Citation:
Xiu-Fang HOU, Chuan BAI, Ya-Lei CAO, Feng FU. Boron Group Ions Direct Conversion of Carbon and Methane into Ethylene in DFT Study[J]. Chinese Journal of Structural Chemistry,
2020, 39(2): 255-262.
doi:
10.14102/j.cnki.0254–5861.2011–2453
Boron Group Ions Direct Conversion of Carbon and Methane into Ethylene in DFT Study
Received Date:
13 May 2019 Accepted Date:
05 September 2019 Available Online:
01 February 2020
Fund Project:
Abstract:
In this study, density functional theory calculations reveal how boron group ions M+ (M = B, Al, Ga, In, and Tl) directly convert carbon and methane into ethylene at room temperature. M+ reacts with the carbon atom to form the cation MC+. Then, the reaction of MC+ with methane leads to the cleavage of metal–carbon bond and the formation of CH2CH2 through C–C coupling, with the ion M+ serving as a leaving group. The cycle then begins again. The mechanism of C/CH4 system catalyzed by five ion types is investigated herein, and the reasons for the different reactivity of five ion types are determined. The moderate strength of the Al+–C bond results in Al+ being the only appropriate catalyst of M+ (M = B, Al, Ga, In, and Tl) that can catalyze methane and carbon into ethylene.
Because of a gradual transformation in the preferred source of global energy, efficient utilization of the C1 molecule has attracted widespread attention[1, 2]. By 2040, the global energy structure will be highly diverse, with oil, natural gas, coal, and nonfossil fuels each accounting for a quarter of all energy sources. Coal demand is declining while demand for natural gas is growing strongly[3]. Natural gas is inconvenient for long-distance transportation because it is difficult to liquefy. In situ conversion of natural gas is also challenging because of the chemical inertness of methane, its main component. The conversion of methane into value-added products, for example, methanol, formaldehyde, and ethylene, has been investigated for decades; however, few conversion processes have been industrialized. The crucial issue of directly and efficiently transforming methane into value-added products in an environmentally friendly manner continues to be explored.
Ethylene is an essential basic raw material. We employ coal and methane as raw materials for producing ethylene. C(3P) in the ground state is weakly reactive with saturated species. Experimental studies[4, 5] have demonstrated that ground state C(3P) does not react with methane, which is in agreement with the theoretical finding that the reaction has a barrier of 51 kJ/mol[6]. In a series of articles, Helmut et al. investigated the activation of C–H bond in methane, for example, [ZnO]+[7], [HgO]+[8], [AuC]+[9], [CuC]+[10], [MO2]+ (M = V, Nb, or Ta)[11], [RuOx]+ (x = 1~3)[12], and [MC]+ (M = 3d)[13]. However, few researches have been conducted on the main group elements and their compounds. Comprising elements of group IIIA in the periodic table, the boron group is boron, aluminum, gallium, indium, thallium, and Uut, the last of which is a synthetic element that is not discussed herein. Aluminum is the most widely distributed metallic element in nature. Herein, we demonstrate M+ (M = B, Al, Ga, In, and Tl)-mediated insertion of a carbon atom into the C–H bond in methane to form ethylene. The origin of the varying reactivity is discussed. This study not only investigates the properties of boron group atoms but also provides a novel prototype for industrial production of ethylene.
2.
COMPUTATIONAL DETAILS
The B3LYP, wB97XD, M06-2X, M06L, B2PLYP, TPSS, and BLYP functionals[14] and 6-311++G**/SDD, 6-311++G**/lanl2dz, Def2-TZVP, and x2c-SV(P)all[15] basis sets are employed. Through analysis and comparison of various functionals and basis sets, full optimization of all stationary points involved in the reaction is achieved using density functional theory (DFT) and the B3LYP functional, the hybrid of Becke's three-parameter exchange functional and the Lee, Yang, and Parr correlation functional[16–18]. The 6-311++G** basis set is employed to describe hydrogen, carbon, boron, and aluminum. Additionally, the relativistic effective core potentials of Stuttgart/Dresden (SDD) are used to describe the metals gallium, indium, and thallium[19]. High-level single-point energy calculations are performed at the stationary points by using the coupled-cluster theory with single, double, and perturbative triple excitations (CCSD(T))[20] approach in the B3LYP geometry. Frequency analysis calculations are performed at the same theoretical level. Furthermore, intrinsic reaction coordinate calculations[21] are performed to confirm that the optimized transition states correctly connect the relevant reactants and products. Natural population analysis is used with natural bond orbital analysis[22, 23]. Orbital interaction diagram analysis and evaluation of the electrostatic potential, localized molecular orbitals, and charge are performed using Multiwfn 3.7[24]. All computations reported herein are conducted using the GAUSSIAN 09 program suite[25].
3.
RESULTS AND DISCUSSION
3.1
Assessment of the levels of theory for describing the C/CH4/M+ system
A reasonable calculation method produces reliable calculation results. To determine which functional and basis set yield reliable results for the C/CH4/M+ system, we compare calculations of the bond dissociation energies (BDEs) of the six species CH3–H, B–C, Al–C, Ga–O, In–CH3, and Tl–O with their previously reported experimental values (No experimental values are available for Ga–C, In–C, and Tl–C)[26]. The DFT-calculated BDEs and relative errors compared with the experimental values are listed in Table S1. First, the B3LYP, wB97XD, M06-2X, M06L, B2PLYP, TPSS and BLYP levels of theory are assessed using the 6-311++G**/SDD basis set. From the data in Table S1, the highest performance is discovered to be that of B3LYP. Next, assuming the B3LYP level of theory, the 6-311++G**/SDD, 6-311++G**/lanl2dz, Def2-TZVP, and x2c-SV(P)all basis sets are compared, and the relative error in BDE calculated using the 6-311++G**/SDD basis set is found to be minimal. Therefore, geometry optimization is performed using the B3LYP/6-311++G**/SDD combination. Finally, the single-point energy of the local minima for the C/CH4/M+ system is calculated using CCSD(T)//B3LYP/6-311++G**/SDD, which is the best level of theory. The result reveals that the computed BDEs of the C–H bond in methane are in agreement with the experimental value, with relative errors within 7% regardless of the functional used. For Tl–O, the variation in the relative error is the greatest, with the error range being 213±84 kJ/mol. According to the differences between the calculated data and experimental results, the CCSD(T)//B3LYP/6-311++G**/SDD level of theory provides the most rotational constants of the BDE of the selected species.
3.2
Mechanistic aspects
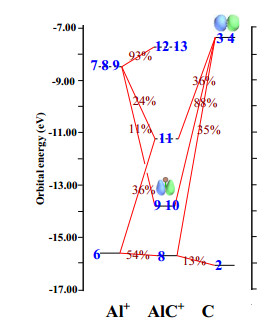
To understand the nature of the molecular orbitals of the cationic carbide MC+, the orbital interaction diagram of AlC+ is plotted, as shown in Fig. 1. The diagrams for the other carbides MC+ (M = B, Ga, In, and Tl) are displayed in Fig. S1. In Fig. 1, the horizontal bars on the left- and right-hand sides correspond to the frontier molecular orbital of Al+ and C fragments, respectively, whereas the bars in the middle correspond to the orbitals of the AlC+ complex. Orbital indices are labeled in blue. The molecular orbitals of the fragments and complex are connected by red lines, with the compositions labeled in the center of the lines. Solid and dashed bars correspond to occupied and virtual molecular orbitals, respectively. For example, the semioccupied molecular orbitals (SOMOs) 9 and 10 of complex AlC+ are constructed from the SOMOs 3 and 4 of the C atom and the lowest unoccupied molecular orbital (LUMO) of the Al atom. The contribution of carbon is 88% and that of aluminum is 11%. The isosurface graphs of SOMOs 9 and 10 of complex AlC+ mainly show carbon. The contribution of carbon is 65% and that of boron is 34% in the SOMO in BC+. However, in GaC+, InC+, and TlC+, the energy level difference between the LUMO of Ga, In, and Tl and the SOMO of the C atom is excessively large, resulting in the SOMOs of the complexes being mainly composed of carbon SOMOs. In addition, Fig. 1 reveals that the unpaired single electrons are located on C, that is, the spin density is high at C, indicating that the C-terminal of AlC+ is beneficial to the transfer of hydrogen atoms.
