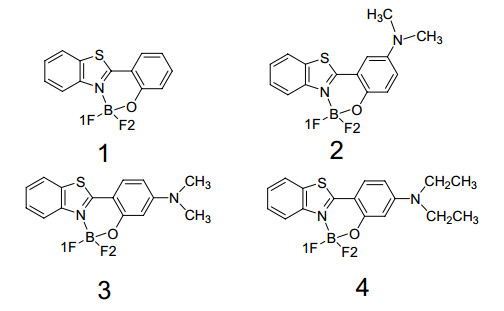
Figure 1.
Molecular structures of BOBTP 1, 2, 3 and 4
Theoretical Insights into the Substituent Effects on the Fluorescence of Boron Complexes of 2-(Benzothiazol-2-yl)phenols: a Quantum Mechanics Study
Hui-Xue LI , Xiao-Feng WANG , Yuan-Cheng ZHU , Kun YUAN , Ling-Ling LV , Zhi-Feng LI

Among various types of luminescent compounds, boron complexes of dipyrromethenes (BODIPYs) have received particular attention due to the excellent photophysical properties of these compounds, such as thermal and photochemical stability, small Stokes shifts, high quantum yields, sharp excitation and emission peaks[1-3]; Further, their photophysical properties can be readily tuned by structural modifications, which provide additional opportunities to meet the different requirements for diverse applications, for instance dyes, nonlinear optical materials and solar cells[4-6].
The core structures of BODIPYs have been modified into its nitrogen-containing heterocyclic compounds, such as aza-boron-dipyrromethene, aza-boron-diindolmethene, aza-boron-diindolmethene[7-11], in which electron delocalization occurs through an oligomethine chain, the core structures give the distinctive photochemical properties of these BODIPY dyes. In addition, a recent study showed that an unsaturated neutral donor is appended to the heteroaromatic ring in a number of boron compounds, the modified complexes exhibit more high photoluminescence quantum yields and good electron-transporting property compared to the traditional BODIPYs[12, 13]. Ulrich et al. synthesized fluorescent borate complexes based on the 2-(20-hydroxyphenyl)benzoxazole core, the optical properties of these complexes have been modeled using timedependent density functional theory. The computations using a range-separated hybrid functional simulated vertical, adiabatic and vibronic energies, as well as analysised the charge-transfer characteristics of each state, the results interpreted the major experimental features[14]. Other boron conjugated complexes with donor and acceptor ligands would be of interest in the development of novel photoluminescent compounds, Kyo et al.[15] synthesized 2-(benzothiazol-2-yl)phenol-based BODIPY (abbreviated as BOBTPs) and investigated the photophysical properties, they found substituents can induce dramatic changes in the photoluminescence properties of N, O-chelated boron complexes. Some BOBTPs can result in bright blue- and orange-emitting phosphors in solution and in the solid state. However, other BOBTPs with different substituents and different position in the ring showed inferior photophysical properties, obviously the substituent effects have done a great deal of influence to photoluminescence and electroluminescence, which offers an important guideline for the development of related photoelectric materials. It is necessary to understand the performance and luminescence mechanism of the BOBTPs. In this paper, we performed a detailed quantum chemistry calculation to obtain a rational explanation of the observed photophysical behaviors, density functional theory (DFT) and time-dependent density functional theory (TDDFT) were employed to calculate the electronic structures, energies and compositions of excited states of BOBTPs, aimed at exploring the effects of these different substituents, which are bonded to different positions of the heterocyclic rings, on the electronic structures and optoelectronic properties.
Using Fermi gold rule, the rates of absorption and emission photon can be gained respectively[16-20], this part can be seen in Supplementary information. The geometry optimization and frequency calculations for BOBTPs were carried out using DFT and TDDFT, hybrid density functional B3LYP and Pople's 6–31+G(d) basis set was employed in all calculations. The standard polarizable continuum solvation model (PCM) was taken to simulate the solvent effects of CH2Cl2. To ensure that each configuration was a minimum on the potential energy surface, the vibrational frequencies of all the BOBTPs were calculated at the same theoretical level. The TDDFT approach was applied to investigate the excited-state electronic properties, the DUSHIN program was used to obtain the reorganization energy on the basis of ground- and excitedstate optimization[21]. All the calculations were performed with the Gaussian 09 program package[22].
The sketch map of the structures is depicted in Fig. 1, the calculated bond lengths and bond angles of the ground states and the excited states for all the BOBTPs are listed in Table 1, the X-ray crystal data of BOBTP 2 and 4[15] are also shown together. Compared with the experiments, the calculated bond length of B–F of 2 and 4 have error of about 0.002 Å, the errors about B–N, B–O, O–C, and C–O for 2 and 4 are less than 0.025 Å, the calculated distance of the fluorine atom and the adjacent hydrogen atom at the benzothiazol ring, which can form intramolecular hydrogen bond, is underestimated by about 0.05 Å for complex 2, and about 0.1 Å for complex 4, respectively, due to the crystal lattice distortion existing in the real molecules and the drawback of hybrid functional B3LYP, the errors of the F…H hydrogen bonds between the experimental and the calculated values are more larger. As for complex 4, the benzothiazol ring, benzene ring, and N, O– chelated boron heterocycle hexatomic ring (abbreviated as X ring) form a approximate plane of aromatic conjugation system theoretically and experimentally, and the calculated dihedral angle of C–C–O–B at X ring is 2.92º, which is close to the experimental value 2.84º, it indicates the measured values is reasonable and acceptable. However, for complex 2 the X ring is not plane whether by computation or from the experiment, the dihedral angle C–C–O–B at X ring is 14.19º from the computaton and 19.51º from the experiment. Unfortunately, the single crystal structure data of the complex 1 and 3 have not been experimentally determined so far, only the calculated bond parameters are listed in the Table 2. One can see the complex 3 is nonplanar skeleton and the dihedral angle C–C–O–B at X ring is 14.19º, it is similar to the complex 2; however, the complex 1 possesses nearly planar skeleton and the dihedral angle C–C–O–B at X ring is 2.92º, which is equal accidentally to one of the complex 4. Because of fluorine atoms with the most largest electronegativity and boron atom with the electron-deficienty, obviously the atomic group BF2 is elecronwithdrawing, the geometries of BOBTP 2, 3 and 4 will reform and optimize again after the electron-donating group NR2 bonding to complex BOBTP, both the B–F bond lengths of all the complexes are unequal, i.e., one is larger compared with the other, it is due to the diversities of the elecron population in bonding orbitals for all complexes, the electron density distributing on the frontier molecular orbitals will be discussed below. In additon, for complexes 1, 2, 3 and 4, the differences of all the bond lengths and bond angles between the ground states and the first excited states are small, however the dihedral angle exists considerable change.
 DownLoad:
CSV
DownLoad:
CSV
| Parameters | BOBTP 2 | BOBTP 4 | ||||
| S0 | S0 exp. | S1 | S0 | S0 exp | S1 | |
| Bond length (Å) | ||||||
| B–N | 1.606 | 1.588 | 1.594 | 1.604 | 1.578 | 1.616 |
| B–O | 1.462 | 1.437 | 1.472 | 1.462 | 1.448 | 1.468 |
| O–C | 1.327 | 1.348 | 1.327 | 1.327 | 1.339 | 1.326 |
| C–N | 1.383 | 1.381 | 1.386 | 1.375 | 1.365 | 1.375 |
| C–H(CH3) | 1.099 | 1.097 | 1.089 | 1.099 | 1.097 | 1.088 |
| B–F(1) | 1.389 | 1.379 | 1.378 | 1.380 | 1.383 | 1.378 |
| B–F(2) | 1.395 | 1.389 | 1.384 | 1.384 | 1.387 | 1.380 |
| F…H | 2.441 | 2.498 | 2.435 | 2.504 | 2.629 | 2.591 |
| Bond angle and dihedral angle (°) | ||||||
| F(1) –B–F(2) | 111.75 | 110.44 | 111.88 | 111.53 | 109.87 | 112.90 |
| N–B–O | 108.24 | 109.39 | 108.61 | 108.51 | 110.18 | 106.97 |
| C–O–B | 125.61 | 123.03 | 125.44 | 126.30 | 125.72 | 123.74 |
| C–C–O–B | 14.19 | 19.51 | 18.51 | 2.92 | 2.84 | 5.73 |
DownLoad:
CSV
| Parameters | BOBTP 1 | BOBTP 3 | ||
| S0 | S1 | S0 | S1 | |
| Bond length (Å) | ||||
| B–O | 1.462 | 1.468 | B–O | 1.462 |
| O–C | 1.327 | 1.326 | O–C | 1.327 |
| C–N | 1.375 | 1.374 | C–N | 1.375 |
| B–F(1) | 1.384 | 1.386 | B–F(1) | 1.384 |
| B–F(2) | 1.389 | 1.394 | B–F(2) | 1.389 |
| F…H | 2.504 | 2.591 | F…H | 2.504 |
| Bond angle and dihedral angle (°) | ||||
| F(1)–B–F(2) | 111.53 | 112.90 | 111.75 | 111.88 |
| N–B–O | 108.51 | 106.97 | 108.24 | 108.61 |
| C–O–B | 126.30 | 123.74 | 125.61 | 125.44 |
| C–C–O–B | 2.92 | 5.73 | 14.19 | 18.51 |
It is significant to examine the nature of the frontier molecular orbitals about these complexes for the electronic spectrum, regardless of the emission spectrum, or the absorption spectrum. One can see the molecular orbitals involved the electron transition from the ground state to the excited state for all the BOBTPs are the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) at Table 3 and Table 4, as well the oscillator strength and main configurations are reported together, furthermore, these important frontier molecular orbitals are plotted in Fig. 2. For all the studied complexes, the patterns of the occupied molecular orbitals and unoccupied molecular orbitals possess the characteristics of π orbital, the calculated absorption spectra of BOBTP 1, 2, 3 and 4 in dichloromethane by B3LYP/6-31+G(d) are in attached Fig. 1, the numbers, locations and shapes of the absorption peaks are consistent with the observed spectrum, especially the spectra of both the BOBTP 3 and 4 look nearly identical, which implies whether the substituent is methyl or ethyl there are almost the same effect on the HOMO and LUMOs when parent molecule and the position of substituent are fixed. The wavelengths of maximum absorption for the lower excited states of BOBTPs in CH2Cl2 by TDDFT are also listed in attached Table 1.
DownLoad:
CSV
| Complex | Oscillator strength | Main configuration | Assignment | λexp (nm) | λcal (nm) |
| 1 | 0.3897 | HOMO→LUMO | 0.6970 | 364 | 363.2 |
| 2 | 0.1252 | HOMO→LUMO | 0.7012 | 456 | 466.3 |
| 3 | 0.8645 | HOMO→LUMO | 0.7014 | 413 | 429.3 |
| 4 | 0.8638 | HOMO→LUMO | 0.7030 | 417 | 425.2 |
DownLoad:
CSV
| Complex | Oscillator strength | Main configuration | Assignment | λexp(nm) | λcal(nm) | μ(Debye) |
| 1 | 0.4948 | HOMO→LUMO | 0.7038 | 433 | 417.1 | 2.385 |
| 2 | 0.1661 | HOMO→LUMO | 0.7075 | 597 | 606.5 | 2.046 |
| 3 | 1.294 | HOMO→LUMO | 0.6120 | 430 | 423.9 | 11.190 |
| 4 | 1.3120 | HOMO→LUMO | 0.7033 | 434 | 422.1 | 11.900 |
| μ: transition electric dipole moments between the ground to excited state 1Debye=8.478×10-30 C • m. | ||||||
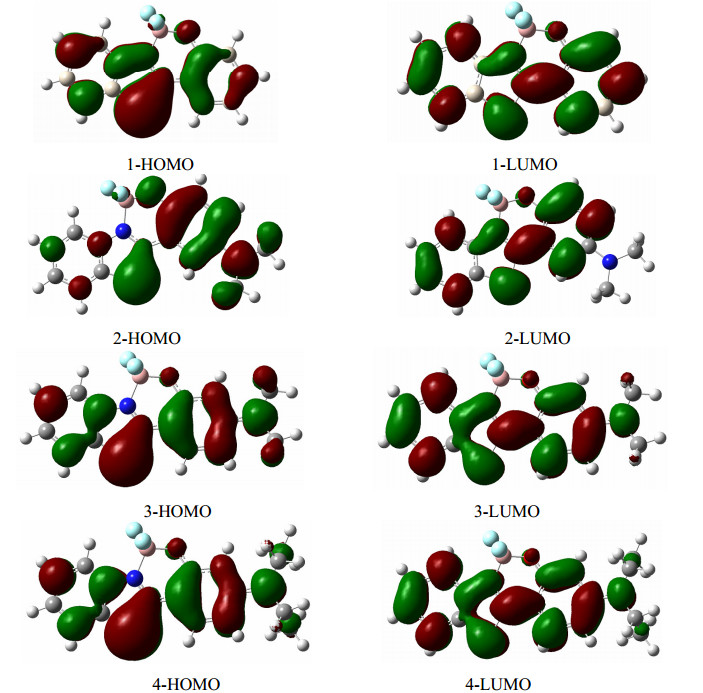
Since the main configurations of electron transition listed in Table 3 and Table 4 for these complexes are between the HOMOs and LUMOs, an effective one-particle model can be used to treat the processes, i.e., it is reasonable that the HOMO and LUMO replace the electronic wave function of the initial state and final state to calculate the molecular properties, however it is correct for a purely oneexcitation process, otherwise, the one-particle model should be cautioned two-excitation process. For the studied complex 3 and 4, the electron densities of all the HOMOs and the LUMOs distribute mainly on the benzothiazol ring, benzene ring and X ring, the overlap extent of electron cloud on the HOMOs and the LUMOs shall be large because they show a delocalized feature, thus the transition electric dipole moments (μ) between the ground and the excited state for complex 3 and 4 must be larger, as well the calculated result shows the μ values of 3 and 4 in Table 4 are more larger and approximately equal, which lead to the large fluorescence quantum yield of both the complexes. In the case of 2, the electron distribution in the HOMO is confined to the phenol ring and the amino moiety, but the electron distribution in the LUMO is shifted to the benzothiazole and the phenol ring, the localization of the electron distribution causes a decrease of the wave function overlap and seems to reduce the fluorescence quantum yield, Osmialowski et al. pointed out that a strong intramolecular charge transfer is present when the amino acts as donor in difluoroboranyl molecule[23], the complex 2 shows the distinct characteristic of intramolecular charge transfer too, all the oscillator strength of the transition for the ground states to the excited states in Table 4 are consistent with all the transition electric dipole moments.
To get more information from the frontier orbitals, the composition of the HOMOs and LUMOs has been calculated. The whole complex molecule was segmented into three moieties, i.e., a benzothiazole-benzen fragment, a BF2O fragment, and a substituent NR2 fragment, all the percentages of the frontier molecular orbitals for each fragment are shown in Table 5. As to the complex 1, the composition of HOMO comes mainly from orbitals of benzothiazole-benzen (93%), the component of LUMO come mainly from benzothiazole-benzen (63%) and BF2O (32%), it implies the wave function overlap of the HOMO and LUMO will be not more effective compared with other complexes, which suggests the transition electric dipole moments is small, thus the absorption coefficient of complex 1 will decrease, the computed data in Table 4 show the corresponding oscillator strength is the smallest. In contrast, the components of HOMOs of complex 2, 3 and 4 come not only from benzothiazole-benzen, but from NR2, similar to the case of the components of LUMOs of these complex, benzothiazole-benzen and NR2 fragments contribute the most to the LUMOs, however, the contribution of NR2 group of complex 2 for the LUMO is almost zero, the above data quantitatively explain the profile of the frontier molecular orbitals.
DownLoad:
CSV
| Fragments | 1 | 2 | 3 | 4 |
| HOMO | LUMO | HOMO | LUMO | |
| benzothiazole-benzen | 0.93 | 0.63 | 0.53 | 0.96 |
| BF2O | 0.03 | 0.32 | 0.03 | 0.02 |
| NR2 | 0.36 | 0 |
The normal-mode (NM) analysis was adopted to estimate the Huang−Rhys (HR) factor and the reorganization energy based on DFT calculations by formula (
HR factor is dimensionless, which can characterize the deformation of a molecule when an electron comes from one electronic state with the ith vibrational level to another with the jth vibrational level. The calculated HR factors for the S1 of the BOBTP molecules are listed in Table 6, and some selected normal modes are presented along with the corresponding HR factors. It can be seen the largest HR factor of BOBTP 1 is 3.1958, the shift of the normal coordinate between the ground state and the excited state is 2.526, which corresponds to the torsion vibration of benzene ring and X ring about complex 1 with the frequency of 46 cm-1, the Dushinsky matrix shows the frequency mainly comes from the mixture of the 20 cm-1 (having account for 17%), 56 cm-1 (having account for 21%) and 81 cm-1 (having account for 10%) vibrational modes of the ground state. For the complex 2, the largest HR factor is 0.5216 and the variabe of the normal coordinate is -1.021, the Dushinsky matrix shows the excited vibrational state with 250 cm-1 mainly comes from the mixture of the 188 cm-1 (having account for 15%) and 254 cm-1 (having account for 19%) vibrational modes of the ground state, as with complex 1, it remains the deformation vibration of benzene and X rings. The largest HR factor is 0.1357 and the shift of the normal coordinate is 0.524 for complex 3, the Dushinsky matrix indicates the excited vibrational state with 28 cm-1 mainly comes from the mixture of the 35 cm-1 (having account for 42%) and 354 cm-1 (having account for 24%) vibrational modes of the ground state. For complex 4 the largest HR factor is 0.1056 and the corresponding shift of the normal coordinate is –0.455, the Dushinsky matrix indicates the excited vibrational state with 18 cm-1 mainly comes from the 27 cm-1 (having account for 42%) vibrational mode of the ground state, the low frequencies of the complex 3 and 4 remain the deformation vibration of benzene and X rings. These results show the torsion of the rings with lower frequencies are hindered instead of the bond stretching for all the BOBTP molecules. However, the HR factor and the change of the normal coordinate of the complex 1 is the largest in all the BOBTPs, which implies the overlap integral between the vibrational wave functions of the ground state and that of the excited state is the smallest, thus the Franck-Condon factor of the formula (7) for complex 1 seems to be minimum value, the rates of emission photon is directly proportional to the square of the Franck-Condon factor of the molecules in formula (7), it surprisingly coincide with the smallest fluorescence quantum yield (0.23) of complex 1, the HR factor and the shift of the normal coordinate of the complex 2 are second only to the complex 1, it has a slightly greater fluorescence quantum yield (0.28). As compared to both the BOBTP molecules, the HR factors and the shifts of the normal coordinate of the complex 3 and 4 are smaller, the fluorescence quantum yields are the highest and nearly 1 in all the complexes. It is well known that NR2 is electron-donating group, the electron density of the BOBTP parent will increase when the amino bonding to the benzene rings in BOBTPs, the deformation about complex 2, 3 and 4 will decrease compared with the complex 1 due to the stronger bond of the BOBTP derivatives, resulting in a more larger HR factor and the shift of the normal coordinate for 1 than those of the other complex 2, 3 and 4. In addition, HR factor plays a important role to determining the internal conversion rate[24], therefore, the energy dissipation via nonradiative channel can be blocked by the substituent effect and the radiative decay became dominant in complex 2, 3 and 4. The observed fluorescence quantum yield of BOBTPs listed in Table 7. The data about Dushinsky matrix for all the complexes are listed in the attached Tables 2~5.
DownLoad:
CSV
| 1 | 2 | ||||||
| ωvib/cm–1 | Di/a.u. | HR | λ/eV | ωvib/cm–1 | Di/a.u. | HR | λ/eV |
| 46 | 2.526 | 3.1958 | 0.018 | 250 | –1.021 | 0.5216 | 0.016 |
| 232 | 1.097 | 0.6013 | 0.017 | 1066 | –0.804 | 0.3231 | 0.043 |
| 1430 | –0.699 | 0.2446 | 0.043 | 478 | 0.721 | 0.2598 | 0.015 |
| 835 | –0.682 | 0.2323 | 0.024 | 686 | 0.661 | 0.2184 | 0.019 |
| 536 | 0.548 | 0.15 | 0.010 | 260 | 0.55 | 0.1515 | 0.005 |
| 547 | –0.523 | 0.1369 | 0.009 | 539 | –0.54 | 0.1458 | 0.010 |
| 135 | 0.519 | 0.1348 | 0.002 | 290 | –0.523 | 0.1369 | 0.005 |
| 1085 | –0.492 | 0.1211 | 0.016 | 1247 | –0.492 | 0.1213 | 0.019 |
| 588 | 0.406 | 0.0823 | 0.006 | 605 | –0.471 | 0.1111 | 0.008 |
| 1052 | 0.38 | 0.0722 | 0.009 | 596 | 0.467 | 0.1091 | 0.008 |
| 25 | 0.353 | 0.06 | 0.000 | 495 | 0.467 | 0.1089 | 0.007 |
| 3 | 4 | ||||||
| ωvib/cm–1 | Di/a.u. | ωvib/cm–1 | Di/a.u. | ωvib/cm–1 | Di/a.u. | ωvib/cm–1 | Di/a.u. |
| 250 | –1.021 | 250 | –1.021 | 250 | –1.021 | 250 | –1.021 |
| 1066 | –0.804 | 1066 | –0.804 | 1066 | –0.804 | 1066 | –0.804 |
| 478 | 0.721 | 478 | 0.721 | 478 | 0.721 | 478 | 0.721 |
| 686 | 0.661 | 686 | 0.661 | 686 | 0.661 | 686 | 0.661 |
| 260 | 0.55 | 260 | 0.55 | 260 | 0.55 | 260 | 0.55 |
| 539 | –0.54 | 539 | –0.54 | 539 | –0.54 | 539 | –0.54 |
| 290 | –0.523 | 290 | –0.523 | 290 | –0.523 | 290 | –0.523 |
| 1247 | –0.492 | 1247 | –0.492 | 1247 | –0.492 | 1247 | –0.492 |
| 605 | –0.471 | 605 | –0.471 | 605 | –0.471 | 605 | –0.471 |
| 596 | 0.467 | 596 | 0.467 | 596 | 0.467 | 596 | 0.467 |
| 495 | 0.467 | 495 | 0.467 | 495 | 0.467 | 495 | 0.467 |
DownLoad:
CSV
| 1 | 2 | 3 | 4 | ||||
| λ/eV | φ | λ/eV | φ | λ/eV | φ | λ/eV | φ |
| 0.262 | 0.23 | 0.232 | 0.28 | 0.081 | 0.98 | 0.079 | 0.96 |
| φ values are from the reference 14 and measured in CH2Cl2 | |||||||
The reorganization energy is associated with HR factor S (
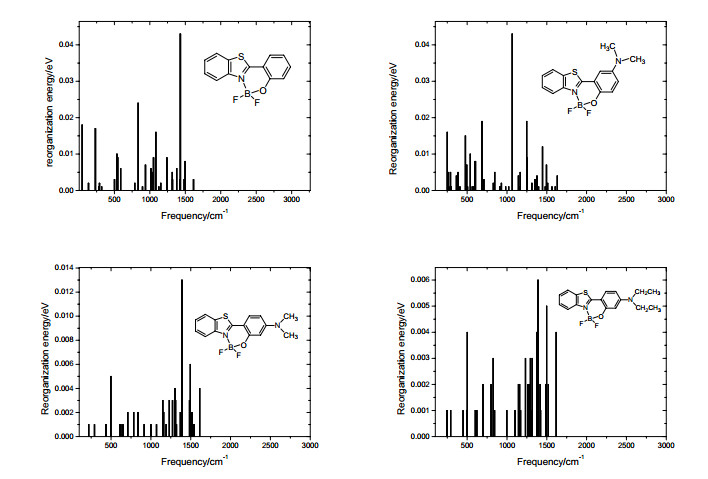
Boltzmann temperature population being taken into account, and displacement harmonic oscillator model being employed, the sum-over-states of the Franck-Condon integrals can be converted into the integrals of the correlation function about the thermal vibration, the formula (6) in supplementary information can transformed by Laguerre polynomials and Fourier transforms as follows[20]:
|
|
(1) |
here
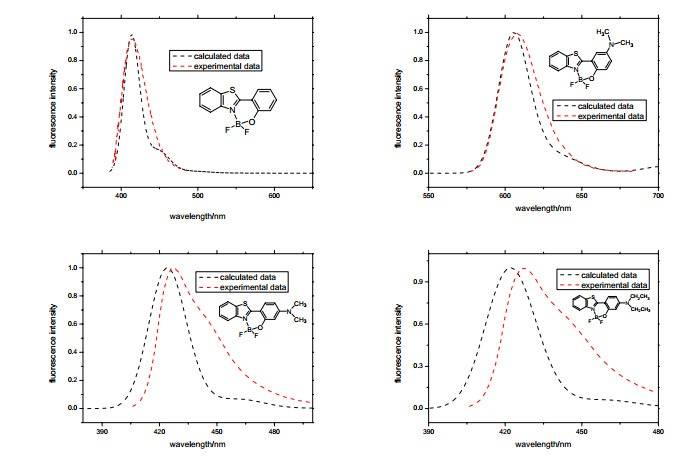
In general, electronic spectra can be simulated approximately by computing vertical excitation energies at the optimized ground electronic state and the first excited state, the line-shape spectra will turn into symmetrical profile using Gaussian or Lorentzian functions as given full width at half maximum (FWHM), this approximation method is efficient and reasonable for rigid molecules although such a treatment neglects completely the spectrum lineshape coming from dynamic effects. The intensities of transitions between two vibronic states can be evaluated as employed the Franck-Condon principle, and the computed vibrationally-resolved electronic spectra are obtained. When the eq. (1) is used to simulated the fluorescence spectra of all the BOBTPs, a Gauss broadening of 800 cm-1 is given to assure convergence of the numerical integration for the correlation function, which eliminate the drawback of the line-spectrum. The total time range for the eq. (1) is set to [-2400 fs, 2400 fs], a time increment Δt is set to 1.2 fs. The emission spectra at room temperature containing the contribution of FC factor are in Fig. 4, one can see, the lineshapes of the BOBTPs for both the experiment and calculation are not in good agreement with each other except BOBTP 2, the experimental lineshape of the complex 1, 3 and 4 have the characteristic of serious deformation, which indicate the Duschinsky and Herzberg–Teller effect on the optical spectra of these molecules can not be ignored at finite temperature, maybe the displaced harmonic approximation model is defective for these complexes, the vibrational modes of complex 1, 3 and 4 have evident impact on their fluorescence spectra, that shows the influence from some of the vibrations can not be ignored, in general, the more larger reorganization energies the more remarkabe the influence, however, due to some factors such as solvation effect, theoretical model, and weak interaction, the simulated spectra in different solvents, which are in good agreement with the observed spectra, are quite difficult, follow-up work will be discussed it.
Furthermore the radiative decay rate (kr) and nonradiative decay rate (knr) for all the BOBTP molecules were carried out theoretically without the Duschinsky mixing effect using MOMAP program[25] and listed in Table 8. As one can see from Table 8 that: (ⅰ) the calculated kr of the BOBTP 2 is 3.06×106 s-1 at room temperature, it is the smallest at all the complexes, the one of the BOBTP 3 is 4.95×108 s-1, which is the maximum and is more than two orders of magnitude compared with 2, the kr of 1 and 4 are in the middle, they are 4.35×107 and 1.52×108 s-1, respectively, all the radiative decay rates are consistent well with the calculated oscillator strengths, the above indicates the change of kr is insensitive for the substituents; (ⅱ) all the calculated knr are close to each other at room temperature, that implies the substituents have relatively little effect on the dissipation of the energy for excited electrons, in comparision with the kr, all the knr are more larger; (ⅲ) all the fluorescence quantum efficiencies have been carried out, for the bobtp 1 and 2 the η is 7.9% and 0.67% respectively, for the bobtp 3 and 4 the one is 35.1% and 37.7%, obviously all the η are smaller than the experimental results, Shuai et al. pointed out[26], due to the natural defect of the single configuration for the TDDFT, the Herzberg-Teller effect[27], the external environment factors, such as the polarity of solvent, the steric hindrance, and the different molecule packing, the nonadiabatic coupling is easily influenced, thus the predicted fluorescence quantum efficiency for all the complexes appeared little errors.
DownLoad:
CSV
| complex | kr/s-1 | knr/s-1 | η |
| 1 | 4.35×107 | 5.07×108 | 7.9% |
| 2 | 3.06×106 | 4.51×108 | 0.67% |
| 3 | 4.95×108 | 9.16×108 | 35.1% |
| 4 | 1.52×108 | 2.51×108 | 37.7% |
To summarize, we theoretically investigated the geometries of the ground state and the first excited state of the BOBTPs complexes, the electron density of the frontier orbitals, the wavelength of maximum absorption and emission of target complexes, and the influence of the substitution effect on reorganization energy, we have found that amine substituent bonding to appropriate positions in the case of BOBTP can reduce the reorganization energy significantly, it is ascribed to electron-donating effect of the amine group, the value of the reorganization energy also plays a important role on the the fluorescence quantum yield of all the BOBTPs, which imply the radiative decay of the complex 3 and 4 is dominant and the non-radiative decay and intersystem crossing rate of both the 1 and 2 can not be ignored. All in all, in order to design organic fluorescent materials with high performance, the substituents and the bonding to positions are important factors to be considered.
Doi, H.; Kinoshita, M.; Kenji Okumoto, A; Shirota, Y. A novel class of emitting amorphous molecular materials with bipolar character for electroluminescence. Chem. Mater. 2016, 15, 1080−1089.
Kamkaew, A.; Lim, S. H.; Lee, H. B.; Kiew, L. V.; Chung, L. Y.; Burgess, K. BODIPY dyes in photodynamic therapy. Chem. Soc. Rev. 2013, 42, 77−88. doi: 10.1039/C2CS35216H
Frein, S. Zhang, Y.; Song, K. H.; Tang, S.; Ravelo, L.; Cusido, J.; Sun, C.; Zhang, H. F.; Raymo, F. M. Far-red photoactivatable bodipys for the super-resolution imaging of live cells. J. Am. Chem. Soc. 2018, 140, 12741−12745. doi: 10.1021/jacs.8b09099
Tang, S.; Zhang, Y.; Dhakal, P.; Ravelo, L.; Anderson, C. L.; Collins, K. M. Raymo, F. M. Photochemical brcodes. J. Am. Chem. Soc. 2018, 140, 4485−4488. doi: 10.1021/jacs.8b00887
Wang, L.; Li, B.; Jiang, C.; Sun, R.; Hu, P.; Chen, S. A BODIPY based fluorescent probe for the rapid detection of hypochlorite. J. Fluoresc. 2018, 28, 933−941. doi: 10.1007/s10895-018-2255-y
Ulrich, G.; Barsella, A.; Boeglin, A.; Niu, S.; Ziessel, R. BODIPY-bridged push–pull chromophores for nonlinear optical applications. ChemPhysChem 2014, 15, 2693−2700. doi: 10.1002/cphc.201402123
Poirel, A.; De Nicola, A.; Ziessel, R. Oligothienyl-BODIPYs: red and near-infrared emitters. Org. Lett. 2012, 14, 5696−5699. doi: 10.1021/ol302710z
Gao, L.; Deligonul, N.; Gray, T. G. Gold(Ⅰ) complexes of brominated azadipyrromethene ligands. Inorg. Chem. 2012, 51, 7682−7688. doi: 10.1021/ic300709n
Kubota, Y.; Tsuzuki, T.; Funabiki, K.; Ebihara, M.; Matsui, M. Synthesis and fluorescence properties of a pyridomethene−BF2 complex. Org. Lett. 2010, 12, 4010−4013. doi: 10.1021/ol101612z
Li, H. J.; Fu, W. F.; Li, L.; Gan, X.; Mu, W. H.; Chen, W. Q.; Duan, X. M.; Song, H. B. Intense one-and two-photon excited fluorescent bis (BF2) core complex containing a 1, 8-naphthyridine derivative. Org. Lett. 2010, 12, 2924−2927. doi: 10.1021/ol1003725
Chibani, S.; Le Guennic, B.; Charaf-Eddin, A.; Maury, O.; Andraud, C.; Jacquemin, D. On the computation of adiabatic energies in Aza-Boron-dipyrromethene dyes. J. Chem. Theory Comp. 2012, 8, 3303−3313. doi: 10.1021/ct300618j
Wang, D.; Liu, R.; Chen, C.; Wang, S.; Chang, J.; Wu, C.; Zhu, H.; Waclawik, E. R. Photophysical and electrochemical properties of aza-boron-diquinomethene complexes. Dyes Pigments 2013, 99, 240−249. doi: 10.1016/j.dyepig.2013.05.009
Loudet, A.; Burgess, K. BODIPY dyes and their derivatives: syntheses and spectroscopic properties. Chem. Rev. 2007, 107, 4891−4932. doi: 10.1021/cr078381n
Le Guennic, B.; Chibani, S.; Charaf-Eddin, A.; Massue, J.; Ziessel, R.; Ulrich, G.; Jacquemin, D. The NBO pattern in luminescent chromophores: unravelling excited-state features using Td-DFT. Phys. Chem. Chem. Phys. 2013, 15, 7534−7540. doi: 10.1039/c3cp50669j
Santra, M.; Moon, H.; Park, M. H.; Lee, T. W.; Kim, Y. K.; Ahn, K. H. Dramatic substituent effects on the photoluminescence of boron complexes of 2-(benzothiazol-2-yl) phenols. Chem. Eur. J. 2012, 18, 9886−9893. doi: 10.1002/chem.201200726
Niu, Y. L.; Peng, Q.; Deng, C. M.; Gao, X.; Shuai, Z. Theory of excited state decays and optical spectra: application to polyatomic molecules. J. Phys. Chem. A 2010, 114, 7817−7831.
Santoro, F.; Lami, A. Improta, R.; Bloino, J.; Barone, V. Effective method for the computation of optical spectra of large molecules at finite temperature including the Duschinsky and Herzberg–Teller effect: The Q[sub x] band of porphyrin as a case study. J. Chem. Phys. 2008, 128, 224311−18. doi: 10.1063/1.2929846
Jankowiak, H. C.; Stuber, J.; Berger, R. Vibronic transitions in large molecular systems: rigorous prescreening conditions for Franck-Condon factors. J. Chem. Phys. 2007, 127, 234101−24. doi: 10.1063/1.2805398
Dierksen, M.; Grimme, S. An efficient approach for the calculation of Franck-Condon integrals of large molecules. J. Chem. Phys. 2005, 122, 244101−10. doi: 10.1063/1.1924389
Scholz, R.; Kobitski, A. Y.; Kampen, T. U.; Schreiber, M.; Zahn, D. T.; Jungnickel, G.; Elstner, M.; Sternberg, M. F. T. Resonant Raman spectroscopy of 3, 4, 9, 10-perylene-tetracarboxylic-dianhydride epitaxial films. Phys. Rev. B. 2000, 61, 13659−13669. doi: 10.1103/PhysRevB.61.13659
Reimers, J. R. A practical method for the use of curvilinear coordinates in calculations of normal-mode-projected displacements and Duschinsky rotation matrices for large molecules. J. Chem. Phys. 2001, 115, 9103−9109. doi: 10.1063/1.1412875
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A. Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N. J.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian, Inc., Wallingford CT 2010, Gaussian 09, Revision C. 01.
Grabarz, A. M.; Laurent, A. D.; Jedrzejewska, B.; Zakrzewska, A.; Jacquemin, D.; Osmialowski, B., The influence of the pi-conjugated spacer on photophysical properties of difluoroboranyls derived from amides carrying a donor group. J. Org. Chem. 2016, 81, 2280−2292. doi: 10.1021/acs.joc.5b02691
Peng, Q.; Yi, Y.; Shuai, Z.; Shao, J. Toward quantitative prediction of molecular fluorescence quantum efficiency : role of duschinsky rotation. J. Am. Chem. Soc. 2007, 129, 9333−9339. doi: 10.1021/ja067946e
Niu, Y.; Li, W.; Peng, Q.; Geng, H.; Yi, Y.; Wang, L.; Nan, G.; Wang, D.; Shuai, Z. Molecular materials property prediction package (momap) 1.0: A software package for predicting the luminescent properties and mobility of organic functional materials. Mol. Phys. 2018, 116, 1078−1090. doi: 10.1080/00268976.2017.1402966
Wu, Q.; Peng, Q.; Niu, Y.; Gao, X.; Shuai, Z. Theoretical insights into the aggregation-induced emission by hydrogen bonding: a Qm/Mm study. J. Phys. Chem. A 2012, 116, 3881−3888. doi: 10.1021/jp3002367
Li, J.; Tian, G.; Luo, Y.; Cao, Z. Theoretical studies on the vibrationally-resolved absorption and fluorescence spectra of H-pyrene+ and H-coronene+. Chem. Phys. Lett. 2015, 641, 57−61. doi: 10.1016/j.cplett.2015.10.034
Table 1. Selected Bond Parameters of Complexes BOBTP 2 and 4 in Optimized Ground- and Excited-state Geometries Obtained by DFT and TDDFT
| Parameters | BOBTP 2 | BOBTP 4 | ||||
| S0 | S0 exp. | S1 | S0 | S0 exp | S1 | |
| Bond length (Å) | ||||||
| B–N | 1.606 | 1.588 | 1.594 | 1.604 | 1.578 | 1.616 |
| B–O | 1.462 | 1.437 | 1.472 | 1.462 | 1.448 | 1.468 |
| O–C | 1.327 | 1.348 | 1.327 | 1.327 | 1.339 | 1.326 |
| C–N | 1.383 | 1.381 | 1.386 | 1.375 | 1.365 | 1.375 |
| C–H(CH3) | 1.099 | 1.097 | 1.089 | 1.099 | 1.097 | 1.088 |
| B–F(1) | 1.389 | 1.379 | 1.378 | 1.380 | 1.383 | 1.378 |
| B–F(2) | 1.395 | 1.389 | 1.384 | 1.384 | 1.387 | 1.380 |
| F…H | 2.441 | 2.498 | 2.435 | 2.504 | 2.629 | 2.591 |
| Bond angle and dihedral angle (°) | ||||||
| F(1) –B–F(2) | 111.75 | 110.44 | 111.88 | 111.53 | 109.87 | 112.90 |
| N–B–O | 108.24 | 109.39 | 108.61 | 108.51 | 110.18 | 106.97 |
| C–O–B | 125.61 | 123.03 | 125.44 | 126.30 | 125.72 | 123.74 |
| C–C–O–B | 14.19 | 19.51 | 18.51 | 2.92 | 2.84 | 5.73 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Selected Bond Parameters of Complexes BOBTP 1 and 3 in Optimized Ground- and Excited-state Geometries Obtained by DFT and TDDFT
| Parameters | BOBTP 1 | BOBTP 3 | ||
| S0 | S1 | S0 | S1 | |
| Bond length (Å) | ||||
| B–O | 1.462 | 1.468 | B–O | 1.462 |
| O–C | 1.327 | 1.326 | O–C | 1.327 |
| C–N | 1.375 | 1.374 | C–N | 1.375 |
| B–F(1) | 1.384 | 1.386 | B–F(1) | 1.384 |
| B–F(2) | 1.389 | 1.394 | B–F(2) | 1.389 |
| F…H | 2.504 | 2.591 | F…H | 2.504 |
| Bond angle and dihedral angle (°) | ||||
| F(1)–B–F(2) | 111.53 | 112.90 | 111.75 | 111.88 |
| N–B–O | 108.51 | 106.97 | 108.24 | 108.61 |
| C–O–B | 126.30 | 123.74 | 125.61 | 125.44 |
| C–C–O–B | 2.92 | 5.73 | 14.19 | 18.51 |
下载: 导出CSV
Table 3. Absorption of BOBTPs in CH2Cl2 Solution According to TDDFT Calculations, together with the Experimental Data
| Complex | Oscillator strength | Main configuration | Assignment | λexp (nm) | λcal (nm) |
| 1 | 0.3897 | HOMO→LUMO | 0.6970 | 364 | 363.2 |
| 2 | 0.1252 | HOMO→LUMO | 0.7012 | 456 | 466.3 |
| 3 | 0.8645 | HOMO→LUMO | 0.7014 | 413 | 429.3 |
| 4 | 0.8638 | HOMO→LUMO | 0.7030 | 417 | 425.2 |
下载: 导出CSV
Table 4. Emissions of BOBTPs in CH2Cl2 Solution According to TDDFT Calculations, together with the Experimental Data
| Complex | Oscillator strength | Main configuration | Assignment | λexp(nm) | λcal(nm) | μ(Debye) |
| 1 | 0.4948 | HOMO→LUMO | 0.7038 | 433 | 417.1 | 2.385 |
| 2 | 0.1661 | HOMO→LUMO | 0.7075 | 597 | 606.5 | 2.046 |
| 3 | 1.294 | HOMO→LUMO | 0.6120 | 430 | 423.9 | 11.190 |
| 4 | 1.3120 | HOMO→LUMO | 0.7033 | 434 | 422.1 | 11.900 |
| μ: transition electric dipole moments between the ground to excited state 1Debye=8.478×10-30 C • m. | ||||||
下载: 导出CSV
Table 5. Molecular Orbital Composition in the Ground State for BOBTPs
| Fragments | 1 | 2 | 3 | 4 |
| HOMO | LUMO | HOMO | LUMO | |
| benzothiazole-benzen | 0.93 | 0.63 | 0.53 | 0.96 |
| BF2O | 0.03 | 0.32 | 0.03 | 0.02 |
| NR2 | 0.36 | 0 |
下载: 导出CSV
Table 6. Part HR Factors (HR > 0.1), Reorganization Energies (λ), Shifts of Mode (Di), and Vibrational Frequencies (ωvib) of the First Excited State for all the BOBTPs
| 1 | 2 | ||||||
| ωvib/cm–1 | Di/a.u. | HR | λ/eV | ωvib/cm–1 | Di/a.u. | HR | λ/eV |
| 46 | 2.526 | 3.1958 | 0.018 | 250 | –1.021 | 0.5216 | 0.016 |
| 232 | 1.097 | 0.6013 | 0.017 | 1066 | –0.804 | 0.3231 | 0.043 |
| 1430 | –0.699 | 0.2446 | 0.043 | 478 | 0.721 | 0.2598 | 0.015 |
| 835 | –0.682 | 0.2323 | 0.024 | 686 | 0.661 | 0.2184 | 0.019 |
| 536 | 0.548 | 0.15 | 0.010 | 260 | 0.55 | 0.1515 | 0.005 |
| 547 | –0.523 | 0.1369 | 0.009 | 539 | –0.54 | 0.1458 | 0.010 |
| 135 | 0.519 | 0.1348 | 0.002 | 290 | –0.523 | 0.1369 | 0.005 |
| 1085 | –0.492 | 0.1211 | 0.016 | 1247 | –0.492 | 0.1213 | 0.019 |
| 588 | 0.406 | 0.0823 | 0.006 | 605 | –0.471 | 0.1111 | 0.008 |
| 1052 | 0.38 | 0.0722 | 0.009 | 596 | 0.467 | 0.1091 | 0.008 |
| 25 | 0.353 | 0.06 | 0.000 | 495 | 0.467 | 0.1089 | 0.007 |
| 3 | 4 | ||||||
| ωvib/cm–1 | Di/a.u. | ωvib/cm–1 | Di/a.u. | ωvib/cm–1 | Di/a.u. | ωvib/cm–1 | Di/a.u. |
| 250 | –1.021 | 250 | –1.021 | 250 | –1.021 | 250 | –1.021 |
| 1066 | –0.804 | 1066 | –0.804 | 1066 | –0.804 | 1066 | –0.804 |
| 478 | 0.721 | 478 | 0.721 | 478 | 0.721 | 478 | 0.721 |
| 686 | 0.661 | 686 | 0.661 | 686 | 0.661 | 686 | 0.661 |
| 260 | 0.55 | 260 | 0.55 | 260 | 0.55 | 260 | 0.55 |
| 539 | –0.54 | 539 | –0.54 | 539 | –0.54 | 539 | –0.54 |
| 290 | –0.523 | 290 | –0.523 | 290 | –0.523 | 290 | –0.523 |
| 1247 | –0.492 | 1247 | –0.492 | 1247 | –0.492 | 1247 | –0.492 |
| 605 | –0.471 | 605 | –0.471 | 605 | –0.471 | 605 | –0.471 |
| 596 | 0.467 | 596 | 0.467 | 596 | 0.467 | 596 | 0.467 |
| 495 | 0.467 | 495 | 0.467 | 495 | 0.467 | 495 | 0.467 |
下载: 导出CSV
Table 7. Molecular Reorganization Energy (λ) and the Fluorescence Quantum Yield (φ) for BOBTPs
| 1 | 2 | 3 | 4 | ||||
| λ/eV | φ | λ/eV | φ | λ/eV | φ | λ/eV | φ |
| 0.262 | 0.23 | 0.232 | 0.28 | 0.081 | 0.98 | 0.079 | 0.96 |
| φ values are from the reference 14 and measured in CH2Cl2 | |||||||
下载: 导出CSV
Table 8. Calculated Radiative Decay Rate (kr) and Nonradiative Decay Rate (knr) from S1 to S0 and the Corresponding Fluorescence Quantum Yield (η) at Different Temperatures for BOBTP Molecules
| complex | kr/s-1 | knr/s-1 | η |
| 1 | 4.35×107 | 5.07×108 | 7.9% |
| 2 | 3.06×106 | 4.51×108 | 0.67% |
| 3 | 4.95×108 | 9.16×108 | 35.1% |
| 4 | 1.52×108 | 2.51×108 | 37.7% |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



