Table 1.
Molecular Structures and Biological Activities of 3-Aroyl-5-substituted Thiophene Derivatives
Citation:
Hui FENG, Chang-Jun FENG, Jing-Pei CAO. Study on the Biological Activity of 3-Aroyl-5-substituted Thiophene Derivatives Based on the CoMFA Method[J]. Chinese Journal of Structural Chemistry,
2020, 39(11): 1978-1984.
doi:
10.14102/j.cnki.0254–5861.2011–2703

Study on the Biological Activity of 3-Aroyl-5-substituted Thiophene Derivatives Based on the CoMFA Method
English
Study on the Biological Activity of 3-Aroyl-5-substituted Thiophene Derivatives Based on the CoMFA Method
Abstract:
A three-dimensional quantitative structure-activity relationship (3D-QSAR) study was conducted to analyze the A1AR density (Bmax) of 56 3-aroyl-5-substituted thiophene derivatives (ASTDs) in human A1 Chinese hamster ovary (hA1CHO) membranes by the comparative molecular field analysis (CoMFA) method. A training set of 45 compounds was used to establish the predictive model, which was verified by the test set of 17 compounds containing template molecule and 5 newly designed molecules. The cross-validation (Rcv2) and non-cross-validation (R2) coefficients of the training set were 0.655 and 0.959, respectively. The model was used to predict the activities of the compounds of the training and test sets, and the results indicated that the models had strong stability and good prediction ability. According to model analysis, the contribution of steric and electrostatic fields was 51.4% and 48.6%, respectively. Based on the 3D contour maps, five excellent ASTDs agonists were designed, which need to be further verified by biomedical experiments.
-
1. INTRODUCTION
Adenosine receptor, one of the G protein-coupled receptors (GPCRs), has unique pharmacological characteristics, tissue distribution and coupling protein. As a drug target, it is mainly used in the treatment of ischemic diseases (brain and heart), sleep disorders, immune and inflammatory disorders, cancer, etc. Among its four subtypes (namely, A1, A2A, A2B and A3[1]), the A1 receptor has attracted the attention of many drug researchers. It mainly acts on central nervous system and peripheral circulatory system, and has the function of promoting immunity. Romagnoli et al.[2] have synthesized a series of new 2-amino-3-aroyl-4-neopentyl-5-substituted thiophene derivatives based on the 2-amino-4-neopentylthiophene skeleton, which were referred to as "3-aroyl-5-substituted thiophene derivatives (ASTDs)". They tested the saturation and competition of selective adenosine A1 agonist [3H]2-chloro-N6-cyclopentyladenosine (abbreviations: [3H]CCPA) on A1 receptor to determine whether the synthesized compounds would change the agonist binding parameters. In the presence of compounds 1~30 in Table 1, A1 adenosine receptor (abbreviation: A1AR) density, expressed as Bmax values, was obtained by [3H]CCPA binding assays in human A1 Chinese hamster ovary (hA1CHO) membranes and of PD81723 (10 μmol·dm-3).
Table 1

 DownLoad:
CSV
DownLoad:
CSV
No. R R4 R5 Bmax[2] LB.exp[2] LBcal 1 4-Cl -CH3 H 798 2.902 2.915 2* 4-Cl -CH2CH(CH3)2 H 833 2.921 3.091 3 4-Cl -C(CH3)3 H 679 2.831 2.858 4 4-Cl -CH2C(CH3)3 H 2038 3.309 3.365 5 4-Cl -CH3 -C6H5 771 2.887 2.925 6 4-Cl -CH3 4-CH3OC6H4 1094 3.038 3.004 7 4-Cl -CH3 -Br 932 2.969 2.922 8* 4-Cl -C5H5 -CH3 731 2.863 2.583 9 4-Cl -C6H5 -C6H5 1126 3.051 3.061 10 3-Cl -CH2C(CH3)3 H 2241 3.350 3.283 11 3, 4-2Cl -CH2C(CH3)3 H 2148 3.331 3.325 12 3-CF3 -CH2C(CH3)3 H 1359 3.133 3.188 13* 3-CH3 -CH2C(CH3)3 H 1726 3.236 3.417 14 4-Cl -CH2C(CH3)3 -Br 3591 3.555 3.442 15 3-Cl -CH2C(CH3)3 -Br 1877 3.273 3.361 16 3, 4-2Cl -CH2C(CH3)3 -Br 2346 3.370 3.404 17 3-CF3 -CH2C(CH3)3 -Br 2286 3.358 3.268 18* 4-CH3 -CH2C(CH3)3 -Br 3351 3.525 3.496 19 4-Cl -CH2C(CH3)3 -C6H5 3649 3.562 3.532 20 3-Cl -CH2C(CH3)3 -C6H5 3286 3.516 3.450 21 2-Cl -CH2C(CH3)3 -C6H5 2863 3.456 3.511 22* 3, 4-2Cl -CH2C(CH3)3 -C6H5 3072 3.487 3.491 23 2, 4-2Cl -CH2C(CH3)3 -C6H5 3603 3.556 3.553 24 3-CF3 -CH2C(CH3)3 -C6H5 2087 3.319 3.351 25 4-CH3 -CH2C(CH3)3 -C6H5 3493 3.543 3.590 26 4-Cl -CH2C(CH3)3 4-CH3OC6H4 3198 3.504 3.533 27 3-Cl -CH2C(CH3)3 4-CH3OC6H4 3139 3.496 3.480 28* 3, 4-2Cl -CH2C(CH3)3 4-CH3OC6H4 3138 3.496 3.522 29 3-CF3 -CH2C(CH3)3 4-CH3OC6H4 2084 3.318 3.333 30 4-CH3 -CH2C(CH3)3 4-CH3OC6H4 3861 3.586 3.600 31 4-Cl -CH2C(CH3)3 3, 4-2CH3-isoxazol-4-yl 735 2.866 2.851 32* 4-Cl -CH2C(CH3)3 1H-pyrazol-4-yl 1308 3.116 3.441 33 4-Cl -CH2C(CH3)3 Thiophen-2-yl 3237 3.510 3.459 34 4-Cl -CH2C(CH3)3 Thiophen-3-yl 3332 3.522 3.459 35 4-Cl -CH2C(CH3)3 Furan-2-yl 2668 3.426 3.433 36* 4-Cl -CH2C(CH3)3 Furan-3-yl 2922 3.465 3.398 37 4-Cl -CH2C(CH3)3 Pyridin-4-yl 2086 3.319 3.348 38 4-Cl -CH2C(CH3)3 Pyridin-3-yl 1928 3.285 3.302 39 4-Cl -CH2C(CH3)3 Pyridin-2-yl 1831 3.262 3.275 40 4-Cl -CH2C(CH3)3 4-F-C6H4 2876 3.458 3.493 41 4-Cl -CH2C(CH3)3 2, 3-2F-C6H3 3176 3.501 3.436 42* 4-Cl -CH2C(CH3)3 2, 4-2F-C6H3 2758 3.440 3.448 43 4-Cl -CH2C(CH3)3 2, 5-2F-C6H3 2768 3.442 3.414 44 4-Cl -CH2C(CH3)3 2, 6-2F-C6H3 2194 3.341 3.392 45 4-Cl -CH2C(CH3)3 4-Cl-C6H4 3022 3.480 3.502 46 4-Cl -CH2C(CH3)3 E-4-Cl-C6H4CH=CH 2509 3.399 3.406 47 4-Cl -CH2C(CH3)3 3, 4-2Cl-C6H3 3143 3.497 3.513 48 4-Cl -CH2C(CH3)3 3-CH3OC6H4 3292 3.517 3.547 49* 4-Cl -CH2C(CH3)3 2-CH3OC6H4 1322 3.121 3.192 50 4-Cl -CH2C(CH3)3 4-CH3O(CH2)2O-C6H4 2879 3.459 3.442 51 4-Cl -CH2C(CH3)3 3, 4-(2OCH2)C6H3 3449 3.537 3.521 52 4-Cl -CH2C(CH3)3 4-CF3OC6H4 3337 3.523 3.517 53 4-Cl -CH2C(CH3)3 4-CH3C6H4 3345 3.524 3.497 54* 4-Cl -CH2C(CH3)3 3-CH3C6H4 2449 3.388 3.457 55 4-Cl -CH2C(CH3)3 2-CH3C6H4 2138 3.329 3.351 56 4-Cl -CH2C(CH3)3 4-(CH3)2CHC6H4 2932 3.467 3.462 The establishment of biological activity database by experimental methods not only needs a great deal of human input, but also material and financial resources. The quantitative structure-activity relationship (QSAR) model[3] based on the relationship between the molecular biological activity and the structure descriptors has been widely employed for predicting biological activity and biological mechanism of molecules with similar structures. QSAR mainly includes two-dimensional QSAR (2D-QSAR) methods[4-9] and three-dimensional QSAR (3D-QSAR) methods[10-18]. Compared with traditional 2D-QSAR, 3D-QSAR methods introduce the three-dimensional conformational properties of bioactive molecules. Therefore, it can reflect the real image of the interaction between bioactive molecules and receptors more accurately, and reveal the mechanism of drug-receptor interaction more deeply. Furthermore, the fitting results of 3D-QSAR are generally better than 2D-QSAR. Recently, comparative molecular field analysis (CoFMA) methods[19-22] have been widely used in the 3D-QSAR study, in which the active conformer and superposition rule for a set of molecules were adopted to provide vivid images of the relationships between the molecular structures of compounds and bio-activity for predicting the potency.
In this work, the A1AR density (Bmax)[2] of 56 ASTDs in hA1CHO membranes cell was studied by the CoMFA method to establish an optimal CoMFA based 3D-QSAR model for these compounds. It is known that the physiological action of drug molecules in vivo results from their interaction with biological macromolecules. Since the receptors and ligands interact through specific binding sites, the specific three-dimensional conformation is of great significance for molecules to exhibit their pharmacodynamic activity. Analyzing the interaction mode of drugs and receptors from the three-dimensional conformation of bioactive small molecules is a common strategy for drug molecule design and selective analysis. We expect that the theoretical results can offer some useful reference information for the experimenters in the design of new ASTDs and the analysis of related action mechanisms.
2. EXPERIMENTAL
Computer-aided drug design can be divided into two methods, i.e., receptor-based and ligand-based drug design. At present, the crystal structure of A1 receptor has not been analyzed, and thus the ligand-based method was used to construct the CoMFA model in this work.
2.1 Studied compounds and their biological activity data
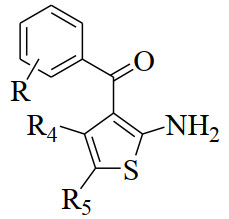
The general structural formula of ASTDs [2] is shown in Fig. 1, and the specific substituents R, R4 and R5 for each compound are listed in Table 1. Their biological activity data (Bmax), in the unit of f mol·(mg⋅protein)-1, are also listed in Table 1.
Figure 1
 Figure 1. Structure of 3-aroyl-5-substituted thiophene derivatives
Figure 1. Structure of 3-aroyl-5-substituted thiophene derivativesThe original bioactivity (Bmax) values[2] were converted to LB by taking the logarithm of Bmax (LB = log(Bmax)), and the LB values (Table 1) were used as dependent variables in the CoMFA study. The data set was randomly divided into a training set of 45 compounds for model construction and a test set of 11 compounds (marked with "*" in Table 1) for model validation. The number of compounds in the test set was determined as one fifth of the total compounds according to 5:1, and the structural diversity and extensive activity were also considered in the data set.
2.2 Minimization and alignment
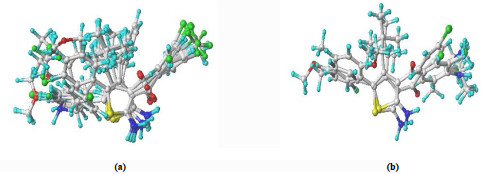
Molecular structures were sketched with sketch module in the latest version Sybyl-x2.1.1, and minimized using Tripos force field by the Gasteiger-Huckel charges and conjugated gradient method, with gradient convergence criteria of 0.05 kcal⋅mol-1. The remaining parameters used in CoMFA were all default unless otherwise stated. Structural alignment is considered as one of the most sensitive parameters in CoMFA analysis. The accuracy of the prediction of CoMFA model and the reliability of contour maps are directly dependent on the structural alignment rule. Molecule 30, with the most potent activity, was used as a template for alignment. Each analog was aligned to the template by rotation and translation using the DATABASE ALIGNMENT command in Sybyl so as to minimize RMSD between atoms in the template and analog. The alignment results based on the above strategy are shown in Fig. 2. These alignments were subsequently used in the calculations of CoMFA probe interaction energy. The superposition databases of training and test sets of all allosteric enhancers are respectively represented as "Tradh.mdb" and "Tesdh.mdb" for modeling.
Figure 2
 Figure 2. 3D view of all the aligned molecules in the training set (a) and test set (b)
Figure 2. 3D view of all the aligned molecules in the training set (a) and test set (b)2.3 Model generation and validation
CoMFA model for biological activity (LB)[2] of ASTDs was established by 45 superposed training set molecules. The Steric (S) and Electrostatic fields (E) on each grid node around the superposed molecules were calculated using Tripos standard force field. The field energy threshold (Cut off value) was set to 125.5 kJ⋅mol-1, and the other parameters were the system default values.
After inputting LB into the dataset of the training set, partial least-squares (PLS) analysis was performed. First, leave-one-out cross-validation method was used to determine the cross-validation Rcv2 and the optimal number (N) of component. When Rcv2 > 0.5[23], the established model has statistical significance at 5% significance level, and its chance correlation probability is less than 5% (the reliability of the model is 95%). Then, non-cross-validation method was employed to further assess the robustness and statistical confidence of the derived 3D-QSAR models. It is generally believed that the model with R2 > 0.9[24] has a good fitting degree, because the model reveals structural factors that affect more than 90% of the biological activity. In addition, the statistical variance ratio F is also a test index to measure the significant level of QSAR model. Finally, the view of CoMFA module is adopted to provide the three-dimensional equipotential map of stereo field and static electric field to directly reflect their contributions to biological activity.
3. RESULTS AND DISCUSSION
3.1 3D-QSAR model
For the 3D-QSAR CoMFA model established in the training set, the best principal component number (N) is 6. The cross-validation coefficient (Rcv2) is 0.655, larger than 0.5, which shows that the model has good prediction ability. The traditional determination coefficient (R2) is 0.959, greater than 0.9, so this model has good fitting ability. At 99% significant level, the F critical value (F0.05(6, 38)) is only 3.35. The F obtained in this model is 146.645 and much larger than 3.35, indicating a close correlation. The steric and electrostatic contributions were found to be 51.4% and 48.6%, respectively. Therefore, the steric field has a greater influence than the electrostatic one. That is, the hydrophobic interaction of substituent groups may be a more important factor for the bioactivity. Excellent correlation between the observed and predicted LB activities for the 11 compounds in test set further verified the reliability of the constructed 3D-QSAR model. It is generally believed that only when the sample change ratio (β, defined as the ratio of number of compounds to number of variables) is ≥5, the model has statistical significance. In this work, the β of this model is 45/6 = 7.5, suggesting it is statistically significant.
For the 3D-QSAR CoMFA model established by all molecules, the optimal principal component number (N) is 6, the cross-validation coefficient (Rcv2) is 0.742, the traditional determination coefficient (R2) is 0.948, and the F value is 146.645. These index values are consistent with those of the above-mentioned 3D-QSAR model, which proves again that the model developed from the training set has good stability.
3.2 Analysis of three-dimensional equipotential diagram
Based on the force field coefficient equipotential map obtained by CoMFA model, the important regions of the steric and electrostatic field related to the activity are determined. Thus, we can effectively explain the molecular structure characteristics affecting the activity of the compound and reveal the underlying molecular mechanism for the compound-receptor interaction.
3.2.1 Steric contour map of biological activity (LB)
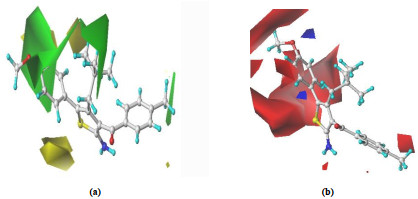
The stereogram of CoMFA model usually includes a green and a yellow regions, showing the introduction of a large and a small group substituents in these regions respectively can enhance the activity of the compound,
Fig. 3(a) shows the steric contour map for the CoMFA model of LB, with the most active compound 30 as a reference. From the spatial distribution of the stereo field, it can be seen that the introduction of larger groups near the 4, 5-position of thiophene (green region) and the presence of smaller groups far from the 4, 5-position (yellow region) are all helpful to improve LB. For example, when the 4-position group (R4) of thiophene is -CH2C(CH3)3, -CH2C(CH3)2 and -C(CH3)3, respectively, the LB decreases in the order of compound No. 4 > 2 > 3 (Table 1). As another example, LB generally shows an increasing trend when the 5-position group (R5) changes from -F to -Cl and -Br (Table 1: No. 40 < 45 < 14).
Figure 3
 Figure 3. CoMFA contour maps of (a) steric field and (b) electrostatic field
Figure 3. CoMFA contour maps of (a) steric field and (b) electrostatic field3.2.2 Electrostatic contour map of biological activity (LB)
In the electrostatic field map, blue and red contours denote the regions that the bioactivity would be increased by the presence of electron-donating and electron-withdrawing groups, respectively.
Fig. 3(b) shows the electrostatic contour map for the CoMFA model of LB, with the most active compound 30 as a reference. From the spatial distribution of electrostatic field, it can be seen that the introduction of a positively charged group at the 4, 5-positions of thiophene (red region) and 4-position of benzene ring (smaller red region) is helpful to improve LB. For example, LB becomes smaller when the 4-position group (R4) of the benzene ring changes from -CH3 to -Cl (Table 1: No. 30 > 26). As another example, LB generally shows an increasing trend when the 5-position group (R5) is -Br, -C6H5 and 4-CH3OC6H4, respectively (Table 1: No. 18 < 25 < 30).
3.3 Theoretical design of new ASTDs with higher bioactivity
The aim of QSAR research is to design molecules from the structural information implied in the established model and to predict the biological activity of the designed molecules by the model, thus providing theoretical basis for the synthesis of new highly bioactive molecules. According to discussions in section 2.2, the introduction of larger groups near the 4, 5-positions and negatively charged groups at the 3, 4-positions of thiophene are all beneficial to enhance the biological activity (LB) of 3-aroyl-5-substituted thiophene derivatives. Based on this rule, five compounds were designed as potential drugs (compound 1~5 in Table 2). The 3D-QSAR model predictions gave large LB values for these compounds, and some of them may have even better biological activity than the most effective drug reported in the literature. These theoretically excellent ASTDs agonists need to be further confirmed by biomedical experiments.
Table 2
Table 2. Predictive Activities of Newly Designed Small Molecules Based on the CoMFA ModelDownLoad:
CSV
No. R R4 R5 Bmax[2] LB.exp[2] LBcal 30 4-CH3 -CH2C(CH3)3 4-CH3OC6H4 3861 3.586 3.600 1 3, 4-2CH3 -CH2C(CH3)3 4-CH3OC6H4 3.612 2 3, 4, 5-3CH3 -CH2C(CH3)3 4-CH3OC6H4 3.638 3 3-CH3, 4-C2H5 -CH2C(CH3)3 4-CH3OC6H4 3.621 4 3-CH3, 4-CH(CH3) -CH2C(CH3)3 4-CH3OC6H4 3.615 5 3, 4-2NH2 -CH2C(CH3)3 4-CH3OC6H4 3.615 4. CONCLUSION
In this paper, a CoMFA model with good internal and external prediction capability was established from the biological activity of a training set of 45 ASTDs, and validated by a test set of 11 molecules. The cross-validated (Rcv2) and non-cross-validated (R2) coefficients of the CoMFA model are 0.655 and 0.959, respectively. The CoMFA model well explains that the difference in bioactivity of ASTDs agonists is mainly due to the effect of substituent groups on the distribution of steric and electrostatic fields of the whole molecule. The following main conclusions are reached. Stereo effect is slightly stronger than the electrostatic effect. When the substituent at the 4, 5-position of thiophene ring is large in volume or weak in electronegativity, the bioactivity of ASTDs would be largely enhanced. According to the established 3D-QSAR model, five excellent ASTDs agonists were designed, which need further verification by biomedical experiments. The information obtained in this study provides an effective tool for predicting the LB of related ASTDs, and for guiding further structural modification and synthesis of new potent ASTDs agonists.
-
-
[1]
Ke, Y. R.; Jin, H. W; Liu, Z. M.; Zhang, L. R. Homology modeling and structure validation of the adenosine A1 receptor. Acta Phys. -Chim. Sin. 2010, 26, 2833–2839. doi: 10.3866/PKU.WHXB20100916
-
[2]
Romagnoli, R.; Baraldi, P. G.; Carrion, M. D.; Cruz-Lopez, O.; Cara, C. L.; Saponaro, G.; Preti, D.; Tabrizi, M. A.; Baraldi, S.; Moorman, A. R.; Vincenzi, F.; Borea, P. A.; Varani, K. Synthesis and biological evaluation of novel 2-amino-3-aroyl-4-neopentyl-5-substituted thiophene derivatives as allosteric enhancers of the A1 adenosine receptor. Bioorgan. Med. Chem. 2014, 22, 148–166. doi: 10.1016/j.bmc.2013.11.043
-
[3]
Feng, C. J. Quantitative Structure-efficacy Relationship and Its Applications. China University of Mining and Technology Press, Xuzhou 2017, p1–26.
-
[4]
Wang, C.; Feng, C. J. QSAR study of the action strength of DOM of phenyl-isopropyl-amine dopes using MLR and BP-ANN. Chin. J. Struct. Chem. 2017, 36, 1720–1728.
-
[5]
Wang, C.; Feng, C. J. QSAR studies on the inhibitory activity of levofloxacin-thiadiazole HDACi conjugates to histone deacetylases. Chin. J. Struct. Chem. 2018, 37, 1679–1688.
-
[6]
Zheng, S. S.; Li, T. T.; Wang, J.; Hu, Y. J.; Zhang, H. X.; Zhao, S. X.; Zhao, Y. H.; Li, C. QSAR models for predicting the aqueous reaction rate constants of aromatic compounds with hydrated electrons. Environ. Chem. 2019, 38, 1005–1013.
-
[7]
Qu, R. J.; Liu, H. X.; Feng, M. B.; Yang, X.; Wang, Z. Y. Investigation on intramolecular hydrogen bond and some thermodynamic properties of polyhydroxylated anthraquinones. J. Chem. Eng. Data 2012, 57, 2442–2455. doi: 10.1021/je300407g
-
[8]
Yang, F.; Wang, M.; Wang, Z. Y. Sorption behavior of 17 phthalic acid esters on three soils: effects of pH and dissolved organic matter, sorption coefficient measurement and QSPR study. Chemosphere 2013, 93, 82–89. doi: 10.1016/j.chemosphere.2013.04.081
-
[9]
Feng, C. J. QSAR studies on the biological activity of substituted triazolo-benzothiazole derivatives. J. Xuzhou Inst. Tech. (Nat. Sci. Ed. ) 2018, 33, 39–44.
-
[10]
Feng, H.; Feng C. J. 3D-QSAR studies on the anti-tumor activity of N-aryl-salicylamide derivatives. Chin. J. Struct. Chem. 2019, 38, 1874–1880.
-
[11]
Tong, J. B.; Qin, S. S.; Lei, S.; Wang, Y. A 3D-QSAR study of HIV-1 integrase inhibitors using RASMS and topomer CoMFA. Chin. J. Struct. Chem. 2019, 38, 867–881.
-
[12]
Shu, M.; Wu, T.; Wang, B. W.; Li, J.; Xu, C. M.; Lin, Z. H. 3D-QSAR and surflex docking studies of a series of alkaline phosphatase inhibitors. Chin. J. Struct. Chem. 2019, 38, 7–16.
-
[13]
Tong, J. B.; Wang, Y.; Lei, S.; Qin, S. S. 3D-QSAR and docking studies of 1, 3, 4-thiazolidinone derivatives using R-group search and surflex-dock. Chin. J. Struct. Chem. 2019, 38, 464–475.
-
[14]
Tong, J. B.; Wang, Y.; Lei, S.; Qin, S. S. Comprehensive 3D-QSAR and binding mode of DAPY inhibitors using R-group search and molecular docking. Chin. J. Struct. Chem. 2019, 38, 25–36.
-
[15]
An, C. H.; Shu, M.; Zai, X. L.; Zhang, B. N.; Li, J.; Chang, Z. C.; Hu, Y.; Lin, Z. H. Combined 3D-QSAR, pharmacophore and docking studies on benzene-sulfonamide derivatives as potent 12-lipoxygenase inhibitors. Lett. Drug Des. Discov. 2017, 14, 74–82.
-
[16]
Tong, J.; Zhan, P.; Bai, M.; Yao, T. Molecular modeling studies of human immunodeficiency virus type 1 protease inhibitors using three-dimensional quantitative structure-activity relationship, virtual screening, and docking simulations. J. Chemometrics 2016, 30, 523–536. doi: 10.1002/cem.2809
-
[17]
Liu, H. X.; Shi, J. Q.; Liu, H.; Wang, Z. Y. Improved 3D-QSPR analysis of the predictive octanol-air partition coefficients of hydroxylated and methoxylated polybrominated diphenyl ethers. Atmos. Environ. 2013, 77, 840–845. doi: 10.1016/j.atmosenv.2013.05.068
-
[18]
Wang, X. Y.; Duan, W. G.; Lin, G. S.; Kang, G. Q.; Shang, M. H.; Lei, F. H. Synthesis and antifungal activity of (Z)-/(E)-verbenone oxime ether compounds. Chem. Ind. Forest Prod. 2019, 39, 27–34. http://en.cnki.com.cn/Article_en/CJFDTotal-LCHX201904007.htm
-
[19]
Baraldi, P. G.; Borea, P. A.; Bergonzoni, M.; Cacciari, B.; Ongini, E.; Recanatini, M.; Spalluto, G. Comparative molecular field analysis (CoMFA) of a series of selective adenosine receptor A2A antagonists. Drug Dev. Res. 1999, 46, 126–133. doi: 10.1002/(SICI)1098-2299(199902)46:2<126::AID-DDR5>3.0.CO;2-7
-
[20]
Feng, C. J. CoMFA model of herbicidal activity of phenyl-sulfonylured derivatives. J. Xuzhou Inst. Tech. (Nat. Sci. Ed. ) 2019, 34, 21–25.
-
[21]
Joshi, S. D.; More, U. A.; Aminabhavi, T. M.; Badiger, A. M. Two-and three-dimensional QSAR studies on a set of antimycobacterial pyrroles: CoMFA, Topomer CoMFA, and HQSAR. Med. Chem. Res. 2014, 23, 107–126.
-
[22]
Cramer, R. D.; Patterson, D. E.; Bunce, J. D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959−5967.
-
[23]
Clark, M.; Cramer, R. D.; Jones, D. M.; Patterson, D. E.; Simeroth, P. E. Comparative molecular field analysis (CoMFA). 2. Toward its use with 3D-structural databases. Tetrahedron Comput. Methodol. 1990, 3, 47–59.
-
[24]
Cramer, R. D.; Burce J. D.; Patterson D. E.; Frank, I. E. Cross-validation, boot strapping, and partial least squares compared with multiple regression in conventional QSAR study. Quant. Struct. -Act. Relat. 1988, 7, 18–25.
-
[1]
-
Figure 2 3D view of all the aligned molecules in the training set (a) and test set (b)
Table 1. Molecular Structures and Biological Activities of 3-Aroyl-5-substituted Thiophene Derivatives
No. R R4 R5 Bmax[2] LB.exp[2] LBcal 1 4-Cl -CH3 H 798 2.902 2.915 2* 4-Cl -CH2CH(CH3)2 H 833 2.921 3.091 3 4-Cl -C(CH3)3 H 679 2.831 2.858 4 4-Cl -CH2C(CH3)3 H 2038 3.309 3.365 5 4-Cl -CH3 -C6H5 771 2.887 2.925 6 4-Cl -CH3 4-CH3OC6H4 1094 3.038 3.004 7 4-Cl -CH3 -Br 932 2.969 2.922 8* 4-Cl -C5H5 -CH3 731 2.863 2.583 9 4-Cl -C6H5 -C6H5 1126 3.051 3.061 10 3-Cl -CH2C(CH3)3 H 2241 3.350 3.283 11 3, 4-2Cl -CH2C(CH3)3 H 2148 3.331 3.325 12 3-CF3 -CH2C(CH3)3 H 1359 3.133 3.188 13* 3-CH3 -CH2C(CH3)3 H 1726 3.236 3.417 14 4-Cl -CH2C(CH3)3 -Br 3591 3.555 3.442 15 3-Cl -CH2C(CH3)3 -Br 1877 3.273 3.361 16 3, 4-2Cl -CH2C(CH3)3 -Br 2346 3.370 3.404 17 3-CF3 -CH2C(CH3)3 -Br 2286 3.358 3.268 18* 4-CH3 -CH2C(CH3)3 -Br 3351 3.525 3.496 19 4-Cl -CH2C(CH3)3 -C6H5 3649 3.562 3.532 20 3-Cl -CH2C(CH3)3 -C6H5 3286 3.516 3.450 21 2-Cl -CH2C(CH3)3 -C6H5 2863 3.456 3.511 22* 3, 4-2Cl -CH2C(CH3)3 -C6H5 3072 3.487 3.491 23 2, 4-2Cl -CH2C(CH3)3 -C6H5 3603 3.556 3.553 24 3-CF3 -CH2C(CH3)3 -C6H5 2087 3.319 3.351 25 4-CH3 -CH2C(CH3)3 -C6H5 3493 3.543 3.590 26 4-Cl -CH2C(CH3)3 4-CH3OC6H4 3198 3.504 3.533 27 3-Cl -CH2C(CH3)3 4-CH3OC6H4 3139 3.496 3.480 28* 3, 4-2Cl -CH2C(CH3)3 4-CH3OC6H4 3138 3.496 3.522 29 3-CF3 -CH2C(CH3)3 4-CH3OC6H4 2084 3.318 3.333 30 4-CH3 -CH2C(CH3)3 4-CH3OC6H4 3861 3.586 3.600 31 4-Cl -CH2C(CH3)3 3, 4-2CH3-isoxazol-4-yl 735 2.866 2.851 32* 4-Cl -CH2C(CH3)3 1H-pyrazol-4-yl 1308 3.116 3.441 33 4-Cl -CH2C(CH3)3 Thiophen-2-yl 3237 3.510 3.459 34 4-Cl -CH2C(CH3)3 Thiophen-3-yl 3332 3.522 3.459 35 4-Cl -CH2C(CH3)3 Furan-2-yl 2668 3.426 3.433 36* 4-Cl -CH2C(CH3)3 Furan-3-yl 2922 3.465 3.398 37 4-Cl -CH2C(CH3)3 Pyridin-4-yl 2086 3.319 3.348 38 4-Cl -CH2C(CH3)3 Pyridin-3-yl 1928 3.285 3.302 39 4-Cl -CH2C(CH3)3 Pyridin-2-yl 1831 3.262 3.275 40 4-Cl -CH2C(CH3)3 4-F-C6H4 2876 3.458 3.493 41 4-Cl -CH2C(CH3)3 2, 3-2F-C6H3 3176 3.501 3.436 42* 4-Cl -CH2C(CH3)3 2, 4-2F-C6H3 2758 3.440 3.448 43 4-Cl -CH2C(CH3)3 2, 5-2F-C6H3 2768 3.442 3.414 44 4-Cl -CH2C(CH3)3 2, 6-2F-C6H3 2194 3.341 3.392 45 4-Cl -CH2C(CH3)3 4-Cl-C6H4 3022 3.480 3.502 46 4-Cl -CH2C(CH3)3 E-4-Cl-C6H4CH=CH 2509 3.399 3.406 47 4-Cl -CH2C(CH3)3 3, 4-2Cl-C6H3 3143 3.497 3.513 48 4-Cl -CH2C(CH3)3 3-CH3OC6H4 3292 3.517 3.547 49* 4-Cl -CH2C(CH3)3 2-CH3OC6H4 1322 3.121 3.192 50 4-Cl -CH2C(CH3)3 4-CH3O(CH2)2O-C6H4 2879 3.459 3.442 51 4-Cl -CH2C(CH3)3 3, 4-(2OCH2)C6H3 3449 3.537 3.521 52 4-Cl -CH2C(CH3)3 4-CF3OC6H4 3337 3.523 3.517 53 4-Cl -CH2C(CH3)3 4-CH3C6H4 3345 3.524 3.497 54* 4-Cl -CH2C(CH3)3 3-CH3C6H4 2449 3.388 3.457 55 4-Cl -CH2C(CH3)3 2-CH3C6H4 2138 3.329 3.351 56 4-Cl -CH2C(CH3)3 4-(CH3)2CHC6H4 2932 3.467 3.462  下载: 导出CSV
下载: 导出CSV
Table 2. Predictive Activities of Newly Designed Small Molecules Based on the CoMFA Model
No. R R4 R5 Bmax[2] LB.exp[2] LBcal 30 4-CH3 -CH2C(CH3)3 4-CH3OC6H4 3861 3.586 3.600 1 3, 4-2CH3 -CH2C(CH3)3 4-CH3OC6H4 3.612 2 3, 4, 5-3CH3 -CH2C(CH3)3 4-CH3OC6H4 3.638 3 3-CH3, 4-C2H5 -CH2C(CH3)3 4-CH3OC6H4 3.621 4 3-CH3, 4-CH(CH3) -CH2C(CH3)3 4-CH3OC6H4 3.615 5 3, 4-2NH2 -CH2C(CH3)3 4-CH3OC6H4 3.615
下载: 导出CSV
-




 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 1
- 文章访问数: 700
- HTML全文浏览量: 8