Figure 1
Figure 1.
Orbital interaction diagram for AlC+. Vertical axis shows the molecular orbital energies in eV. Solid and dashed horizontal bars correspond to occupied and virtual orbitals, respectively. If two labels occur on the same bar, the corresponding states are doubly degenerate
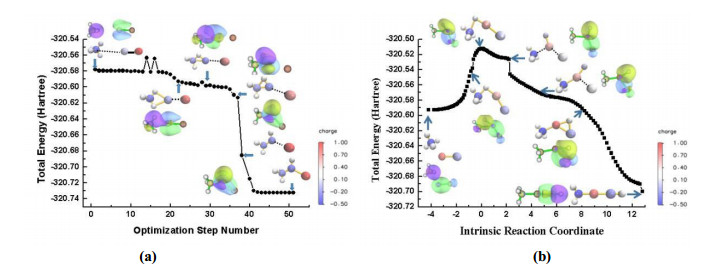
The ground state of the aluminum ion is a singlet, and the energy of the first excited triplet state is 426.25 kJ/mol higher than that of the ground state. The energy level difference of M+ with M = B, Ga, In, and Tl is 449.9, 544.7, 487.4, and 626.5 kJ/mol, respectively. Therefore, we only examine the singlet state M+ herein. The energy level difference between the first excited singlet state and triplet ground state of the carbon atom is 151.26 kJ/mol, which is slightly higher than 121.8 kJ/mol reported in the literature[4]. When 1Al+ and 3C form 3AlC+, 36.8 kJ/mol of energy is released. 3AlC+ can approach methane through two routes: the carbon-end approach (path A) and aluminum-end approach (path B). When the C-terminal of 3AlC+ is close to methane, the hydrogen transfer product 3M2 is directly produced through a barrier-free process. To clarify the origin of the marked difference between paths A and B, the details of the configuration changes within the process are illustrated in Fig. 2. It is a full geometry optimization in Fig. 2a.
Figure 2
Figure 2.
Configuration, LMOs, and charge changes as 3AlC+ approaches the C–H in methane. The C-terminal of 3AlC+ is close to methane in (a), whereas the Al-terminal is displayed in (b). Positive and negative atomic charges are expressed by atomic color, which ranges from blue to red, corresponding to charges from −0.5 to 1.0 |e|
Fig. 2 displays several types of information: changes in the configurations, energies, localized molecular orbitals (LMOs), and charge as 3AlC+ approaches the C–H in methane. Figs. 2a and 2b respectively show the approach of the C- and Al-terminal of 3AlC+ to methane. Analysis of the configuration changes reveals that H from methane migrates to the C' of AlC+ in both paths. In terms of energy change, Fig. 2a shows a barrier-free process, whereas the process illustrated in Fig. 2b has a high barrier. We also evaluate the variation of the LMOs to visualize the variation in the chemical bonds. In Fig. 2a, the C–H bond in methane breaks and a H–C' (AlC) bond forms; additionally, the C'–Al bond changes. In the first hypothetical configuration in Fig. 2a, the C–C' bond is 4.0 Å. The purple-yellow isosurface shows the typical covalent character of the C–H bond; the green-blue isosurface represents the SOMO of AlC. As the reaction proceeds, the length of the C–C bond decreases to 2.3 Å because of electrostatic attraction. The electrons in LMO around C–H have delocalized slightly to the C' atom in the second configuration. In the third configuration, the electrons have been further delocalized to the C' atom, and the C–H bond in methane and C'–Al bond are breaking. Then, a H–C' bond forms. The H–C' and C–Al bonds are fully formed in the sixth configuration. In Fig. 2a, the purple-yellow isosurface shows the typical C–H bond in methane, whereas the green-blue isosurface represents the bonding in AlC. As the reaction proceeds, the C–H' bond breaks and a H'–Al bond forms. The migrated H' then continues to move to C' in the AlC fragment, forming the linear complex CH3AlCH+, named 3M2' in Fig. 3.
