School of Chemistry and Life Science, Changchun University of Technology, Changchun 130012, China
b.
State Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China
Corresponding author:
ZHAO Zhenbo, professor; Tel:0431-85716671; E-mail:1710229261@qq.com; Research interests:catalytic materials; ZHAO Fengyu, professor; Tel:0431-85262410; E-mail:zhaofy@ciac.ac.cn; Research interests:heterogeneous catalysis
Received Date:
15 April 2020 Accepted Date:
09 June 2020 Revised Date:
30 April 2020 Available Online:
10 November 2020
Abstract:
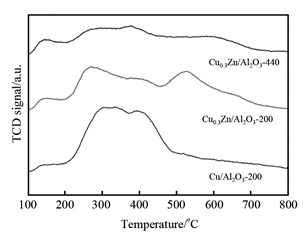
Biomass conversion is one of the most effective ways for alleviating the energy and environmental crisis. A series of CuZn/Al2O3 catalysts was prepared by co-precipitation method and the structure and properties of the catalysts obtained were characterized by several techniques such as inductively coupled plasma (ICP), X-ray diffraction (XRD), transmission electron microscopy (TEM), and CO2-temperature programmed desorption (TPD). We focused our attention on the effects of reduction temperature on the catalytic performances of CuZn/Al2O3 catalysts in γ-valerolactone hydrogenation. It is found that reduction of temperature significantly influences the activity and selectivity of CuZn/Al2O3 catalysts, that is, the larger reduction temperature benefits for the formation of 1, 4-pentanediol, and a high selectivity of 98% is achieved over the catalyst at 440℃, while it is only 71% on the catalyst reduced at 200℃. Based on the structure analysis of the catalyst, it is concluded that the high reduction temperature can promote the reduction of ZnO and generate new surface active sites and vary the surface basic-acid properties, thus result in the improvement of 1, 4-pentanediol selectivity.
Mosier N, Wywan C, Dale B. Features of Promising Technologies for Pretreatment of Lignocellulosic Biomass[J]. Bioresour Technol,
2005, 96(6):
673-686.
doi: 10.1016/j.biortech.2004.06.025
[2]
林鹿, 何北海, 孙润仓. 木质生物质转化高附加值化学品[J]. 化学进展,
2007,19,(Z2): 1206-1216.
LIN Lu, HE Beihai, SUN Runcang. High Value Chemicals from Lignocellulosic Biomass[J]. Prog Chem,
2007, 19(Z2):
1206-1216.
[3]
余强, 庄新姝, 袁振宏. 木质纤维素类生物质制取燃料及化学品的研究进展[J]. 化工进展,
2012,31,(4): 784-791.
YU Qiang, ZHUANG Xinshu, YUAN Zhenhong. Research Progress on Fuel and Chemicals Production from Lignocellulose Biomass[J]. Chem Ind Eng Proc,
2012, 31(4):
784-791.
[4]
Girisuta B, Janssen L P B M, Heeres H J. Green Chemicals:A Kinetic Study on the Conversion of Glucose to Levulinic Acid[J]. Chem Eng Res Des,
2006, 84(5):
339-349.
doi: 10.1205/cherd05038
[5]
Rackemann D W, Doherty W O. The Conversion of Lignocellulosics to Levulinic Acid[J]. Biofuel Bioprod Biorefin,
2011, 5(2):
198-214.
doi: 10.1002/bbb.267
[6]
Banerjee B, Singuru R, Kundu S K. Towards Rational Design of Core-Shell Catalytic Nanoreactor with High Performance Catalytic Hydrogenation of Levulinic Acid[J]. Catal Sci Technol,
2016, 6(13):
5102-5115.
doi: 10.1039/C6CY00169F
[7]
Feng H, Li X, Qian H. Efficient and Sustainable Hydrogenation of Levulinic-acid to gamma-Valerolactone in Aqueous Solution over Acid-resistant CePO4/Co2P Catalysts[J]. Green Chem,
2019, 21(7):
1743-1756.
doi: 10.1039/C9GC00482C
[8]
Li W, Xie J, Lin H. Highly Efficient Hydrogenation of Biomass-Derived Levulinic Acid to γ-Valerolactone Catalyzed by Iridium Pincer Complexes[J]. Green Chem,
2012, 14(9):
2388-2390.
doi: 10.1039/c2gc35650c
[9]
Mamun O, Saleheen M, Bond J Q. Investigation of Solvent Effects in the Hydrodeoxygenation of Levulinic Acid to γ-Valerolactone over Ru Catalysts[J]. J Catal,
2019, 379:
164-179.
doi: 10.1016/j.jcat.2019.09.026
[10]
Pinto B P, Fortuna A L L, Cardoso C P. Hydrogenation of Levulinic Acid (LA) to γ-Valerolactone (GVL) over Ni-Mo/C Catalysts and Water-Soluble Solvent Systems[J]. Catal Lett,
2017, 147(3):
751-757.
doi: 10.1007/s10562-017-1977-9
[11]
Upare P P, Lee J M, Hwang D W. Selective Hydrogenation of Levulinic Acid to γ-Valerolactone over Carbon-Supported Noble Metal Catalysts[J]. J Ind Eng Chem,
2011, 17(2):
287-292.
doi: 10.1016/j.jiec.2011.02.025
[12]
Ahn Y C, Han J. Catalytic Production of 1, 4-Pentanediol from Corn Stover[J]. Bioresour Technol,
2017, 245(Pt A):
442-448.
[13]
Alonso D M, Wettstein S G, Dumesic J A. Gamma-Valerolactone, A Austainable Platform Molecule Derived from Lignocellulosic Biomass[J]. Green Chem,
2013, 15(3):
584-595.
doi: 10.1039/c3gc37065h
[14]
Corbel-demailly L, Ly B K, Minh D P. Heterogeneous Catalytic Hydrogenation of Biobased Levulinic and Succinic Acids in Aqueous Solutions[J]. ChemSusChem,
2013, 6(12):
2388-2395.
doi: 10.1002/cssc.201300608
[15]
Mehdi H, Fabos V, Tuba R. Integration of Homogeneous and Heterogeneous Catalytic Processes for a Multi-step Conversion of Biomass:From Sucrose to Levulinic Acid, γ-Valerolactone, 1, 4-Pentanediol, 2-Methyl-tetrahydrofuran, and Alkanes[J]. Top Catal,
2008, 48(1/2/3/4):
49-54.
[16]
Pagliaro M, Ciriminna R, Kimura H. From Glycerol to Value-Added Products[J]. Angew Chem Int Ed,
2007, 46(24):
4434-4340.
doi: 10.1002/anie.200604694
[17]
Climent M J, Corma A, Iborra S. Conversion of Biomass Platform Molecules into Fuel Additives and Liquid Hydrocarbon Fuels[J]. Green Chem,
2014, 16(2):
516-547.
doi: 10.1039/c3gc41492b
[18]
Lange J P, Price R, Ayoub P M. Valeric Biofuels:A Platform of Cellulosic Transportation Fuels[J]. Angew Chem Int Ed,
2010, 49(26):
4479-4483.
doi: 10.1002/anie.201000655
[19]
Obregon I, Gandarias I, Al-shaal M G. The Role of the Hydrogen Source on the Selective Production of γ-Valerolactone and 2-Methyltetrahydrofuran from Levulinic Acid[J]. ChemSusChem,
2016, 9(17):
2488-2495.
doi: 10.1002/cssc.201600751
[20]
Pace V, Hoyos D P, Castoldi L. 2-Methyltetrahydrofuran (2-MeTHF):A Biomass-Derived Solvent with Broad Application in Organic Chemistry[J]. ChemSusChem,
2012, 5(8):
1369-1379.
doi: 10.1002/cssc.201100780
[21]
Sun D, Saito T, Yamada Y. Hydrogenation of γ-Valerolactone to 1, 4-Pentanediol in a Continuous Flow Reactor[J]. Appl Catal A,
2017, 542:
289-295.
