
Figure 1.
Mechanism of the coordination of CO2 with a transition metal atom

The accelerated consumption of fossil energy led to the growth of carbon oxide, and utilization of carbon dioxide has become the focus of world attention[1-3]. The value of the products from the fixation of carbon dioxide nor is the only consideration prompting research in this area[4, 5]. Under normal conditions, carbon dioxide is one of the most thermodynamically stable and inert triatomic molecules. Therefore, its activation and conversion into useful organic compounds requires a great deal of energy input[6-8].
The interaction between simple molecules and transition metal is easy to form complexes because transition metal contains d orbitals unfilled. Moreover, the coordination of CO2 with a transition metal atom is deemed to be a key step in its activation. For the reaction of CO2 with transition metal, three kinds of mechanisms are accepted (as shown in Fig. 1), in which η1-side-on (addition mechanism of C) and η1-C (insertion mechanism) have been confirmed by X-ray crystallography. Only spectroscopic data are available to suggest that the η1-O-end-on (addition mechanism of O) bound geometry occurs in some complexes[9].
By means of the test on transition metal complexes in the CO2 hydrogenation, Solymosi et al.[10] and Weatherbee et al.[11] found that Ru is the best metal for the activity of catalysts. Through the study on relativistic DFT, Xian-Yang Chen et al. discussed the reaction mechanism of transition metal Ru with CO2, and showed that it is easier to insert into a C-O bond yielding ORuCO species[12]. Catalytic hydrogenation of carbon dioxide produces various kinds of chemicals or fuels such as methanol and formaldehyde[13-15]. Several articles have reviewed about the hydrogenation of carbon dioxide. To clearly explain the catalytic mechanism for carbon dioxide hydrogenation, there are commonly referenced reactions[16, 17]:
Transition metal complexes are usually containing more than one unpaired electrons, so spin inversion more likely happens to change the spin states by spin-orbital coupling (SOC), and intersystem crossing (ISC) occurs between the potential energy surfaces of different spin states[18-21]. Schröder et al.[22], denoted this as "two-state reactivity" (TSR) for the reactions involving a spin inversion that enables the system to find and follow low-energy reaction pathways. To understand how metal atoms can react with CO2 and what the reaction mechanisms in the catalytic processes are, as well as how the two-state reactions of spin-forbidden happen, here we present a detailed study of the reaction mechanism in gas phase of ruthenium atom catalyst for the hydrogenation of CO2 at the singlet, triplet and quintet states. We found the spin orbital coupling (SOC) located at the crossing points (CPs), calculated out the intersystem crossing (ISC) occurring with a large probability, and determined the minimum energy reaction pathway in a two-state reactivity.
In order to study the reaction mechanism of ruthenium catalyst of carbon dioxide hydrogenation, computations were carried out with the Gaussian03 program package[23]. All calculations use density functional theory (DFT) program under the B3LYP level[24, 25]. The standardized 6-311++G (3df, 3pd) basis set was used for the H, C and O[26] and the LANL2DZ pseudo potential basis was applied for Ru. The IRC was then calculated and used to track the minimum energy path from transition states to the corresponding minima, probe the reaction path, and check if the correct transition state is located. In this paper, we have adopted the method which Yoshizawa et al.[27] reported about the intrinsic coordinate single point vertical excited to find out the crossing point (CP) between different spin states. The minimum energy crossing point (MECP) between the multistate hypersurfaces is based on CP and obtained by using the energy gradient optimization method of Harvey et al[28].
The coupling of nuclear motion and the electronic movement will induce transitions between different potential energy surfaces. The Landau-Zener approximate formula[29-31] has been successfully applied to calculate the thermally-averaged spin transition probabilities between two different electronic states[32]. The results calculated show that the probability of intersystem crossing (ISC) is higher, which is from one state to the other state. The probability of surface hopping through the crossing point is studied by the Landau-Zener model given in the Computational Details section. In eq. 1, HSOC is the spin-orbital coupling energy between the two states, and ▽E1 and ▽E2 are the mass-weighted gradients at the point of crossing and ν is the velocity of the molecule at MECP[32].
|
|
where Si and Lik are the orbital and spin angular momentum operators of an electron; Zk* is the effective nuclear charge; rik is the distance between the nucleus k and the electrons i. $\frac{{{\alpha }^{2}}}{2}=\frac{{{e}^{2}}}{2m_{e}^{2}{{c}^{2}}}$ is the fine constant[33].
In the non-adiabatic reaction, there are two transitions that would occur in kinds of traversal to avoid cross region. Probability of the first traversal involving crossing is PISC in the forward direction, and the probability of the second traversal involving crossing is PISC (1-PISC) in the reverse direction. The sum of the two probabilities for crossing point in the forward and reverse directions is expressed in the parameter κISC [34].
|
|
As for Ru + CO2 reaction mechanisms, there are two rival reaction channels: one is an addition mechanism, and the other is an insertion mechanism. However, as the high energy barrier is required for addition mechanism, it doesn't merit any additional discussion here.
With Ru atom, the ground state of Ru atom is 5F (4d75s1), the configuration of the lowest energy orbital of Ru (5F) is (5s)1(4d0)1(4d±1)2(4d2)2(4d-2)2; the difference between triplet 3F (4d85s0) and the ground state 5F is only one electron transition from s to dz2. 3F excited states are (5s)0(4d0)2 (4d±1)2 (4d2)2 (4d-2)2 and 1F excited states are (5s)0 (4d0)2 (4d±1)4 (4d2)2(4d-2)0.
For the optimized geometries of various compounds and transition states, see attached Drawing No. 1. Fig. 3 provides the energy of stagnation points of different spin states relative to Ru (5F) + CO2 + 3H2 potential energy diagrams at the B3LYP method. Table 1 presents vibrational frequencies (cm-1) of various transition states. For the hydrogen transfer reaction, the absolute value of virtual frequency is bigger than the non-hydrogen transfer reaction. And that is very likely causing the tunnel effect[35].
 DownLoad:
CSV
DownLoad:
CSV
| Triplet | 521.33i | 1420.68i | 1015.85i | 664.83i | 430.25i | 147.49i | 192.02i | 1252.68i |

The calculation indicates that the energies of singlet and triplet states are higher than the quintet state by 49.56 and 19.6 kcal/mol in the initial reaction Ru + CO2 + H2 (Fig. 3a), respectively. At the beginning of the reaction, Ru atom attaches to the C-O bond to form a planar ηC, O2 -coordination model of Ru (CO) O (3A) without any barrier, and the symmetry of CO2 (D∞h) is broken. The C-O bond length in 1/3/5IM0 intermediate is elongated to 1.26, 1.25 and 1.21 Å, respectively. The ∠OCO is changed from 180° to 142.8°, 144.1° and 151.9°, respectively. The triplet state has lower energy than the quintet state by 2.33 kcal/mol. For the quintet, NBO analysis shows 0.99 electronic in the C-Ru bonding. Composition is (44.34%) 0.66C (sp4.64) + (55.66%) 0.746Ru (sp0.03d12.57). It is the polar σ bond.
Computations of charge decomposition analysis were carried out using the Multiwfn program[36]. For the initial product 3IM0, the number of electrons donated from CO2 to Ru is 0.08, and Ru to CO2 is 0.13, and electrons involved in repulsive polarization are -0.1. For the initial product 5IM0, the number of electrons donated from CO2 to Ru is 0.14, and Ru to CO2 is 0.11. Electrons involved in repulsive polarization are -0.28. We can see from Table 2 and Fig. 4 that the frontier molecular orbital of 3IM0 is composed of atomic orbitals of Ru. The frontier molecular orbital of 5IM0 consists of Ruthenium atomic orbitals and CO2 molecular orbitals, in which LUMO includes 37.85% CO2 and 61.89% Ru and HOMO contains 27.23% CO2 and 72.76% Ru.
DownLoad:
CSV
| Quintet | 18 | –207.08 | 0.8088 | 0.81 | 0.1912 | 0.19 |
| 19 | –200.8 | 0.9199 | 0.92 | 0.0802 | 0.08 | |
| 20(HOMO) | –194.53 | 0.4929 | 0.49 | 0.5071 | 0.51 | |
| 21(LUMO) | –131.78 | 0.7276 | 0.73 | 0.2723 | 0.27 | |
| 18 | –156.88 | 0.9186 | 0.92 | 0 | 0 | |
| 19 | –156.88 | 0.8621 | 0.86 | 0 | 0 | |
| Triplet | 20 | –150.6 | 0.8076 | 0.81 | 0 | 0 |
| 21(HOMO) | –75.3 | 0.8523 | 0 | 0 | 0 | |
| E represents the energy of each orbital. | ||||||

The Ru atom is inserted when the TS1 change to IM1 from IM0 intermediate, via transition state TS1 to forms the IM1, which is confirmed by IRC calculation. This reaction step has 23.94 kcal/mol barrier height and exothermic energy of 29.49 kcal/mol on triplet. The Ru-O and Ru-C bonds in TS1 compared to that in IM0 are further shortened by about 0.1 Å; IM1 is more stable with 31.13 kcal/mol lower than that of the initial reactants. The process of IM1 hydrogenation form IM2 has exothermic caloric value of 24.79 kcal/mol on singlet. Finally, H shifts into O and dehydrate to form Ru-CO. The overall reactions on this pathway releases 54.55 and 53.56 kcal/mol heat on the singlet and triplet states, respectively. However, it requires 1.0 kcal/mol energy on quintet. The second stage is the formation of CH2O molecule (Fig. 3b). The first is Ru-CO hydrogenation to form IM5, which releases 33.97 and 7.31 kcal/mol energy on singlet and triplet. However, the quintet need absorb heat of 2.20 kcal/mol, this is adverse on the thermodynamics and kinetics. The third stage is (CH2O) Ru hydrogenation forming IM9 and then the hydrogen shifts into O and C forming CH3OH (Fig. 3c). This stage requires 30.81 and 27.41 kcal/mol heat on singlet and triplet, separately. Interestingly, the quintet is exothermic with caloric value of 8.45 kcal/mol. The reaction of CO2 Hydrogenation Catalyzed by Ru forming formaldehyde, which need absorb 13.84 kcal/mol quantities of heat. Experimental data shows that the reaction of CO2 hydrogenation to form CH2O requires 13.69 kcal/mol heat. However, the formation process of CH3OH releases exothermal energy of 4.87 kcal/mol. Experimental data also shows that the formation process of CH3OH with 4.92 kcal/mol, which is in good agreement with the theoretical calculation.
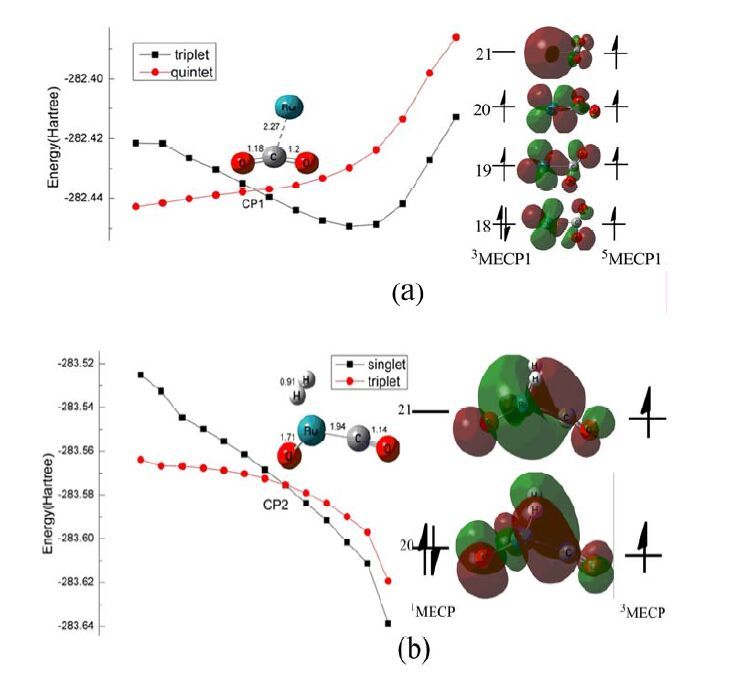
The spin-orbital coupling (SOC) matrix element is equal to zero if the spins for the left sagittal and ket vector differ by more than unity (.S' - S". > 1) by the selection rule and the probability of intersystem crossing will be swept aside[37]. For the whole catalytic cycle, three crossing points (CP1, CP2 and CP3) were obtained. Among them, CP1 locates between the quintet and triplet states, while CP2 and CP3 lie in the singlet-triplet states potential energy surfaces; they may play a key role in the nonadiabatic processes for the whole reaction path. As shown in Fig. 3, the relative potential energy of the reactants on quintet are more stable with 19.6 kcal/mol than the reactants on triplet. However, the energy of IM0 on the triplet is 2.33 kcal/mol lower than the quintet. Therefore, there is an energy crossing point (CP1) in the process of the formation of IM0, and the existence of MECP1 leads to intersystem crossing (ISC). The values of the spin densities of Ru at MECP1 are different on both PESs. For the MECP1, spin densities of Ru is 3.81 on the quintet state, and on that the triplet state is 2.19. Obviously, spin inversion occurs mainly on the Ru atom. Fig. 5a shows that the twenty-first orbital is the 5s orbital (82.8%) of Ru, which is the highest single occupied orbital (HOMO) of the quintet state, but it is also the lowest empty orbital (LUMO) of the triplet state. The twentieth orbital is the dxz orbital (94.8%) of Ru, which is the lowest single occupied orbital (SOMO) of the quintet state and the highest single occupied orbital (HOMO) of the triplet state. At this point, the spin density of Ru on the triplet occupies 2.08 a.u. and that on the quintet occupies 4.04 a.u, indicating that the spin flip occurs on Ru. The α single electron hooping from the s to the dxz orbital will undergo spin inversion. Compound 3IM0 is more stable than 5IM0. Similarly, in the process of IM1 hydrogenation, the relative potential energy of 3IM1 is more stable by 9.6 kcal/mol than 1IM1, but energy of IM2 on the singlet is 15.22 kcal/mol lower than the triplet. There is an energy crossing point (CP2) in the process of forming IM2. For the triplet state, the energy value of the spin densities of Ru is 1.28 a.u. at the MECP1. Namely, the spin inversion occurs on Ru. Fig. 5b shows that the twenty-first orbital is the dxy orbital (26.4%) of Ru, which is the highest single occupied orbital (HOMO) in triplet state, but is the lowest empty orbital (LUMO) on singlet state. The twentieth orbital is the lowest singly occupied orbital (SOMO) composed of dxz (30.2%) on the triplet state, which is the highest double occupied orbital (HOMO) on the singlet state. The α single electron leaps from the dxy orbital to the dxz orbital will undergo spin inversion. Compound 1IM2 is more stable than 3IM2. The step of IM2 to IM3 involves the transfer of H atoms from Ru to the O atom. For the triplet, reaction releases 34.29 kcal/mol in the whole process, and it needs to overcome a 4.72 kcal/mol barrier. For the singlet, reaction releases exothermal energy of 4.76 kcal/mol in the whole process, which needs to conquer a 24.97 kcal/mol barrier. The lowest-energy structure of the reac tant and the transition are located in the singlet state, whereas the lowest-energy structures for the product lie in triplet states. Spin-surface inversion occurs after the transition state, so the spin inversion has little effect on the kinetics.
In the present work, an estimate of the probability of ISC from one state to the other can be obtained using a semiclassical picture based on potential energy surfaces, where the probability of surface hopping through the crossing point is given by the Landau-Zener model (given in the theoretical calculations section). All parameters of the intersections are shown in Table 3. The large values of the spin transmission coefficient κISC show that the intersystem crossing is likely to occur.
DownLoad:
CSV
| MECP2 | 1826.81 | 0.0083 | 86.59 | 0.312 | 0.527 |
| SOC is the spin orbital coupling constant, HSOC(cm–1) is the spin-orbital coupling energy between the two states,ΔF(kcal/mol·Å) is the gradient of the two potential energy surfaces at the point of crossing. | |||||
The cycle reaction of carbon dioxide hydrogenation catalyzed by Ru in the gas phase has been studied at the B3LYP/6-311++G (3df, 3pd) ∪ LANL2DZ level of theory, consisting of three parts: carbon dioxide hydrogenation dehydration, the formation of formaldehyde and the formation of methanol. Dehydration part has three intersections (CP1, CP2 and CP3) and the results show that the reaction has typical two-state reactivity (TSR). For MECP, the large value of the spin transmission coefficient (κISC) was estimated, which shows that ISC occurs with great probability. The spin inversion exists among the singlet, triplet and quintet PESs, with the minimum energy reaction paths found as follows:
5Ru + CO2 + H2 → MECP1 →3IM0→
3TS1 → 3IM1 → MECP2 →
1IM2 → 1TS2 → MECP3 → 3IM3 →
3TS3 → 3IM4 → 3RuCO + H2O
This reaction step releases about 33.96 kcal/mol. The triplet reaction path is the minimum energy reaction path for the formation of formaldehyde and methanol. The formaldehyde synthesis from CO2 hydrogenation absorbs about 13.845 kcal/mol. However, the methanol synthesis releases 4.87 kcal/mol. That is to say, the reaction is more advantageous to form methanol.
Shi H, Chen G, Zhang C, Zou Z. Polymeric g-C3N4 coupled with NaNbO3 nanowires toward enhanced photocatalytic reduction of CO2 into renewable fuel[J]. Acs Catalysis, 2014, 4: 3637-3643. doi: 10.1021/cs500848f
Raksakoon C, Maihom T, Probst M, Limtrakul J. Hydration of carbon dioxide in copper-alkoxide functionalized metal-organic frameworks: a DFT study[J]. J. Phys. Chem. C, 2015, 119: 3564-3571. doi: 10.1021/jp511185p
Daza Y. A, Kent R. A, Yung M. M, Kuhn J. N. Carbon dioxide conversion by reverse water-gas shift chemical looping on perovskite-type oxides[J]. Ind. Eng. Chem. Res, 2014, 53: 5828-5837. doi: 10.1021/ie5002185
Zall C. M, Linehan J. C, Appel A. M. A molecular copper catalyst for hydrogenation of CO2 to formate[J]. ACS Catalysis, 2015, 5: 5301-5305. doi: 10.1021/acscatal.5b01646
Yang X, Kattel S, Senanayake S. D, Boscoboinik J. A, Nie X, Graciani J. Low pressure CO2 hydrogenation to methanol over gold nanoparticles activated on a CeOXTiO2 interface[J]. J. Am. Chem. Soc, 2015, 137: 10104-10107. doi: 10.1021/jacs.5b06150
Kobayashi K, Tanaka K. Reactivity of CO2 activated on transition metals and sulfur ligands[J]. Inorg. Chem, 2015, 54: 5085-5095. doi: 10.1021/ic502745u
Miller A. J. M, Labinger J. A, Bercaw J. E. Trialkylborane-assisted CO2 reduction by late transition metal hydrides[J]. Organomet. Chem, 2011, 30: 4308-4314. doi: 10.1021/om200364w
Mondal B, Neese F, Ye S. Control in the rate-determining step provides a promising strategy to develop new catalysts for CO2 hydrogenation: a local pair natural orbital coupled cluster theory study[J]. Inorg. Chem, 2015, 54: 7192-7198. doi: 10.1021/acs.inorgchem.5b00469
Jessop P. G, Ikariya T, Noyori R. Homogeneous hydrogenation of carbon dioxide[J]. Chem. Rev, 1995, 95: 259-272. doi: 10.1021/cr00034a001
Solymosi F, Erdöhelyi A. Hydrogenation of CO2 to CH4 over alumina-supported noble metals[J]. J. Mol Catal Rev, 1980, : 8471-8474.
Weatherbee G. D, Bartholomew C. H. Hydrogenation of CO2 on group VIII metals: IV[J]. Specific activities and selectivities of silica-supported Co, Fe, and Ru. J. Catal, 1984, 87: 352-362.
Chen X. Y, Zhao Y. X, Wang S. G. Relativistic DFT study on the reaction mechanism of second-row transition metal Ru with CO2[J]. J. Phys. Chem. A, 2006, 110: 3552-3558. doi: 10.1021/jp053296+
Wang W. H, Himeda Y, Muckerman J. T, Manbeck G. F, Fujita. E. CO2 Hydrogenation to formate and methanol as an alternative to photo- and electrochemical CO2 reduction[J]. Chem. Rev, 2015, 115: 12936-12973. doi: 10.1021/acs.chemrev.5b00197
Declercq R, Bouhadir G, Bourissou D, Légaré M. A, Courtemanche M. A, Nahi K. S. Hydroboration of carbon dioxide using ambiphilic phosphine-borane catalysts: on the role of the formaldehyde adduct[J]. ACS Catalysis, 2015, 5: 2513-2520. doi: 10.1021/acscatal.5b00189
Karamad M, Hansen H. A, Rossmeisl J, Norskov J. K. Mechanistic pathway in the electrochemical reduction of CO2 on RuO2[J]. ACS Catalysis, 2015, 5: 4075-4081. doi: 10.1021/cs501542n
Tominaga K. I, Sasaki Y, Kawai M, Watanabe T, Saito M. Ruthenium complex catalysed hydrogenation of carbon dioxide to carbon monoxide, methanol and methane[J]. J. Chem. Soc. Chem. Commun, 1993, 7: 629-631.
Tsuchiya K, Huang J. D, Tominaga K. Reverse water-gas shift reaction catalyzed by mononuclear Ru complexes[J]. ACS Catalysis, 2013, 3: 2865-2868. doi: 10.1021/cs400809k
Harvey J. N, Poli R, Smith K. M. Understanding the reactivity of transition metal complexes involving multiple spin states[J]. Coord. Chem. Rev, 2003, 238: 347-361.
Shaik S. Spin-orbital coupling in the oxidative activation of H-H by FeO+. Selection rules and reactivity effects[J]. J.Am. Chem. Soc, 1997, 119: 1773-1786. doi: 10.1021/ja963033g
Nian J, Wang Y, Ma W, Ji D, Wang C, La M. Theoretical investigation for the cycle reaction of N2O (x1Σ+) with CO (1Σ+) catalyzed by IrO n+(n = 1 2) and utilizing the energy span model to study its kinetic information[J]. J.Phys. Chem. A, 2011, 115: 11023-11032.
Ma W. P, Wang Y. C, Lv L. L, Jin Y. Z, Nian J. Y, Ji D. F, Wang Q. A theoretician’s view of the Ce+ mediated activation of the N-H bond in ammonia[J]. Comput. Theor. Chem, 2011, 977: 69-77. doi: 10.1016/j.comptc.2011.09.016
Schröder D, Shaik S, Schwarz H. Two-state reactivity as a new concept in organometallic chemistry[J]. Acc. Chem. Res, 2000, 33: 139-145. doi: 10.1021/ar990028j
Frisch, M. J[J]. , , : .
Becke A. D. Density-functional thermochemistry[J]. III. The role of exact exchange. J. Chem. Phys, 1993, 98: 5648-5652.
Lee C, Yang W, Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J]. Phys. Rev. B, 1988, 37: 785-789. doi: 10.1103/PhysRevB.37.785
Frisch M. J, Pople J. A, Binkley J. S. Self-consistent molecular orbital methods 25[J]. Supplementary functions for Gaussian basis sets. J. Chem. Phys, 1984, 80: 3265-3269.
Yoshizawa K, Shiota Y, Yamabe T. Intrinsic reaction coordinate analysis of the conversion of methane to methanol by an iron-oxo species: a study of crossing seams of potential energy surfaces[J]. J. Chem. Phys, 1999, 111: 538-545. doi: 10.1063/1.479333
Harvey J. N, Aschi M, Schwarz H. The singlet and triplet states of phenyl cation[J]. A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces. Theor. Chem. Acc, 1998, 99: 95-99.
Coveney P. V, Child M. S, Barany A. The two-state S matrix for the Landau-Zener potential curve crossing model: predissociation and resonant scattering[J]. J. Phys. B: At. Mol. Phys, 1985, 18: 4557-4580. doi: 10.1088/0022-3700/18/23/009
Zhu C. Y, Nakamura H. Theory of nonadiabatic transition for general two-state curve crossing problems[J]. II. Landau-Zener case. J. Chem. Phys, 1995, 102: 7448-7461.
Wittig C. The Landau-Zener Formu[J]. J.Phys. Chem. B, 2005, 109: 8428-8430. doi: 10.1021/jp040627u
Goodrow A, Bell A. T, Head-Gordon M. Are spin-forbidden crossings a Bottleneck in methanol oxidation[J]. J.Phys. Chem. C, 2009, 113: 19361-19364. doi: 10.1021/jp906603r
Jin Y. Z, Wang Y. C, Geng Z. Y, Wang H. J, Gan Y. Z. Competitive activation of C-H and C-F bonds in gas phase reaction of Ir+ with CH3F: a DFT study[J]. J. Organomet. Chem, 2012, 717: 195-201. doi: 10.1016/j.jorganchem.2012.07.017
Steinfeld J I, Francisco J. S, Hase W. L. Chemical kinetics and dynamics[J]. Prentice Hall, 1999, : .
Shavitt . On the problem of calculating the rate constants of elementary reactions[J]. Chem. Phys, 1959, 31: 1359-1367.
Lu T, Chen F. Multiwfn: a multifunctional wavefunction analyzer[J]. J. Comp. Chem, 2012, 33: 580-592. doi: 10.1002/jcc.v33.5
Fedorov D. G, Koseki S, Schmidt M. W, Gordon M. S. Spin-orbital coupling in molecules: chemistry beyond the adiabatic approximation[J]. Int. Rev. Phys. Chem, 2003, 22: 551-592. doi: 10.1080/0144235032000101743

Figure 3 Potential energy diagrams of the whole reaction pathway on singlet, triplet and quintet states (Relative energy is relative to the sum of electronic and zero-point energies of 5Ru+CO2+3H2. a is carbon dioxide hydrogenation dehydration part, b is the formation of formaldehyde and c is the formation of methanol)

Figure 4 Molecular orbital interaction diagram (The electrons distributed in both sides of the graphics of atom and molecular orbital are the same electron, which just make the graphics more intuitive and beautiful.)

Figure 5 Potential energies crossing points diagram along the reaction potential energy surfaces (PESs) and frontier molecular orbitals interaction analyses.
Table 1. Vibrational Frequencies (cm-1) of Various Transition States Calculated at the B3LYP Level Using the 6-311++G (3df, 3pd) ∪LANL2DZ Bases Set
| Triplet | 521.33i | 1420.68i | 1015.85i | 664.83i | 430.25i | 147.49i | 192.02i | 1252.68i |
 下载: 导出CSV
下载: 导出CSV
Table 2. Molecular Orbital Energy and Component of the Main Orbital of the Initial Product IM0
| Quintet | 18 | –207.08 | 0.8088 | 0.81 | 0.1912 | 0.19 |
| 19 | –200.8 | 0.9199 | 0.92 | 0.0802 | 0.08 | |
| 20(HOMO) | –194.53 | 0.4929 | 0.49 | 0.5071 | 0.51 | |
| 21(LUMO) | –131.78 | 0.7276 | 0.73 | 0.2723 | 0.27 | |
| 18 | –156.88 | 0.9186 | 0.92 | 0 | 0 | |
| 19 | –156.88 | 0.8621 | 0.86 | 0 | 0 | |
| Triplet | 20 | –150.6 | 0.8076 | 0.81 | 0 | 0 |
| 21(HOMO) | –75.3 | 0.8523 | 0 | 0 | 0 | |
| E represents the energy of each orbital. | ||||||
下载: 导出CSV
Table 3. Serial Parameters of Intersections
| MECP2 | 1826.81 | 0.0083 | 86.59 | 0.312 | 0.527 |
| SOC is the spin orbital coupling constant, HSOC(cm–1) is the spin-orbital coupling energy between the two states,ΔF(kcal/mol·Å) is the gradient of the two potential energy surfaces at the point of crossing. | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