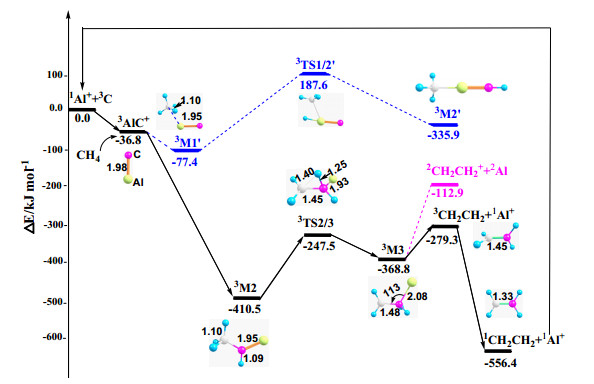
Figure 3
Figure 3.
PESs and relevant structural information for Al+/C/CH4, calculated at the CCSD(T)//B3LYP/6-311++G**, SDD level of theory. Relative energies are given in kJ/mol. Bond lengths are given in Å and bond angles in degree. The C atom in AlC+ is red and Al is yellow; carbon in CH4 is white and H is green. The PESs of M+ (M = B, Ga, In, and Tl)/C/CH4 are presented in Fig. S2 in the supporting information
Atomic point charge is vital to understanding the properties of a molecule, the possible reaction direction, and the state of atoms in a molecule. Atomic charge is unobservable, and its definition is not unique. Charges on the complexes during the first H migration along paths A and B are presented as the atomic-dipole-corrected Hirshfeld atom population (ADCH)[27] in Fig. 3. The ADCH is an improved version of Hirshfeld charge. Positive and negative atomic charges are reflected by atomic color, which ranges from blue to red, corresponding to charge −0.5 to 1.0 |e|. In Fig. 2a, no energy barrier exists and the C' end of AlC+ approaches methane. The initial configuration is our hypothetical configuration, and the charge of C' near Al is 0.002, expressed by light blue, whereas that of Al is 0.99, expressed by red. Because the electronegativity of carbon is greater than that of hydrogen, the charge of carbon (−0.37) in the methane fraction is negative, the charge of H near the CAl fraction is 0.06, and that of the other two H atoms is 0.12. During the process of H' migrating from methane to C' (six configurations in Fig. 2a), the charge of H' is 0.06, 0.14, 0.19, 0.19, 0.18, and 0.20 and that of C' is 0.00, −0.10, −0.22, −0.22, −0.24, and −0.28, respectively. The charge of H' is positive and increases gradually, whereas that of C' is negative and decreases gradually. The heterocharged ions attract each other, and the electrostatic force (or Coulomb force) is the main force driving the barrier-free process. In Fig. 2b, the Al (red) end of AlC+ approaches methane, which is a process that requires overcoming the 265.0 kJ/mol barrier. The primary complex in Fig. 2b is 3M1'. During the process of H' migrating from methane to C' (six configurations in Fig. 2b), the charge of H' is 0.13, 0.18, 0.09, 0.006, 0.03, 0.18, and 0.25 and that of Al is 0.81, 0.55, 0.67, 0.58, 0.59, 0.84, and 0.79, respectively. The charges of H' and Al are all positive; thus, Coulomb repulsion is the main reason why this process has a large energy barrier. Another phenomenon that the attraction of the negatively charged C' to H' causes the H' to transfer to Al and then continues to migrate to C' also occurs. The product of this step is the seventh complex displayed in Fig. 2b, which is labelled as 3M2' in Fig. 3. This step reaction is impractical because of the high barrier.