doi: 10.1016/j.apcata.2017.05.034
[22]
Zhai X J, Li C, Di X. Preparation of Cu/MgO Catalysts for γ-Valerolactone Hydrogenation to 1, 4-Pentanediol by MOCVD[J]. J Fuel Chem Technol,
2017, 45(5):
537-546.
doi: 10.1016/S1872-5813(17)30028-2
[23]
Du X, Bi Q, Liu Y. Tunable Copper-Catalyzed Chemoselective Hydrogenolysis of Biomass-Derived γ-Valerolactone into 1, 4-Pentanediol or 2-Methyltetrahydrofuran[J]. Green Chem,
2012, 14(4):
935-939.
doi: 10.1039/c2gc16599f
[24]
Obregon I, Gandarias I, Ocio A. Structure-Activity Relationships of Ni-Cu/Al2O3 Catalysts for γ-Valerolactone Conversion to 2-Methyltetrahydrofuran[J]. Appl Catal B,
2017, 210:
328-341.
doi: 10.1016/j.apcatb.2017.04.006
[25]
Li M, Li G, Li N. Aqueous Phase Hydrogenation of Levulinic Acid to 1, 4-Pentanediol[J]. Chem Commun,
2014, 50(12):
1414-1416.
doi: 10.1039/c3cc48236g
[26]
Wu J, Gao G, Sun P. Synergetic Catalysis of Bimetallic CuCo Nanocomposites for Selective Hydrogenation of Bioderived Esters[J]. ACS Catal,
2017, 7(11):
7890-7901.
doi: 10.1021/acscatal.7b02837
[27]
Xie Z, Chen B, Wu H. Highly Efficient Hydrogenation of Levulinic Acid into 2-Methyltetrahydrofuran over Ni-Cu/Al2O3-ZrO2 Bifunctional Catalysts[J]. Green Chem,
2019, 21(3):
606-613.
doi: 10.1039/C8GC02914H
[28]
Xu Q, Li X, Pan T. Supported Copper Catalysts for Highly Efficient Hydrogenation of Biomass-Derived Levulinic Acid and γ-Valerolactone[J]. Green Chem,
2016, 18(5):
1287-1294.
doi: 10.1039/C5GC01454A
[29]
Kanai Y, Watanabe T, Fujitani T. Evidence for the Mmigration of ZnOx in a Cu/ZnO Methanol Synthesis Catalyst[J]. Catal Lett,
1994, 27(1/2/3/4):
67-78.
[30]
Kuld S, Conradsen C, Moses P G. Quantification of Zinc Atoms in a Surface Alloy on Copper in an Industrial-Type Methanol Synthesis Catalyst[J]. Angew Chem Int Ed,
2014, 53(23):
5941-5945.
doi: 10.1002/anie.201311073
[31]
Kuld S, Thorhauge M, Falsig H. Quantifying the Promotion of Cu Catalysts by ZnO for Mmethanol Synthesis[J]. Science,
2016, 352(6288):
969-974.
doi: 10.1126/science.aaf0718
[32]
Tisseraud C, Comminges C, Belin T. The Cu-ZnO Synergy in Methanol Synthesis from CO2, Part 2:Origin of the Methanol and CO Selectivities Explained by Experimental Studies and a Sphere Contact Quantification Model in Randomly Packed Binary Mixtures on Cu-ZnO Coprecipitate Catalysts[J]. J Catal,
2015, 330:
533-544.
doi: 10.1016/j.jcat.2015.04.035
[33]
Valant A L, Comminges C, Tisseraud C. The Cu-ZnO Synergy in Methanol Synthesis from CO2, Part 1:Origin of Active Site Explained by Experimental Studies and a Sphere Contact Quantification Model on Cu+ZnO Mechanical Mixtures[J]. J Catal,
2015, 324:
41-49.
doi: 10.1016/j.jcat.2015.01.021
图 1
GVL加氢制1, 4-PDO和2-MTHF的反应路径。
Figure 1
The reaction path of GVL hydrogenation to 1, 4-PDO and 2-MTHF

 下载:
下载:





 下载:
下载:






