg M. Y. Electrocatalysis of hydrogen production by active site analogues of the iron hydrogenase enzyme: structure /function relationships[J]. Dalton Trans., 2003, 21: 4158-4163.
-
[15]
Capon J. F, Gloaguen F, Schollhammer P, Talarmin J. Electrochemical proton reduction by thiolate-bridged hexacarbonyldiiron clusters[J]. J. Electroanal. Chem., 2004, 566: 241-247. doi: 10.1016/j.jelechem.2003.11.032
-
[16]
Song L. C, Yang Z, Bian H. Z, Hu Q. M. Novel single and double diiron oxadithiolates as models for the active site of[J]. Organometallics, 2004, 13: 3082-3084.
-
[17]
Si Y, Charreteur K, Capon J. F, Gloaguen F. Pétillon, F. Y, Schollhammer, P. Talarmin, J. Non-innocent bma ligand in a dissymetrically disubstituted diiron dithiolate related to the active site of the[J]. J. Inorg. Biochem., 2010, 104: 1038-1042. doi: 10.1016/j.jinorgbio.2010.05.011
-
[18]
Cui H. H, Wu N. N, Wang J. Y, Hu M. Q, Wen H. M, Chen C. N. Pyridyl- and pyrimidyl-phosphine-substituted[J]. J. Organomet. Chem., 2014, 767: 46-53. doi: 10.1016/j.jorganchem.2014.04.026
-
[19]
Capon J. F, Gloaguen F, Schollhammer P, Talarmin J. Activation of proton by the two-electron reduction of a di-iron organometallic complex[J]. J. Electroanal. Chem., 2006, 595: 47-52. doi: 10.1016/j.jelechem.2006.06.005
-
[20]
Felton G. A. N, Vannucci A. K, Chen J. Z, Lockett L. T, Okumura N, Petro B. J, Zakai U. I, Evans D. H, Glass R. S, Lichtenberger D. L. Hydrogen generation from weak acids: electrochemical and computational studies of a diiron hydrogenase mimic[J]. J. Am. Chem. Soc., 2007, 41: 12521-12530.
-
[21]
Chen L, Wang M, Gloaguen F, Zheng D. H, Zhang P. L, Sun L. C. Multielectron-transfer templates via consecutive two-electron transformations: iron-sulfur complexes relevant to biological enzymes[J]. Chem. Eur. J., 2012, 18: 13968-13973. doi: 10.1002/chem.v18.44
-
[22]
Chen L, Wang M, Gloaguen F, Zheng D. H, Zhang P. L, Sun L. C. Tetranuclear iron complexes bearing benzenetetrathiolate bridges as four-electron transformation templates and their electrocatalytic properties for proton reduction[J]. Inorg. Chem., 2013, 52: 1798-1806. doi: 10.1021/ic301647u
-
[23]
Kang D. M, Kim S. G, Lee S. J, Park J. K, Park K. M, Shin S. C. Synthesis, characterization, and absorption spectra of metallamacrocycles,[J]. Soc., 2005, 9: 1390-1394.
-
[24]
Sevillano P, Fuhr O, Hampe O, Lebedkin S, Neiss C, Ahlrichs R, Fenske D, Kappes M. M. Synthesis, characterization and quantum mechanical calculations of[J]. J. Inorg. Chem., 2007, 33: 5163-5167.
-
[25]
Stott T. L, Wolf M. O, Patrick B. O. Structural and electronic properties of phosphino(oligothiophene) gold(I) complexes[J]. Inorg. Chem., 2005, 3: 620-627.
-
[26]
Brown, J. M.; Lucy, A. R. Trans-bis(diphenylphosphino)cyclopropane; a ligand selective for binuclear complexation with ca. 4.5 ? intermetallic separation. J. Organomet. Chem. 1986, 1-2, 241-246.
-
[27]
Hourihane R, Gray G, Spalding T, Deeney T. Synthesis and spectroscopic characterisation of compounds with formula[J]. Chem., 2002, 642: 40-47.
-
[28]
Li P, Wang M, He C. J, Li G. H, Liu X. Y, Chen C. N, Akermark B, Sun L. C. Influence of tertiary phosphanes on the coordination configurations and electrochemical properties of iron hydrogenase model complexes: crystal structures of[J]. J. Inorg. Chem., 2005, 12: 2506-2513.
-
[29]
Messelhauser J, Lorenz I. P, Haug K, Hiller W. Synthesis and structure of the ethenedithiolato complex[J]. Z. Naturforsch Teil. B, 1985, 40: 1064-1067.
-
[30]
Stott T. L, Wolf M. O. Spectroscopic study of phosphine-substituted oligothiophenes[J]. J. Phys. Chem. B, 2004, 108: 18815-18819. doi: 10.1021/jp047037g
-
[31]
Sheldrick, G. M. SHELXS97, Program for the Solution of Crystal Structure. University of G?ttingen, Germany 1997.
-
[32]
Sheldrick, G. M. SHELXL97, Program for the Refinement of Crystal Structure. University of G?ttingen, Germany 1997.
-
[33]
Zhao X; Georgakaki I. P, Miller M. L, Mejia-Rodriguez R, Chiang C. Y, Darensbourg M. Y. Catalysis of H2/D2 scrambling and other H/D exchange processes by[J]. Inorg. Chem., 2002, 15: 3917-3928.
-
[34]
Ott S, Borgstrom M, Kritikos M, Lomoth R, Bergquist J, Akermark B, Hammarstrom L, Sun L. C. Model of the iron hydrogenase active site covalently linked to a ruthenium photosensitizer: synthesis and photophysical properties[J]. Inorg. Chem., 2004, 15: 4683-4692.
-
[35]
Gloaguen F, Lawrence J. D, Rauchfuss T. B, Benard M, Rohmer M. M. Bimetallic carbonyl thiolates as functional models for Fe-Only hydrogenases[J]. Inorg. Chem., 2002, 25: 6573-6582.
-
[36]
Gao W. M, Liu J. H, Akermark B, Sun L. C. Bidentate phosphine ligand based Fe2S2-containing macromolecules: synthesis, characterization, and catalytic electrochemical hydrogen production[J]. Inorg. Chem., 2006, 23: 9169-9171.
-
[37]
Matthews S. L, Heinekey D. M. A carbonyl-rich bridging hydride complex relevant to the Fe-Fe hydrogenase active site[J]. Inorg. Chem., 2010, 49: 9746-9748. doi: 10.1021/ic1017328
-
[38]
Gao W. M, Ekstrom J, Liu J. H, Chen C. N, Eriksson L, Weng L. H, Akermark B, Sun L. H. Binuclear iron-sulfur complexes with bidentate phosphine ligands as active site models of Fe-hydrogenase and their catalytic proton reduction[J]. Inorg. Chem., 2007, 46: 1981-1991. doi: 10.1021/ic0610278
-
[1]
-
Figure 1 ORTEP drawing ofcomplex 1 with thermal ellipsoids at 30% probability. Hydrogen atoms are omitted for clarity
Figure 2 Packing diagram of complex 1,showing the formation of the three-dimensional supramolecular framework
Figure 3 UV-visabsorption spectra of Fe2(μ-PDT)(CO)6 and 1 recorded in theCH2Cl2 solution
Figure 4 Cyclic voltammogram of 1 (1.0 mM) in the presence of CH3COOH at a scan rateof 100 mV s-1 in 0.1 M n-Bu4NPF6 CH2Cl2 solution
Table 1. Selected Bond Lengths (Å) and Bond Angles (°)
Bond Dist. Bond Dist. Bond Dist. Fe(1) -C(7) 1.769(7) Fe(2) -S(1) 2.252(2) S(2) -C(1) 1.813(8) Fe(1) -P(1) 2.2347(16) P(1) -C(21) 1.822(6) S(3) -C(21) 1.719(6) Fe(1) -S(1) 2.2603(18) P(1) -C(9) 1.835(7) O(11) -H(11A) 0.8469 Fe(1) -Fe(2) 2.5114(13) P(1) -C(15) 1.845(7) Fe[a]-CCO,ba[b] 1.1300(26) Fe(2) -C(5) 1.799(8) S(1) -C(3) 1.853(9) Angle (°) Angle (°) Angle (°) C(7) -Fe(1) -C(8) 90.9(3) C(5) -Fe(2) -C(6) 89.4(4) C(21) -P(1) -Fe(1) 116.7(2) C(7) -Fe(1) -P(1) 98.1(2) C(4) -Fe(2) -C(6) 100.8(4) C(9) -P(1) -Fe(1) 116.4(2) S(1) -Fe(1) -S(2) 84.59(7) S(1) -Fe(2) -S(2) 85.23(7) C(15) -P(1) -Fe(1) 117.2(2) P(1) -Fe(1) -S(2) 110.48(7) C(4) -Fe(2) -S(2) 97.7(3) C(5) -Fe(2) -Fe(1) 100.6(3) C(7) -Fe(1) -Fe(2) 100.4(2) C(6) -Fe(2) -Fe(1) 105.9(3) C(8) -Fe(1) -Fe(2) 96.7(2) S(1) -Fe(1) -Fe(2) 56.01(5) S(1) -Fe(2) -Fe(1) 56.34(5) H(11A)-O(11) -H(11B) 126.1 P(1) -Fe(1) -Fe(2) 155.51(6) C(4) -Fe(2) -Fe(1) 144.6(3) C(3) -S(1) -Fe(1) 114.0(3) Fe(2) -S(1) -Fe(1) 67.64(6) Fe(2) -S(2) -Fe(1) 67.46(6) C(1) -S(2) -Fe(1) 116.1(3) C(1) -S(2) -Fe(2) 109.7(3) C(3) -S(1) -Fe(2) 110.0(3) [a] The Fe atoms are the ones only in one [Fe2S2] unit. [b] Average of five Fe-CCO,ba bonds  下载: 导出CSV
下载: 导出CSV
Table 2. Hydrogen Bond Lengths (Å) and Bond Angles (°)
D-H···A d(D-H) d(H···A) d(D···A) ∠DHA O(11) -H(11A)···O(11) a 0.8462(5) 1.9549(5) 2.8013(7) 176.2(3) O(11) -H(11B)···O(4) 0.8435(6) 2.5494(6) 3.3919(8) 176.6(1) Symmetry code: (a) -x,-y+2,-z+1
下载: 导出CSV
Table 3. Characteristic IR Absorption Peaks of v(CO) Bands
Complexes v(CO) KBr/cm-1 ve1st(CO)KBr/cm-1 Fe2(μ-PDT)(CO)6 2074,2029,1988 0 1 2044,1984,1953,1935 30 [(μ-pdt)Fe2(CO)5]2(PNP)d 2041,1977,1985,1933 33 [(μ-pdt)Fe2(CO)5]2(PNNP)d 2044,1983,1957,1935 30 d reported by our group[18]. e v1st(CO) = ν1st(CO)1 -ν1st(CO)mono-substituted
下载: 导出CSV
-





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 4656
- HTML全文浏览量: 113