The potential-energy surfaces (PESs) of the most favorable reaction pathways and structural information on the relevant species in the Al+/C/CH4 system are presented in Fig. 3. Methane initially coordinates axially in a η2 manner with the Al atom of AlC+, forming the complex 3M1' and releasing 77.4 kJ/mol of energy. The energy gained in generating 3M1' is less than sufficient to cross the transition state 3TS1/2'. Therefore, the complex 3M2' cannot form. According to the analysis in Fig. 2, this step is impractical because of Coulomb repulsion. When the C-terminal of AlC+ coordinates with methane, complex 3M2 is produced directly. Another H on the methyl group then migrates to the C' of the AlC fragment through the transition state TS2/3, resulting in complex 3M3. This step occurs easily because the energy of the transition state TS2/3 is lower than that of the reaction asymptote (247.5 kJ/mol). The complex 3M3 decomposes in two manners: decomposition into 2CH2CH2+ and 2Al, which requires 245.9 kJ/mol of energy, and decomposition into 3CH2CH2 and 1Al+, which needs only 89.5 kJ/mol. Triple ethylene is unstable and converts to singlet ethylene. This is the most energetically favorable pathway. The product 1Al+ continues to catalyze. Interestingly, although the Al+ ion is not directly involved in the C–H bond breaking of methane and may thus be considered as an innocent spectator ligand, its presence alters the environment around C and modifies the reaction mechanism between carbon and methane. In summary, C and CH4 can produce ethylene directly under the catalysis of an aluminum ion.
In the B+/C/CH4 system, CH4 initially coordinates with the B atom of 3BC+ to form the encounter complex 3M1' (−346.4 kJ/mol; Fig. S2). Next, a H in CH4 transfers through transition state 3TS1/2' to form the intermediate 3M2'. Subsequently, the second hydrogen transfer forms complex 3M3'. Compound 3M3' is a stable intermediate, and continuing the reaction to produce ethylene products is difficult. When CH4 initially coordinates with the C atom of 3BC+, the reaction mechanism is similar to that of the C-terminal in the AlC+ system; namely, complex 3M2 is formed directly. Next, the second H transfer occurs through transition state 3TS2/3 to form the intermediate 3M3. The decomposition of 3M3 is different from that in the AlC+ system. The product of B and 2CH2CH2+ is more stable than that of 1B+ and 3CH2CH2 because of the lack of electrons in B. Therefore, B+ cannot catalyze C and CH4 to produce ethylene. The PES of Ga+/C/CH4 is displayed in Fig. S2. Forming 3GaC+ is difficult because the energy level difference between Ga and C is too large, as illustrated by the orbital interaction diagram presented in Fig. S1. The situation for system In+/C/CH4 is similar to that of system Ga+/C/CH4. In Tl+/C/CH4, although the energy level difference between Tl and C is large, the remarkable relativistic effect[8] of Tl leads to the formation of the relatively stable 3TlC+. Subsequently, the reaction mechanism of TlC+/CH4 is similar to that of AlC+/CH4. Therefore, C and CH4 can produce ethylene directly under catalysis by thallium ions, although the reaction efficiency is less than that when aluminum ions are employed.
The cause of the remarkable differences in reaction mechanism among M+ (M = B, Al, Ga, In, and Tl)/C/CH4 can be traced to the distinctly different M–C bonding features of B+, Al+, Ga+, In+, and Tl+. The strength of the metal–carbon bond follows the order of B, Al, Ga and In; owing to the strong relativistic effects, the Tl–C bond is slightly stronger than that of In–C. The B–C bond is too strong, whereas the Ga–C and In–C bonds are too weak; only the Al–C bond is of moderate strength. Therefore, Al+ is the only appropriate catalyst for this system.
4.
CONCLUSION
According to theoretical calculations, of all M+ (M = B, Al, Ga, In, and Tl), only Al+ ions can catalyze the reaction of methane and carbon to produce ethylene. The reaction mechanism is investigated in detail in this study. The electron-deficient nature of B leads to the reaction of BC+ with methane to form the CH2BCH2+ complex. Because the energy level differences of Ga and In with C are too large, the formation of MC+ is difficult and the reaction with methane is impossible. Although Tl+ can catalyze the reaction to produce ethylene, its efficiency is poor. The cause of the remarkable differences in the reaction mechanism among M+ (M = B, Al, Ga, In, and Tl)/C/CH4 can be traced to the distinctly different intrinsic properties of the metal ions. In summary, in this computational study, we discover that aluminum ions can directly convert carbon and methane into ethylene at room temperature. This is partly because of the moderate-strength Al–C bond and electrophilic nature of C.
[1]
Ravi, M.; Ranocchiari, M.; Jeroen, A. van Bokhoven. The direct catalytic oxidation of methane to methanol-a critical assessment. Angew. Chem. Int. Ed.2017, 56, 16464−16483. doi: 10.1002/anie.201702550
Leonori, F.; Skouteris, D.; Petrucci, R.; Casavecchia, P.; Rosi, M.; Balucani, N. Combined crossed beam and theoretical studies of the C(1D) + CH4 reaction. J. Chem. Phys.2013, 138, 024311−11. doi: 10.1063/1.4773579
[5]
Jeong, G. H.; Klabunde, K. J.; Pan, O. G.; Paul, G. C.; Shevlin, P. B. Reactions of carbon atoms/clusters with methane, methyl bromide, and water at 10 and 77 K. J. Am. Chem. Soc.1989, 111, 8784−8790. doi: 10.1021/ja00206a003
[6]
Kim, G. S.; Nguyen, T. L.; Mebel, A. M.; Lin, S. H.; Nguyen M. T. Ab Initio/RRKM study of the potential energy surface of triplet ethylene and product branching ratios of the C(3P) + CH4 reaction. J. Phys. Chem. A2003, 107, 1788−1796.
[7]
Yue, L.; Li, J. L.; Zhou, S. D.; Sun, X. Y.; Schlangen, M.; Shaik, S.; Schwarz, H. Control of product distribution and mechanism by ligation and electric field in the thermal activation of methane. Angew. Chem. Int. Ed.2017, 56, 10219−10223. doi: 10.1002/anie.201703485
[8]
Yue, L.; Zhou, S.; Sun, X.; Schlangen, M.; Schwarz, H. Direct room-temperature conversion of methane to protonated formaldehyde: the gas-phase chemistry of mercury among the zinc triad oxide cations. Angew. Chem. Int. Ed.2018, 57, 3251−3255. doi: 10.1002/anie.201712405
[9]
Li, J.; Zhou, S.; Schlangen, M.; Weiske, T.; Schwarz, H. Hidden hydride transfer as a decisive mechanistic step in the reactions of the unligated gold carbide [AuC]+ with methane under ambient conditions. Angew. Chem.2016, 128, 13266−13269. doi: 10.1002/ange.201606707
[10]
Geng, C.; Li, J.; Weiske, T.; Schlangen, M.; Shaik, S.; Schwarz, H. Electrostatic and charge-induced methane activation by a concerted double C−H bond insertion. J. Am. Chem. Soc.2017, 139, 1684−1689. doi: 10.1021/jacs.6b12514
[11]
Zhou, S.; Li, J.; Schlangen, M.; Schwarz, H. Differences and commonalities in the gas-phase reactions of closed-shell metal dioxide clusters [MO2]+ (M = V, Nb, and Ta) with methane. Chem. Eur. J.2016, 22, 7225−7228. doi: 10.1002/chem.201600498
[12]
Sun, X.; Zhou, S.; Yue, L.; Schlangen, M.; Schwarz, H. Thermal activation of CH4 and H2 as mediated by the ruthenium-oxide cluster ions [RuOx]+ (x = 1~3): on the influence of oxidation states. Chem. Eur. J.2019, 25, 1−11. doi: 10.1002/chem.201806054
[13]
Geng, C.; Weiske, T.; Li, J.; Shaik, S.; Schwarz, H. On the intrinsic reactivity of diatomic 3d transition-metal carbides in the thermal activation of methane: striking electronic structure effects. J. Am. Chem. Soc.2018, 141, 599−610.
[14]
Kang, Y. K.; Park, H. S. Assessment of CCSD(T), MP2, DFT-D, CBS-QB3, and G4(MP2) methods for conformational study of alanine and proline dipeptides. Chem. Phy. Lett.2014, 600, 112−117. doi: 10.1016/j.cplett.2014.03.067
[15]
Pollak, P.; Weigend, F. Segmented contracted error-consistent basis sets of double- and triple‑ζ valence quality for one- and two-component relativistic all-electron calculations. J. Chem. Theory Comput.2017, 13, 3696−3705. doi: 10.1021/acs.jctc.7b00593
[16]
Becke, A. D. Density-functional thermochemistry. Ⅲ. The role of exact exchange. J. Chem. Phys.1993, 98, 5648−5652. doi: 10.1063/1.464913
[17]
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A1988, 38, 3098−3100. doi: 10.1103/PhysRevA.38.3098
[18]
Lee, C.; Yang, W. T.; Parr, R. G. Development of the Colle-Salvetti correlation energy formula into a functional of the electron density. Phys. Rev. B1988, 37, 785−789. doi: 10.1103/PhysRevB.37.785
[19]
Andrae, D.; Häubermann, U.; Dolg, M.; Stoll, H.; Preub, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta1990, 77, 123−141. doi: 10.1007/BF01114537
[20]
John, A.; Martin, H. G. Quadratic configuration interaction: a general technique for determining electron correlation energies. J. Chem. Phys.1987, 87, 5968−5975. doi: 10.1063/1.453520
[21]
Gonzalez, C.; Schlegel, H. B. An improved algorithm for reaction path following. J. Chem. Phys.1989, 90, 2154−2161. doi: 10.1063/1.456010
[22]
Reed, A. E.; Weinstock, R. B.; Weinhold, F. Natural population analysis. J. Chem. Phys.1985, 83, 735−746. doi: 10.1063/1.449486
[23]
Carpenter, J. E.; Weinhold, F. Analysis of the geometry of the hydroxymethyl radical by the different hybrids for different spins natural bond orbital procedure. J. Mol. Struct. (Theochem. ) 1988, 169, 41−62. doi: 10.1016/0166-1280(88)80248-3
[24]
Lu, T.; Chen, F. Multiwfn: a multifunctional wave function analyzer. J. Comput. Chem.2012, 33, 580−592. doi: 10.1002/jcc.22885
[25]
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Annenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian, Inc., Wallingford CT 2013, Gaussian 09, Revision E. 01.
[26]
Luo, Y. R. Comprehensive Handbook of Chemical Bond Energies. CRC Press Inc, Boca Raton, FL 2007, 1−1687.
[27]
Lu, T.; Chen, F. Atomic dipole moment corrected Hirshfeld population method. J. Theor. Comput. Chem.2012, 11, 163−183. doi: 10.1142/S0219633612500113
Figure 1
Orbital interaction diagram for AlC+. Vertical axis shows the molecular orbital energies in eV. Solid and dashed horizontal bars correspond to occupied and virtual orbitals, respectively. If two labels occur on the same bar, the corresponding states are doubly degenerate
Figure 2
Configuration, LMOs, and charge changes as 3AlC+ approaches the C–H in methane. The C-terminal of 3AlC+ is close to methane in (a), whereas the Al-terminal is displayed in (b). Positive and negative atomic charges are expressed by atomic color, which ranges from blue to red, corresponding to charges from −0.5 to 1.0 |e|
Figure 3
PESs and relevant structural information for Al+/C/CH4, calculated at the CCSD(T)//B3LYP/6-311++G**, SDD level of theory. Relative energies are given in kJ/mol. Bond lengths are given in Å and bond angles in degree. The C atom in AlC+ is red and Al is yellow; carbon in CH4 is white and H is green. The PESs of M+ (M = B, Ga, In, and Tl)/C/CH4 are presented in Fig. S2 in the supporting information

 DownLoad:
DownLoad:

 下载:
下载:




