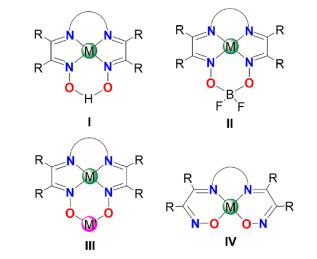
Figure 1.
Coordination modes of diimine-dioxime
Unusual Coordination Mode of Tetradentate Diiminedioxime Ligand in a Mononickel(Ⅱ) Complex: Synthesis, Characterization, and Computational Study
Hao-Miao LI , Jia-Le ZHAO , Deng-Meng SONG , Qing SHI , Ning WANG , Jun LI , Wen-Hua XU

Due to the coexistence of mildly acidic hydroxyl group and slightly basic imine unit, the name of oxime is derived from the combination of hydroxyl and imine, C=NOH. Since Chugaev discovered the reaction between Ni(Ⅱ) salts and dimethylglyoxime to form the [4N]-coordinated complex in 1905, a great deal of work on oxime and their coordination complexes has been published[1-3]. Subsequently, complexes of Co(Ⅲ) vic-dioximes have been extensively investigated as a result of their similarity to vitamin B12[4]. In recent years, the oxime-containing transition metal complexes have drawn great attention as catalysts for electro- and photocatalytic H2-evolving reaction (HER), not only due to the relatively high turnover frequencies and moderate overpotentials, but also to their designable and tunable structures[5-7]. Especially, these complexes containing alkyl bridged diimine-dioxime ligands exhibit much better stabilities and activities than those of bis-glyoximato complexes[8-11]. The redox properties of model complexes could be adjusted via modifying the alkyl bridge and the R groups on C=N unites.
As shown in Fig. 1, the bridged diimine-dioxime ligands could coordinate with metal atoms through N and/or O atoms in versatile coordination modes. In most complexes, the tetradentate ligand just coordinates via four N atoms, and the two oxygen atoms do not participate. The H-bridged coordination mode I and BF2-analogue mode Ⅱ are quite common[12-14]. O-binding is generally found in binuclear and multinuclear complexes as bridging oximato mode Ⅲ, wherein the N atoms of oxime units are also involved in bonding[15, 16]. Only a limited number of complexes are known to belong to mode IV at present, in which the oximate anions are linked to the metal atom through O atoms of oxime and N atoms of imine[17-19]. However, most of these complexes were merely characterized by UV-vis and IR spectra, without more structure details such as 1H NMR or single crystal structure by X-ray diffraction.
Herein, a diimine-dioxime ligand PhdoenH2 was synthesized from α-benzilmonoxime and ethylenediamine. The Ni(Ⅱ) complex 1 via the complexation of PhdoenH2 with Ni(Ⅱ) salts (Ni(ClO4)2, Ni(BF4)2, NiCl2, Ni(acac)2 and NiSO4) exhibits the rare coordination mode IV which is different from those reported Ni(Ⅱ) complexes, such as [Ni(MedoenH)]+ (2; MedoenH2 = 1, 8-dihydroxyimino-1, 2, 7, 8-tetramethyl-3, 6-diazocta-2, 6-diene)[20]. The structure of complex 1 has been carefully characterized by 1H NMR spectrum, elemental analysis, and X-ray single-crystal diffraction. Theoretical density functional theory (DFT) investigations revealed that the coordination mode should be attributed to the different relative thermodynamic stabilities between the oxime cis- and trans-isomers of PhdoenH2 and MedoenH2.
α-Benzilmonoxime and [Ni(MedoenH)]ClO4 were prepared according to the literature methods, respectively[20, 21]. Ethylenediamine and Ni(Ⅱ) salts were purchased from Aladdin Reagent Inc. (China).
1H NMR spectra were collected in deuterated dimethyl sulfoxide (DMSO-d6) using Bruker AC-400 at room temperature, and chemical shifts were reported in ppm units with respect to the reference frequency of TMS. Mass spectrometry (MS) analyses were carried out on an electrospray ionization time-of-flight mass spectrometer (ESI-TOF MS, UltiMate 3000, Dionex Company). UV-vis spectra were measured on a Shimadzu UV1800 spectro-photometer. Elemental analyses (C, H and N) were performed by Vario EL-Ⅲ CHNOS instrument. Infrared spectra were obtained on a BEQ UZNDX 550-FTIR spectrophotometer (Bruker) with KBr pellets. Thermal gravimetric analysis was performed on a NETZSCH STA449C instrument.
α-Benzilmonoxime (2.25 g, 10 mmol) and ethylenediamine (0.30 g, 5 mmol) were refluxed in 15 mL methanol for 48 h. After cooling to room temperature, the white solid precipitated was filtered, washed with methanol, and dried under vacuum (1.49 g, yield: 63%). 1H NMR (400MHz, DMSO-d6): δ, ppm, 11.73 (s, 2H, 2 OH), 7.75~7.25 (m, 20H, 4 Ph), 3.90~3.50 (m, 4H, CH2CH2).
[(PhdoenH2)] (0.47 g, 1 mmol) was added into 10 mL EtOH, and the mixture was brought to reflux. Then the solution of Ni(ClO4)2·6H2O (0.37 g, 1 mmol) in 10 mL EtOH was added dropwise into the suspension. The mixture was refluxed overnight. After evaporation of solvents, the obtained solid was purified by chromatography on silica gel with CH2Cl2/EtOH (50:1, v/v) as eluent. An orange solid was obtained from the collected red band after removing the solvent (0.16 g, yield: 30%). Single crystals suitable for X-ray diffraction were obtained by layering n-hexane over a CH2Cl2 solution containing the crude complex at room temperature. 1H NMR (400MHz, DMSO-d6): δ, ppm, 7.40~7.00 (m, 20H, 4 Ph), 3.09 (s, 4H, CH2CH2). Elemental analysis (%): calcd. for C30H24N4NiO2: C, 67.83; H, 4.55; N, 10.55. Found: C, 67.72, H, 4.35, N, 10.21. HR-MS (ESI): m/z+ = 531.1326 (M+H+), calcd.: 530.1253.
The diffraction data were collected using a graphite-monochromated CuKα radiation (λ = 1.54178 Å) at 100.00(10) K with a SuperNova, Dual, Cu at zero, AtlasS2 diffractometer. The structure was solved by direct methods using the SHELXS and refined with SHELXL of the SHELXTL package[22]. Anisotropic thermal factors were assigned to all non-hydrogen atoms. H atoms attached to C were added theoretically. Complex 1 is of monoclinic system, space group P21/c with a = 12.8234(4), b = 13.0403(4), c = 15.7134(6) Å, β = 106.622(4)º, V = 2517.81(15) Å3, Z = 4, S = 1.034, F(000) = 1104, R = 0.0577 and wR = 0.1484 (I > 2σ(I)). R = Σ||Fo| − |Fc||/Σ|Fo|; wR = [Σ[w(Fo2 − Fc2)2]/Σw(Fo2)2]1/2. The selected bond lengths and bond angles are given in Table 1.
 DownLoad:
CSV
DownLoad:
CSV
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| Ni(1)–O(1) | 1.853(2) | Ni(1)–N(3) | 1.844(2) | N(1)–O(1) | 1.309(3) | ||
| Ni(1)–O(2) | 1.826(2) | Ni(1)–N(4) | 1.853(2) | N(2)–O(2) | 1.313(3) | ||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| O(1)–Ni(1)–O(2) | 81.98(9) | O(1)–Ni(1)–N(3) | 94.72(10) | Ni(1)–O(1)–N(1) | 127.38(17) | ||
| N(3)–Ni(1)–N(4) | 88.31(10) | O(2)–Ni(1)–N(4) | 94.90(9) | Ni(1)–O(2)–N(2) | 129.01(17) |
Electrochemistry measurements were carried out with a CHI650E electrochemical workstation. A glassy carbon disk (diameter 3 mm) was used as the working electrode, a platinum column as the counter electrode, and Ag/AgNO3 electrode as the reference electrode. All potentials were referenced against ferrocene/ferrocenium as an external standard. The solution was purged with argon for 15 min before measurement. A solution of 0.1 mol·L−1 n-Bu4NPF6 in MeCN was used as the supporting electrolyte.
The geometries were optimized at density functional TPSS[23] level of theory. The SDD type ECP together with the valence basis functions[24] was chosen for the metal element and def2-SVP for the rest (TPSS/SDD/def2-SVP)[25]. Solvation effects in ethanol were treated by the implicit solvation model IEFPCM[26]. Study showed that the hybrid functional with small percentage of exchange performed well for late transition metal compounds[27]. The geometry was optimized at the TPSSh/SDD/def2-SVP level of theory in acetonitrile solution. The def2-SVP part of basis set was replaced by def2-TZVP in the TD-DFT calculations. The natural transition orbital (NTO) analysis was performed on the excited states[28]. The free energies of the compounds and their reduction species were obtained at the TPSS/SDD/def2-TZVP level of theory using the ideal-gas approximation at 298 K and 1 atm. SMD model was employed to compute the solvation energy[29]. The global minimum search was performed with Spartran'10 program[30]. The low-lying conformers were first picked up using both the systematic and Monte Carlo search methods and the energies were evaluated with the MMFF94 force field method[31]. The configurations with molecular mechanics energies within 5 kcal·mol−1 were selected for further DFT B3LYP-D3/6-31G(d) optimizations and finally DLPNOCCSD(T)/cc-pVTZ single point energy calculations (E) at the B3LYP-D3/6-31G(d) level[32, 33]. The solvation energies (Esol) with SMD model at the DFT optimized geometries were computed at the B3LYP-D3/6-311+G(d) level and free energy corrections (∆G) at the B3LYP-D3/6-31G(d) level with the ideal gas approximation. The free energy is finally expressed as G = E + Esol + ∆G. The EDA calculation was performed with ADF 2016 program and bond orders with Multiwfn 3.6[34]. ORCA 4.2 was used for wavefunction calculations[35, 36]. All other calculations were performed with the Gaussian09 program[37].
In 1964, Uhlig and Friedrich reported the diimine-dioxime ligand MedoenH2 reacted with Ni(ClO4)2·6H2O to form complex 2 in coordination mode I ([NiN4]+)[20]. When the analogue ligand PhdoenH2 was used to react with Ni(Ⅱ), it is interesting to find that the only isolable product is [Ni(Phdoen)] belonging to the rare coordination mode IV ([NiN2O2]) (Scheme 1). In the 1H NMR spectrum of 1, the OH proton and hydrogen bridge (O–H···O) band were not observed, which indicates that coordination took place via the oxygen atoms of oxime group rather than nitrogen atoms. And the result of elemental analyses is also in good agreement with the theoretical value.
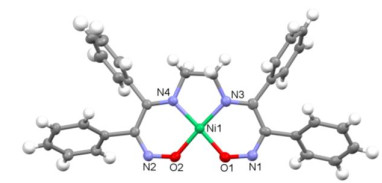
The structure of complex 1 was further confirmed by X-ray diffraction analysis of single crystal obtained by layering n-hexane over a CH2Cl2 solution. Fig. 2 shows the structure of monomeric unit of 1. The nickel left of 1 is coordinated with two N and two O atoms of the ligand, and any atoms of solvent molecules or adjacent units do not bind to the metal left from the axial sites. The NiN2O2 unit displays a slightly distorted square planar geometry with a 5-6-5 ring pattern, and Ni(1) lies 0.012 Å from the N2O2 plane. The X-ray crystal structure of complex 2 which features a non-symmetrical hydrogen-bond has been reported by Peters[16]. In complex 2, the nickel left is also in the distorted square planar environment, but is ligated by four N atoms of the diimine-dioxime ligand. The distances of Ni–Nimine bonds in complex 1 are slightly longer than those in 2. The distances of N–O– bonds in 1 are almost identical with that of N(2)–O(2) bond in the deprotonated oximate unit of 2, and significantly shorter than the N(1)–O(1) bond distance of oxime unit in 2. The O(1)−Ni(1)−O(2) angle in 1 is constrained in the present structure to the square 81.98(9)°, while the N(1)−Ni(1)−N(2) angle in 2 is 102.45(14)°.
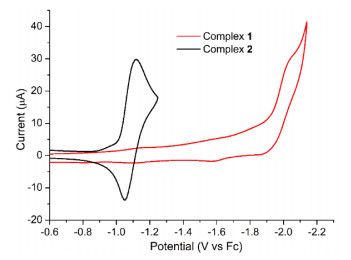
Redox properties of complexes 1 and 2 were studied by cyclic voltammetry in MeCN under an Ar atmosphere. The ferrocene (Fc) was used as the reference, and the potentials are given versus Fc0/+. The cyclic voltammogram (CV) of 2 exhibits a reduction peak at Epc = −1.12 V and an associated re-oxidation peak at Epa = −1.04 V, as shown in Fig. 3. The peak-to-peak separation for this couple is ΔEp = 72 mV at a scan rate of 100 mV·s−1, while ΔEp is also 75 mV for the couple of Fc0/+ under the same condition. This process is attributed to the one-electron redox of NiⅡ/NiI couple. For comparison, the cyclic voltammogram (CV) of 1 exhibits an irreversible reduction peak at Epc = −2.05 V, which is more negative than that of 2. It indicates the strong electrondonating ability of O− atoms renders the reduction of nickel core in 1 more difficultly. The coordination bonds were analyzed for complexes 1 and 2. The energy decomposition shows that Ni–O/N bonds have balanced contributions from both ionic and covalent interactions. The bond orders are less than 1. The Mulliken charges on Ni are +0.20 and +0.39 e for 1 and 2, respectively. The smaller positive charges of Ni atom in 1 make it difficult to be reduced than those in 2, and this result is consistent with the phenomena of cyclic voltammetry. The calculated values of the relative reduction potentials are −1.819 and −0.935 V for 1 and 2, respectively, which are in good agreement with the experimental values. Note that the spin density of the Ni(I) complex is delocalized over the ligand (Fig. S10).
The different coordination modes of 1 and 2 can be attributed to the different geometry properties of ligands. The C=N double bond of the oxime group leads to either cis or trans isomers. The coordination mode of complexes 1 or 2 requires the ligand to be a cis or trans isomer, respectively. Consider the hypothetical reaction of mode I/IV interconversion:
|
|
(I) |
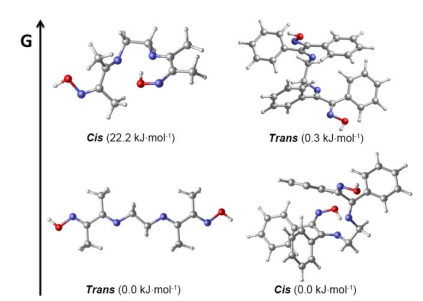
For ligand MedoenH2, the free energy (ΔG) of reaction I is 9.47 kJ⋅mol−1, indicating that the complex [NiN4]+ is more stable. Meanwhile, the global minimization shows that the energy of cis isomer of MedoenH2 is 22 kJ⋅mol−1 higher than that of the trans isomer (Fig. 4). Therefore, the trans isomer would be the dominant existence of MedoenH2, which results in the formation of 2 with coordination mode I in the reaction of MedoenH2 and Ni(Ⅱ). In contrast, ΔG is negative (−0.11 kJ⋅mol−1) for the ligand PhdoenH2, implying that complex [NiN2O2] is preferred. However, the energy gap between the cis- and trans-isomers of PhdoenH2 is only 0.8 kJ⋅mol−1, which means the coexistence of these two isomers. So, with very small reaction free energies, the reaction between PhdoenH2 and Ni(Ⅱ) would be quite complicated, accompanied with the formation of various products, where [NiN2O2] is the most abundant one. This result is consistent with the experimental phenomena.
Herein, the synthesis and structural characterization of a mononickel complex 1 were reported, in which the diimine-dioxime ligand links to the Ni atom through two O atoms of oxime and two N atoms of imine. As far as we know, it is the first time that the crystal structure and 1H NMR spectra of this unusual [2N2O]-coordination mode were reported. Recently, the alkyl bridged diimine-dioxime ligands have been widely applied into the synthesis of molecular catalysts for HER. However, when designing the [4N]-coordinated transition-metal complexes, the effect of diimine-dioxime ligand should be considered. There are two isomers, cis and trans, for the C=N double bond of the oxime group. And the [2N2O]- and [4N]-coordination modes require the ligands to be a cis or trans isomer of the C=N double bond in oxime group, respectively. The equilibrium abundance of each isomer is determined by the relative free energies.
Tschugaeff, L. Ueber ein neues, empfindliches reagens auf nickel. Ber. Dtsch. Chem. Ges. 1905, 38, 2520‒2522. doi: 10.1002/cber.19050380317
Chakravorty, A. Structural chemistry of transition metal complexes of oximes. Coord. Chem. Rev. 1974, 13, 1‒46. doi: 10.1016/S0010-8545(00)80250-7
Kukushkin, V. Y.; Tudela, D.; Pombeiro, A. J. L. Metal-ion assisted reactions of oximes and reactivity of oxime-containing metal complexes. Coord. Chem. Rev. 1996, 156, 333‒362. doi: 10.1016/0010-8545(95)01234-6
Schrauzer, G. N. Organocobalt chemistry of vitamin B12 model compounds (cobaloximes). Acc. Chem. Res. 1968, 1, 97‒103. doi: 10.1021/ar50004a001
Kaeffer, N.; Chavarot-Kerlidou, M.; Artero, V. Hydrogen evolution catalyzed by cobalt diimine-dioxime complexes. Acc. Chem. Res. 2015, 48, 1286‒1295. doi: 10.1021/acs.accounts.5b00058
Hu, X.; Brunschwig, B. S.; Peters, J. C. Electrocatalytic hydrogen evolution at low overpotentials by cobalt macrocyclic glyoxime and tetraimine complexes. J. Am. Chem. Soc. 2007, 129, 8988‒8998. doi: 10.1021/ja067876b
Song, D.; Li, B.; Li, X.; Sun, X.; Li, J.; Li, C.; Xu, T.; Zhu, Y.; Li, F.; Wang, N. Orthogonal supramolecular assembly triggered by inclusion and exclusion interactions with cucurbit[7]uril for photocatalytic H2 evolution. ChemSusChem. 2019, 13, 394‒399.
Andreiadis, E. S.; Jacques, P. A.; Tran, P. D.; Leyris, A.; Chavarot-Kerlidou, M.; Jousselme, B.; Matheron, M.; Pécaut, J.; Palacin, S.; Fontecave, M.; Artero, V. Molecular engineering of a cobalt-based electrocatalytic nanomaterial for H2 evolution under fully aqueous conditions. Nat. Chem. 2012, 5, 48‒53.
McCrory, C. C. L.; Uyeda, C.; Peters, J. C. Electrocatalytic hydrogen evolution in acidic water with molecular cobalt tetraazamacrocycles. J. Am. Chem. Soc. 2012, 134, 3164‒3170. doi: 10.1021/ja210661k
Basu, D.; Mazumder, S.; Niklas, J.; Baydoun, H.; Wanniarachchi, D.; Shi, X.; Staples, R. J.; Poluektov, O.; Schlegel, H. B.; Verani, C. N. Evaluation of the coordination preferences and catalytic pathways of heteroaxial cobalt oximes towards hydrogen generation. Chem. Sci. 2016, 7, 3264‒3278. doi: 10.1039/C5SC04214C
Huo, P.; Uyeda, C.; Goodpaster, J. D.; Peters, J. C.; Miller, T. F. Breaking the correlation between energy costs and kinetic barriers in hydrogen evolution via a cobalt pyridine-diimine-dioxime catalyst. ACS Catal. 2016, 6, 6114‒6123. doi: 10.1021/acscatal.6b01387
Cui, L.; Ono, T.; Morita, Y.; Hisaeda, Y. Electrocatalytic reactivity of imine/oxime-type cobalt complex for direct perfluoroalkylation of indole and aniline derivatives. Dalton Trans. 2020, 49, 7546‒7551. doi: 10.1039/D0DT01377C
Guttentag, M.; Rodenberg, A.; Kopelent, R.; Probst, B.; Buchwalder, C.; Brandstätter, M.; Hamm, P.; Alberto, R. Photocatalytic H2 production with a rhenium/cobalt system in water under acidic conditions. Eur. J. Inorg. Chem. 2012, 2012, 59‒64. doi: 10.1002/ejic.201100883
Simándi, L. I.; Simándi, T. M.; May, Z.; Besenyei, G. Catalytic activation of dioxygen by oximatocobalt(Ⅱ) and oximatoiron(Ⅱ) complexes for catecholase-mimetic oxidations of o-substituted phenols. Coord. Chem. Rev. 2003, 245, 85‒93. doi: 10.1016/S0010-8545(03)00057-2
Uyeda, C.; Peters, J. C. Selective nitrite reduction at heterobimetallic CoMg complexes. J. Am. Chem. Soc. 2013, 135, 12023‒12031. doi: 10.1021/ja4053653
Uyeda, C.; Peters, J. C. Access to formally Ni(Ⅰ) states in a heterobimetallic NiZn system. Chem. Sci. 2013, 4, 157‒163. doi: 10.1039/C2SC21231E
Aly, M. M.; Stephanos, J. J. Factors influencing linkage isomerism of the oximato group in the Ni(Ⅱ), Co(Ⅱ) and Cu(Ⅱ) complexes of vicinal oxime-imine ligands. J. Mol. Struct. 1993, 293, 75‒76. doi: 10.1016/0022-2860(93)80018-Q
Emam, M. E. M.; Bekheit, M. M.; Moussa, M. N. H.; El-Hendawy, A. E. N. A. A physicochemical study of Schiff base metal complexes derived from α-benzylmonoxime. Transition Met. Chem. 1994, 19, 117‒118.
Bhula, R.; Weatherburn, D. C.; Gainsford, G. J. The crystal and molecular structure of 4, 4΄-(1, 2-ethanediyldinitrilo)-bis[(2, 3-pentanedione)-3, 3΄-dioximato] (2−)N4, N4΄, O3, O3΄ copper(Ⅱ). Inorg. Chim. Acta 1987, 128, L7‒L9. doi: 10.1016/S0020-1693(00)84680-0
Uhlig, E.; Friedrich, M. Untersuchungen an oximkomplexen. Ⅲ. nickelchelate des bis-(diacetylmonoxim-imino)-propans-1, 3 und des bis-(diacetylmonoxim-imino)-äthans-1, 2. Anorg. Allg. Chem. 1966, 343, 299‒307. doi: 10.1002/zaac.19663430510
Soleimani, E.; Taheri, S. A. N. Synthesis and characterization of monomeric and dimeric dihydroxo-bridged complexes of iron(Ⅱ) with α-benzilmonoxime. Asian J. Chem. 2011, 23, 1561‒1563.
Sheldrick, G. M. SHELXS-97, Program for X-ray Crystal Structure Solution. University of Göttingen, Germany 1997.
Tao, J.; Perdew, J. P.; Staroverov, V. N.; Scuseria, G. E. Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401‒4. doi: 10.1103/PhysRevLett.91.146401
Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Phys. Chem. 1987, 86, 866‒872. doi: 10.1063/1.452288
Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297‒3305. doi: 10.1039/b508541a
Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999‒3094. doi: 10.1021/cr9904009
Moltved, K. A.; Kepp, K. P. Chemical bond energies of 3d transition metals studied by density functional theory. J. Chem. Theory Comput. 2018, 14, 3479‒3492. doi: 10.1021/acs.jctc.8b00143
Martin, R. L. Natural transition orbitals. J. Phys. Chem. 2003, 118, 4775‒4777. doi: 10.1063/1.1558471
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378‒6396. doi: 10.1021/jp810292n
Spartan'10, Wavefunction, Inc., Irvine, CA.
Halgren, T. A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490‒519. doi: 10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P
Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Phys. Chem. 2013, 138, 034106‒18. doi: 10.1063/1.4773581
Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E. F.; Neese, F. Sparse maps-a systematic infrastructure for reduced-scaling electronic structure methods. Ⅱ. linear scaling domain based pair natural orbital coupled cluster theory. J. Phys. Chem. 2016, 144, 024109‒10. doi: 10.1063/1.4939030
Lu, T.; Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580‒592. doi: 10.1002/jcc.22885
Neese, F. The ORCA program system. Wiley Interdiscip. Rev. : Comput. Mol. Sci. 2011, 2, 73‒78.
Neese, F. Software update: the ORCA program system, version 4.0. Wiley Interdiscip. Rev. : Comput. Mol. Sci. 2017, 8, e1327‒6.
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03, Revision B. 01, Gaussian, Inc., Pittsburgh PA 2003.

Figure 2 ORTEP drawing of complex 1 showing thermal ellipsoids at the 50% probability level

Figure 3 CVs of 1 and 2 in MeCN under Ar atmosphere. Conditions: 1 mM analyte in 0.1 mol⋅L−1 n-Bu4NPF6/MeCN; glassy carbon working electrode, Pt wire counter electrode, Ag/AgNO3 reference electrode; referenced to internal Fc standard; scan rate 100 mV⋅s−1

Figure 4 The lowest trans and cis isomers of ligand MedoenH2 (left) and PhdoenH2 (right). Relative energies are in kJ⋅mol−1
Table 1. Selected Bond Lengths (Å) and Bond Angles (°)
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| Ni(1)–O(1) | 1.853(2) | Ni(1)–N(3) | 1.844(2) | N(1)–O(1) | 1.309(3) | ||
| Ni(1)–O(2) | 1.826(2) | Ni(1)–N(4) | 1.853(2) | N(2)–O(2) | 1.313(3) | ||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| O(1)–Ni(1)–O(2) | 81.98(9) | O(1)–Ni(1)–N(3) | 94.72(10) | Ni(1)–O(1)–N(1) | 127.38(17) | ||
| N(3)–Ni(1)–N(4) | 88.31(10) | O(2)–Ni(1)–N(4) | 94.90(9) | Ni(1)–O(2)–N(2) | 129.01(17) |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

