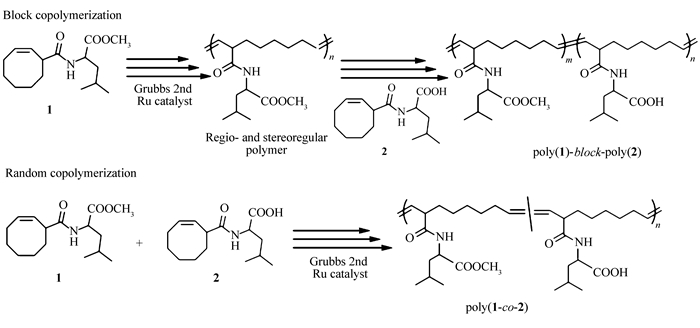
Scheme 1.
Synthesis of amino acid-derived cyclooctene monomers

Designing and synthesizing of polymers with good biocompatibility and biomimetics to mimic biomacromolecules with multifarious biological functional activities is an important research field[1-14]. Within the past decade, biomimetic or bioinspired materials have been increasingly used in a broad range of biomedical applications[15]. Amino acids and peptides containing polymers are definitely important biomimetic materials due to their outstanding properties such as biocompatibility and bioactivity[16-21]. Several studies have focused on designing polymers to mimic biological systems for cellular adhesion and bacterial chemotaxis, through introducing amino acids or peptides as pendent functional groups to the abiotic backbone[16]. The typical and efficient way for synthesizing these polymers is through controlled/living radical polymerization (CRP). Extensive efforts have been paid for controlling the molecular mass, polydispersity and architecture of the resulting polymers[18, 20, 22-23]. However, due to the intrinsic nature of radical polymerization, precise control of the regio- and stereostructure of final polymers, which are possibly comparable to that of biopolymers, remains one of the formidable unsolved goals.
Alternatively, coordination polymerization of olefin and heterocyclic monomers mediated by organometallic complexes can provide regio- and stereoregular polymers due to the steric hindrance effect between the monomers and bulk ligand of complexes as reported in many literatures[24-26]. Among the abundant organometallic complexes system, ruthenium catalysts have attracted considerable attentions in the last few years because of the unprecedented functional group tolerance[24]. A large amount of amino acid-functionalized norbornenes and oxa-norbornenes monomers have been applied to synthesize biomimic polymers via ruthenium catalysts mediated ring-opening metathesis polymerization (ROMP)[27-29]. However, the primary structure of resulting polymer from these monomers cannot be completely controlled due to restricted steric hindrance effect between the monomers and bulk ligand of complex during polymerization[24]. Therefore, it is difficult to relate the biological properties of these polymers to specific structural characteristics.
Recently, ROMP of 3-substituted cyclooctene has been demonstrated to afford polymers with highly head-to-tail regioregularity and trans-stereoregularity[30-36]. Inspired by these elegant works, we designed and synthesized a range of 3-glycine-subtituted and 5-glycine-substituted cyclooctenes. The final amino acid-bearing polymers from ROMP afford different regio- and stereostructure depended on the substituent position in original cyclooctene[37]. Systematically studies of the effect of microstructures on materials performances demonstrated that the polymer properties are strongly influenced by the regularity of the side-chains located along the polymer backbone. Notably, above research works are all about the homopolymerization of 3-substituted cyclooctene monomers, no work has been exploited for the copolymerization of 3-substituted cyclooctenes via ROMP, especially for amino acids functionalized cyclooctenes. Generally, copolymers will exhibit specific properties that cannot be afforded from the single component[38]. Hence, we envisioned that amino acids substituted on the 3-position of the cyclooctene ring might undergo ruthenium-catalyzed ring-opening metathesis homopolymerization and copolymerization to achieve high regio- and stereochemical control. Nonetheless, it was not obvious that the monomers with more functional groups would give analogous results, because active groups of amino acids might coordinate metathesis catalysts[39].
In this paper, we first designed two new 3-substitued cyclooctenes monomers, namely (cyclooct-2-ene-1-carbonyl)-L-leucine methyl ester (1) and (cyclooct-2-ene-1-carbonyl)-L-leucine (2) (Scheme 1). Polymerizaiton of monomers 1 or 2 with Grubbs second generation catalyst was conducted in a regio- and stereoselective manner to afford leucine-derived homopolymers with relatively low polydispersity, high head-to-tail regioregularity and high trans-stereoregularity, which provided precise regulation of the primary structures of the resulting polymers (Scheme 2). Then we employed direct copolymerization to obtain original amino acid based cyclooctenes block copolymers with leucine and leucine methyl ester groups as hydrophilic and hydrophobic elements, respectively. We also investigated the difference of the amphihilicities between the random and block copolymers and the self-assembly of the block copolymers.
Tetrahydrofuran (THF) and diethyl ether (Et2O) were purchased from Beijing Chemical Works and refluxed with sodium and distilled under nitrogen before use. Ethyl acetate (EtOAc), dichloromethane (DCM) and petroleum ether were purchased from Beijing Chemical Works and freshly distilled from CaH2 before use. All the other reagents and solvents were purchased from Energy Chemical and used as received without further purification unless otherwise stated.
1H nuclear magnetic resonance (NMR) spectra and 13C NMR spectra were recorded on a Bruker AV-300 spectrometer and a Bruker AV-400 spectrometer, respectively. Number-average relative molecular mass (Mn) and polydispersity indexes (PDI) were determined by gel permeation chromatography (GPC) on a Waters 1515 high performance liquid chromatography (HPLC) pump equipped with Waters Styragel HT3, HT4 columns and a Waters 2414 Refractive index Detector (eluent: dimethylfumarate (DMF) containing 0.02 mol/L LiBr; flow rate:1 mL/min; temperature:50 ℃; standard: PMMA in the relative molecular mass range from 1860 to 3.3×105). Fourier transform infrared(FT-IR) spectra were performed on a Bruker TENSOR-27 spectrophotometer. Ultraviolet-visible (UV-Vis) spectra was measured on a U-4100 spectrometer. Dynamic laser scattering (DLS) measurement was performed on a WyattQELS instrument with a vertically polarized He-Ne laser (DAWN EOS, Wyatt Technology, USA). The scattering angle was fixed at 90°. Thermogravimetric analyses (TGA) were obtained at a heating rate of 10 ℃/min in N2 with a Perkin-Elemer TGA-2 thermogravimetric analyzer. The tapping mode atomic force microscopy (AFM) images were obtained with a MultiMode Scanning Probe Microscope coupled to a Nanoscope Ⅲa controller (Digital Instruments/Veeco, Santa Barbara, CA, USA) under ambient conditions by spinning coating dilute acetone solution of poly(1)50-block-poly(2)50 on a mica surface.
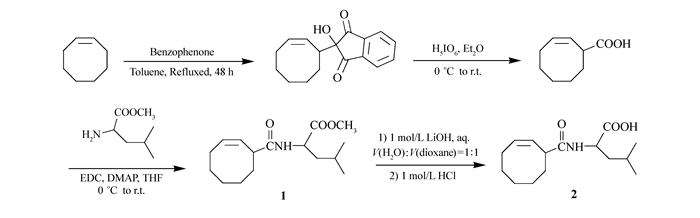
Synthesis of 2-(cyclooct-2-en-1-yl)-2-hydroxy-1H-indene-1, 3(2H)-dione The ene adduct was synthesized according to the literature[40]. The residue was recrystallized from the CCl4. Yield:86%. 1H NMR(400 MHz, CDCl3), δ:7.98~8.02(2H, m), 7.88~7.93(2H, m), 5.83~5.92(1H, m), 5.59~5.65(1H, t), 3.14~3.22(1H, t), 2.87(1H, bs), 2.00~2.24(2H, m), 1.11~1.66(8H, m).
Synthesis of (±)-cyclooct-2-ene-1-carboxylic acid The cyclooct-2-ene-1-carboxylic acid was synthesized according to the literature[41]. The residue was purified by column chromatography using V(petroleum ether):V(EtOAc):V(HAc)=75:25:1 as eluent to afford the product as a colourless oil (7.1 g, 85%). 1H NMR(400 MHz, CDCl3), δ:11.50(1H, bs), 5.64~5.84(2H, m), 3.45~3.53(1H, m), 1.23~2.21(10H, m).
Synthesis of (cyclooct-2-ene-1-carbonyl)-L-leucine methyl ester To a solution of cyclooct-2-ene-1-carboxylic acid (4.10 g, 26.6 mmol) in dry DCM (100 mL), was added L-leucine methyl ester (4.60 g, 31.7 mmol) and 4-dimethylaminopyridine (0.38 g, 3.1 mmol). The solution was cooled in an ice bath to 0 ℃ and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (6.07 g, 31.7 mmol) was added during 5 min. After that the ice bath was removed and the reaction mixture was stirred overnight at ambient temperature. The 1-(3-(dimethylamino)propyl)-3-ethylurea (EDU) was removed by filtration through a fritted Büchner funnel (G3). The filtrate was washed with two 50 mL portions of 0.5 mol/L hydrochloric acid and two 50 mL portions of saturated sodium bicarbonate solution. The organic solution was dried over anhydrous sodium sulfate and concentrated with a rotary evaporator. The residue was purified by column chromatography using V(petroleum ether):V(EtOAc)=4:1 as eluent to afford the product as an white solid (5.23 g, 70%). 1H NMR(400 MHz, CDCl3), δ:5.79~5.93(2H, m), 5.57~5.65(1H, m), 4.58~4.61(1H, m), 3.71(3H, s), 3.28~3.35(1H, m), 2.13~2.15(2H, m), 1.25~1.94(11H, m), 0.95~0.97(6H, m). 13C NMR(100 MHz, CDCl3), δ:21.92, 21.94, 22.80, 24.83, 24.85, 25.18, 25.20, 26.45, 26.48, 26.51, 26.55, 29.18, 29.19, 32.74, 33.07, 41.16, 41.17, 43.85, 43.95, 50.48, 50.51, 52.22, 127.99, 128.22, 131.88, 131.94, 173.75, 173.82, 174.81. ESI[M-H+]:calcd for C16H26NO3, 280.20; found, 280.20.
Synthesis of (cyclooct-2-ene-1-carbonyl)-L-leucine To a solution of (cyclooct-2-ene-1-carbonyl)-L-leucine methyl ester 0.84 g(3 mmol) in V(H2O):V(dioxane)=1:1(10 mL) was added 1 mol/L lithium hydroxide(8 mL, 8 mmol) and the mixture stirred at ambient temperature for 3 h. The reaction mixture was acidified to pH=2 with 1 mol/L hydrochloric acid and extracted three times with DCM. The organic solution was dried over anhydrous sodium sulfate and concentrated with a rotary evaporator to obtain a white solid(0.72 g, 90%). 1H NMR(400 MHz, CDCl3), δ:11.66(1H, bs), 5.92~6.00(1H, m), 5.78~5.87(1H, m), 5.57~5.65(1H, m), 4.58~4.62(1H, m), 3.28~3.35(1H, m), 2.12~2.16(2H, m), 1.14~1.88(11H, m), 0.95~0.97(6H, m). 13C NMR(100 MHz, CDCl3), δ:21.90, 21.92, 22.84, 24.86, 25.16, 25.18, 26.45, 26.50, 29.16, 32.79, 33.14, 41.29, 41.37, 43.86, 43.98, 50.81, 127.64, 127.86, 132.05, 132.10, 175.85, 176.58. ESI[M-H+]:calcd for C15H24NO3, 266.18; found, 266.20.
Homopolymerization Polymerizations were carried out in a glass tube equipped with a three-way stopcock under nitrogen. Monomer 1(98.5 mg, 0.35 mmol) and Grubbs second generation Ru catalyst(3.0 mg, 3.5×10-3 mmol) were dissolved in THF(0.5 mL) separately. The catalyst solution was added to the monomer solution and the resulting mixture was vigorously stirred. It was kept in a water bath at 30 ℃ for 4 h, during which the color of the polymerization mixture gradually changed from pink to pale brown. Then, ethyl vinyl ether was added to the mixture to quench the reaction. The mixture was poured into a large amount of diethyl ether to precipitate a polymer (precipitation-dissolution 3 times) and finally dried overnight under vacuum at 50 ℃ until constant mass.
Random copolymerization It was carried out using monomer mixtures at set ratios in a manner similar to the homopolymerization. THF was used as a solvent.
Block copolymerization Monomer 1 was polymerized for 2~4 h in THF, which was confirmed by the 1H NMR measurement of a sample portion drawn from the polymerization mixture. Monomer 2 was added to the mixture, and then the resulting mixture was stirred for another 24 h. The polymer was disposed in a manner similar to the homopolymerization.
Density functional theory (DFT) calculations were performed using the Amsterdam Density Functional program[42-44]. All the structures calculations were based on the Becke-Perdew exchange-correlation functional[45-47]. A triple slater-type orbital (STO) basis set was employed for Ru, while all other atoms were described by a double-ζ plus polarization STO basis. The 1s22s22p63s23p63d104s24p6 configuration on vanadium, the 1s22s22p6 configuration on chlorine and the 1s2 configuration of carbon, nitrogen, and oxygen were assigned to the core and treated by the frozen-core approximation.
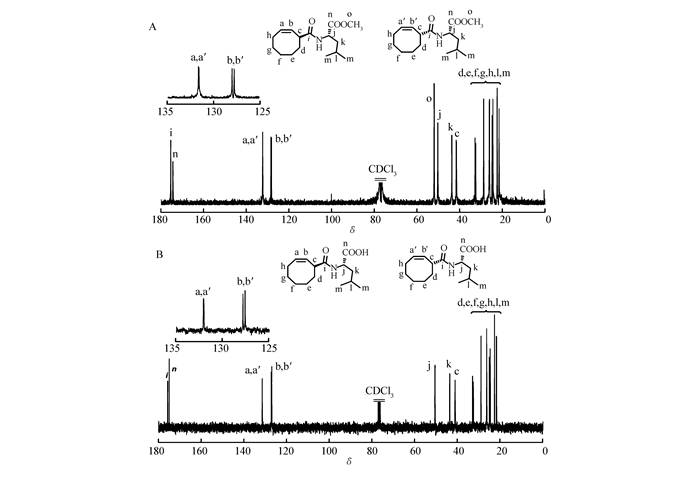
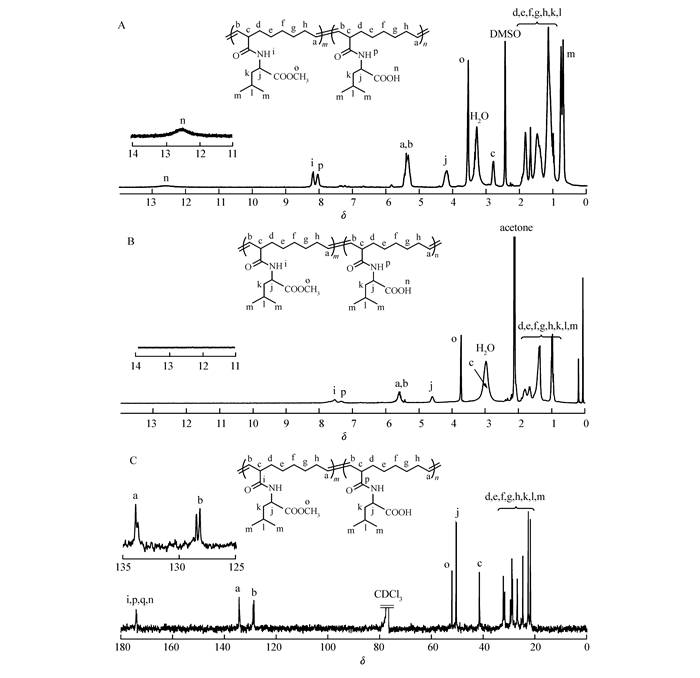
We initially envisioned a general approach to 3-carboxylic acid substituted cyclooctene, bromide of cyclooctene with N-bromosuccinimide produced 3-bromocyclooctene, which subsequently reacted with Grignard reagent to give target 3-carboxylic acid substituted cyclooctene. Unfortunately, coupling reaction between 3-bromocyclooctene easily occurred in the presence of Grignard reagent[48]. Alternatively, as shown in Scheme 1, synthesis of 3-carboxylic acid substituted cyclooctenes was accomplished through another reaction route according to the literature[40-41]. Monomer 1 was then synthesized by the reaction of 3-carboxyl substituted cyclooctenes with 2 times stoichiometric amount of L-leucine methyl ester hydrochloride in 75% yield (Scheme 1). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) was used as a condensation agent due to the easy purified process. Monomer 2 was synthesized by lithium hydroxide hydrolysis of monomer 1 in 80% yields. The structures of the monomers were confirmed by 1H NMR, 13C NMR and electrospray ionization mass spectrometry (ESI-MS). 1H NMR spectra (see Fig.S3 in Supporting Information) analysis of two monomers showed the characteristic peak at 3.71 and 10.5, ascribed to methyl ester and carboxyl group, respectively. As shown in Fig. 1, the 13C NMR spectrum of monomers also revealed the expected resonance and further verified successful synthesis of target monomers.
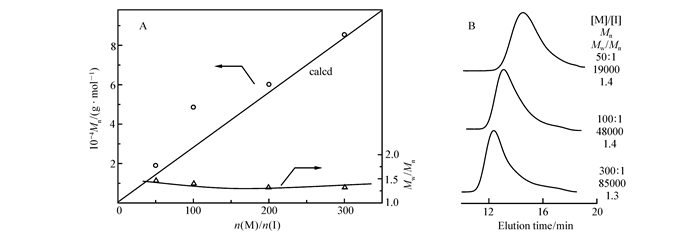
After obtaining monomers 1 and 2 with high purity, we initially attempted to polymerize in THF at 30 ℃ using Grubbs second generation catalyst, which is widely used for ROMP of various functional groups including the carboxy group. As shown in Fig. 2, the Mn values of the obtained poly(1) at monomer-to-initiator(M/I) molar ratios of 50 and 300 were 1.9×104 and 8.5×104, respectively, both of which were consistent with the calculated Mn values (1.4×104 and 8.4×104, respectively), suggesting that Grubbs second generation catalyst showed significant control for the ROMP of monomer 1 and gave the polymer with the expected relative molecular mass. Size exclusion chromatography (SEC) traces showed a single peak with small PDI (around 1.3~1.4) at different M/I ratios. Monomer 2 also underwent homopolymerization to afford polymers with Mn of 3.8×104 and Mw/Mn of 1.5(Table 1), respectively. These results suggested that the ROMP of leucine-substituted cyclooctene using Grubbs′ catalyst has proceeded in a living way to some extent. Table 1 also listed the results of the random copolymerization of monomers 1 and 2. The random copolymers with Mn′s of 1.42×104~3.31×104(Mw/Mn=1.4~1.6) were produced in high yields (73%~82%). The constituent of the copolymer could be calculated from 1H NMR(see Fig.S4 in Supporting Information). As shown in Table 1, the 1/2 unit ratio in the copolymer was similar to the initial feed ratio.
 下载:
导出CSV
下载:
导出CSV
| Feed ratio n(1):n(2) |
Yieldb/% | Mnc | Mw/Mnc | Unit ratiod n(1):n(2) |
| 100:0 | 66 | 48 600 | 1.40 | 100:0 |
| 75:25 | 82 | 21 700 | 1.39 | 72:28 |
| 50:50 | 73 | 14 200 | 1.45 | 58:42 |
| 25:75 | 77 | 33 100 | 1.60 | 24:76 |
| 0:100 | 70 | 37 500 | 1.51 | 0:100 |
| a.Conditions: [M]total=0.35 mol/L in THF; Grubbs second generation(G2) as catalyst, [M]total/[G2]=100:1; 30 ℃; 4 h; b.diethyl ether-isoluble part; c.determined by GPC(DMF, PMMA calibration); d.detemined by 1H NMR(DMSO-d6). | ||||
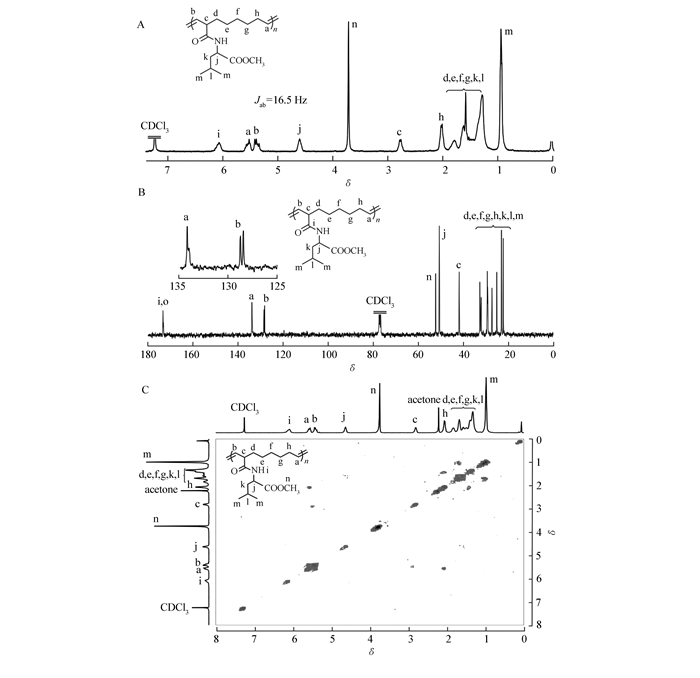
The regio- and stereo-structure of poly(1) was prudentially characterized by 1H NMR, 13C NMR, 1H-1H-cosy (Fig. 3) and FTIR analysis (see Fig.S6 in Supporting Information). As we can see in Fig. 3, the 13C NMR displayed two sets of distinct olefinic resonance peaks located at 128.5 and 134.3, which forcefully verified high regioregularity of the obtained polymer (head-to-head and tail-to-tail or alternating head-to-tail). Moreover, each group of olefinic signal in 13C NMR spectra of poly(1) was split to double peaks. This is because monomer 1 has two epimers, as evidenced by 13C NMR spectra of monomer 1 (Fig. 1A). Meanwhile, the distinct correlation of the olefinic protons Ha and Hb in the poly(1) strongly confirmed the perfectly head-to-tail (HT) regioregularity rather than head-to-head and tail-to-tail(HH/TT) regioregularity. The 1H NMR and 1H-1H cosy analysis shown in Fig. 3, demonstrated a distinct trans-double-bond of olefinic signals, in which the coupling constant was 16.5 Hz for the double-bond resonance peaks. The FTIR spectra of poly(1) showed obvious absorptions at 966 cm-1, also confirmed the trans configuration of the olefin. The poly(2) also gave similar regio- and stereoregularities results according to the NMR and FTIR analysis (see Fig.S6-S8 in Supporting Information).
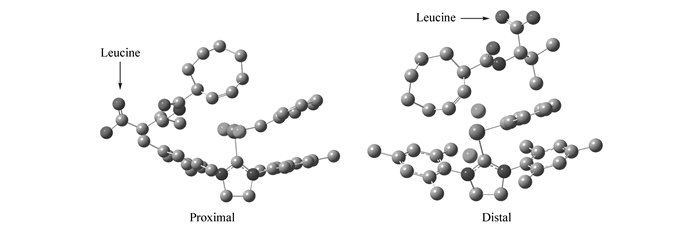
To better disclose the regioselectivity during ROMP, we carried out quantum chemical calculations of the conformation of monomer 2 during adding step to Ru catalyst. As shown in Fig. 4, two possible geometries of monomer 2 can be considered, namely, leucine substituent being proximal to the metal center and distal to it. The calculations revealed that the structure of proximal was more stable than that of distal over 4.60 kJ/mol, which indicating that it was energetically favorable for adding from distal of metal center.
Compared to conventional block copolymers, amino acid-based block polymers can be designed to control the hydrophilicity patterns and secondary structures through the reasonable selection and arrangement of amino acid moieties, leading to bioengineered tailor-made materials[49-50]. As stated in the introduction, the active groups of amino acids might coordinate metathesis catalysts during the polymerization. Here, monomer 2 with carboxy groups successfully underwent polymerization to obtain a polymer with a relatively low PDI. Based on the results above, we tested the block copolymerization of monomers 1 and 2 by sequential monomer addition. In the first stage of the polymerization, monomer 1 was compeletly converted within 4 h, which was monitored by the 1H NMR measurement of a small portion withdraw. Subsequently, a solution of monomer 2 was quickly injected to the mixture, and then the reaction mixture was stirred for another 24 h. As listed in Table 2, a series of well-defined block copolymers was prepared with with Mn′s of 1.45×104~1.78×104(Mw/Mn=1.4~1.6) in high yields (52%~79%). The block copolymer displayed a methyl ester proton signal at 3.7 and a distinct carboxy proton signal at 12.5 in DMSO-d6(Fig. 5A), demonstrating the formation of amphiphilic leucine-bearing block copolymer. 13C NMR spectroscopy confirmed the perfectly HT regioregularity of the resultant block copolymer(Fig. 5C).
下载:
导出CSV
| Feed ratio n(1):n(2) |
Unit ratiod n(1):n(2) |
Yieldb/% | Mnc | Mw/Mnc |
| 75:25 | 64 | 17 300 | 1.59 | 74:26 |
| 60:40 | 52 | 14 500 | 1.40 | 62:38 |
| 50:50 | 57 | 17 800 | 1.46 | 53:47 |
| 40:60 | 79 | 13 500 | 1.49 | 44:56 |
| 25:75 | 75 | 17 800 | 1.51 | 34:66 |
| a.Conditions: [M]total=0.35 mol/L in THF; Grubbs second generation(G2) as catalyst, [M]total/[G2]=100:1; 30 ℃; 4 h; b.diethyl ether-isoluble part; c.determined by GPC(DMF, PMMA calibration); d.detemined by 1H NMR(DMSO-d6). | ||||
Amphiphilic diblock copolymers having a carboxy block exhibt interesting properties, including pH-response, variable solubility and ability of assembly to form micelles, not only in aqueous media but also in organic solvents[51-53]. We envisioned that leucine-based block copolymer consisting of acidic blocks would exhibit unique properties as well. With block copolymers in hand, we investigated their physical properties. Table 3 showed the solubility of the homopolymers and copolymers in different solvent. Both of poly(1) and poly(2) were well soluble in THF, DMF, MeOH and DMSO. The copolymers also have good solubility in these solvents without regard to the unit ratios. Moreover, poly(1) was soluble in CH2Cl2, CHCl3, and acetone, while poly(2) cannot be dissolved in these solvents. By examining the solubility of the copolymers carefully, we found an interesting distinction in solubility between the random and block copolymers in acetone. That is, the block copolymers composed of 1/2 with unit molar ratios of 75:25, 60:40, and 50:50 can be dissolved in acetone, while the random copolymers with the same ratios were partially insoluble in the solvent. This is very encouraging because a significant difference in solubility of the block copolymers may lead to its adjustable phase behavior in solution.
下载:
导出CSV
| Polymer | PE | Et2O | DCM | CHCl3 | THF | Acetone | DMF | MeOH | DMSO |
| poly(1) | - | - | + | + | + | + | + | + | + |
| poly(2) | - | - | - | - | + | - | + | + | + |
| poly(1-co-2) | |||||||||
| 75:25 | - | - | + | + | + | ± | + | + | + |
| 50:50 | - | - | ± | + | + | ± | + | + | + |
| 25:75 | - | - | - | ± | + | - | + | + | + |
| poly(1)-block-poly(2) | |||||||||
| 75:25 | - | - | + | + | + | + | + | + | + |
| 60:40 | - | - | + | + | + | + | + | + | + |
| 50:50 | - | - | ± | + | + | + | + | + | + |
| 40:60 | - | - | - | + | + | ± | + | + | + |
| 25:75 | - | - | - | ± | + | ± | + | + | + |
| a.(-)insoluble; (±)partly soluble; (+)soluble. | |||||||||
On the other hand, though the block copolymers with 1/2 unit molar ratios of 60:40 and 50:50 were soluble in acetone, the acetone solutions were slightly turbid. We assumed that these block copolymers might undergo self-assemble to forming reverse micelles in acetone. We then started to investigate the 1H NMR spectra of poly(1)50-block-poly(2)50 in acetone-d6 to explore the micelle formation. As shown in Fig. 5B, the block copolymer exhibited a methyl ester proton signal, but no carboxylic acid proton signal can be found. Generally, the self-assembly of block copolymer in solution would lead to reduced or negligible proton signals of the core part. Therefore, the absence of carboxylic acid proton signal in acetone-d6 was attributed to the formation of a reverse micelle composed of a hydrophilic core of poly(2) and a hydrophobic shell of poly(1).
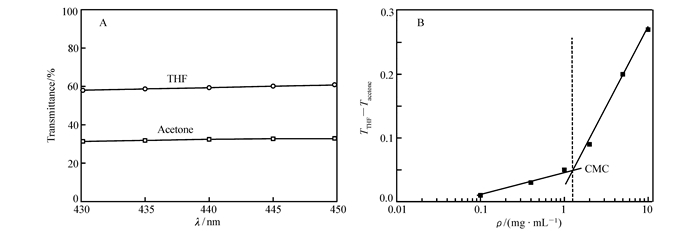
Turbidity is a simple and efficient parameter to confirm aggregates, because it reflects the variation of polymer particles′ size in solution. Here, the turbidity of polymer solutions was determined by using UV-Vis spectroscopy. Fig. 6A showed the transmittance of 10 mg/mL solutions of poly(1)50-block-poly(2)50 in acetone and THF. As listed in Table 3, THF is a good solvent that both hydrophilic and hydrophobic blocks can be dissolved well, while acetone is a solvent that can be benefit for reverse micelle formation as described above. The transmittance of the polymer solutions in THF at 400~700 nm was higher than that in acetone, which further manifests that larger size particles (micelles) exist in acetone(Fig. 6A). The critical micelle concentration (CMC) is defined as the lowest concentration of polymeric amphiphiles to form micelles. Many physical properties show sharp changes at CMC such as solubilization, surface tension and turbidity. In this study, we measured the CMC of poly(1)50-block-poly(2)50 in acetone using turbidity measurement, which can be defined by the difference between the transmittance of the THF and acetone solutions of polymers at 435 nm. As shown in Fig. 6B, it clearly displayed a inflection point around mass concentration(ρ) of 1.2 mg/mL, which could be considered as the CMC. The DLS measurement of poly(1)50-block-poly(2)50 in acetone solution also confirmed the presence of aggregates with an average hydrodynamic radius(Rh) around 30 nm (Fig. 7A). TEM was employed to visualize the morphology of the reverse micelle. As shown in Fig. 7B, spherical shapes with diameters mainly around 50~140 nm were clearly observed by TEM. The size distribution of particles is relatively wide in TEM, which may be due to the fact that the polymer is not very stable after forming a small size primary micelle in acetone, and will aggregate to form a large size secondary micelle during spinning process.

In summary, ring-opening metathesis polymerization (ROMP) of amino acid-substituted cyclooctene monomer 1 or 2 was regio- and stereoselective and the polydispersity index (PDI) of obtained leucine-based polymers ranged from 1.3~1.6. Amphiphilic block copolymers were easily prepared through the successive polymerization of monomers 1 and 2. Interestingly, the block copolymers with 1:2 unit molar ratios of 50:50 were soluble in acetone, forming reverse micelles with radius around 30 nm in acetone composed of a hydrophilic core of poly(2) and a hydrophobic shell of poly(1), while the random copolymers with the same ratios were partially insoluble in the solvent. These polymers could be used as biomimetic materials when well-defined regio- and stereoregular structures are advantageous.
Supporting information [reaction formula, NMR spectra] is available free of charge via internet at http://yyhx.ciac.jl.cn.
Laursen J S, Harris P, Fristrup P. Triangular Prism-Shaped β-Peptoid Helices as Unique Biomimetic Scaffolds[J]. Nat Commun, 2015, 6: 7013-7022. doi: 10.1038/ncomms8013
Lai H, Chen X, Lu Q. A New Strategy to Synthesize Bottlebrushes with a Helical Polyglutamate Backbone via N-Carboxyanhydride Polymerization and RAFT[J]. Chem Commun, 2014, 50: 14183-14186. doi: 10.1039/C4CC06575A
陶友华. 基于内酰胺开环聚合的氨基酸聚合新方法[J]. 高分子学报, 2016(9): 1151-1159. TAO Youhua. New Polymerization Methodology of Amino Acid Based on Lactam Polymerization[J]. Acta Polym Sin, 2016, (9): 1151-1159.
Li M, Tao Y, Tang J. Synergetic Organocatalysis for Eliminating Epimerization in Ring-Opening Polymerizations Enables Synthesis of Stereoregular Isotactic Polyester[J]. J Am Chem Soc, 2019, 141(1): 281-289. doi: 10.1021/jacs.8b09739
Wang S X, Tao Y, Wang J. A Versatile Strategy for the Synthesis of Sequence-Defined Peptoids with Side-Chain and Backbone Diversity via Amino Acid Building Blocks[J]. Chem Sci, 2019, 10: 1531-1538. doi: 10.1039/C8SC03415J
Wang S, He W, Xiao C. Synthesis of Y-Shaped OEGylated Poly(amino acid)s:The Impact of OEG Architecture[J]. Biomacromolecules, 2019, 20(4): 1655-1666. doi: 10.1021/acs.biomac.9b00026
Tao Y, Wang S, Zhang X. Synthesis and Properties of Alternating Polypeptoids and Polyampholytes as Protein-Resistant Polymers[J]. Biomacromolecules, 2018, 19(3): 936-942. doi: 10.1021/acs.biomac.7b01719
He W, Tao Y, Wang X. Functional Polyamides:A Sustainable Access via Lysine Cyclization and Organocatalytic Ring-Opening Polymerization[J]. Macromolecules, 2018, 51(20): 8248-8257. doi: 10.1021/acs.macromol.8b01790
Chen J, Li M, He W. Facile Organocatalyzed Synthesis of Poly(ε-lysine) under Mild Conditions[J]. Macromolecules, 2017, 50(23): 9128-9134. doi: 10.1021/acs.macromol.7b02331
Zhang X, Wang S, Liu J. Ugi Reaction of Natural Amino Acids:A General Route Toward Facile Synthesis of Polypeptoids for Bioapplications[J]. ACS Macro Lett, 2016, 5(9): 1049-1054. doi: 10.1021/acsmacrolett.6b00530
Tao Y, Wang Z, Tao Y H. Polypeptoids Synthesis Based on Ugi Reaction:Advances and Perspectives[J]. Biopolymers, 2019, 110(3): .
Zhang H, Chen J, Zhang X. Multidentate Comb-Shape Polypeptides Bearing Trithiocarbonate Functionality:Synthesis and Application for Water-Soluble Quantum Dots[J]. Biomacromolecules, 2017, 18(3): 924-910. doi: 10.1021/acs.biomac.6b01760
Tao Y, Chen X, Jia F. New Chemosynthetic Route to Linear ε-Poly-lysine[J]. Chem Sci, 2015, 6: 6385-6391. doi: 10.1039/C5SC02479J
Bellomo E G, Wyrsta M D, Pakstis L. Stimuli-Responsive Polypeptide Vesicles by Conformation-Specific Assembly[J]. Nat Mater, 2004, 3: 244-248. doi: 10.1038/nmat1093
Deming T J. Synthetic Polypeptides for Biomedical Applications[J]. Prog Polym Sci, 2007, 32: 858-875. doi: 10.1016/j.progpolymsci.2007.05.010
Kiessling L L, Fishman J M. Biologically Active Polymers[B]. Handbook of Metathesis; Wiley-VCH Verlag GmbH & Co.KGaA: 2015: 169-206.
Chen X, Lai H, Xiao C. New Bio-renewable Polyester with Rich Side Amino Groups from L-Lysine via Controlled Ring-Opening Polymerization[J]. Polym Chem, 2014, 5: 6495-6502. doi: 10.1039/C4PY00930D
Mori H, Endo T. Amino-Acid-Based Block Copolymers by RAFT Polymerization[J]. Macromol Rapid Commun, 2012, 33: 1090-1107. doi: 10.1002/marc.201100887
Ayres L, Hans P, Adams J. Peptide Polymer Vesicles Prepared by Atom Transfer Radical Polymerization[J]. J Polym Sci Polym Chem, 2005, 43: 6355-6366. doi: 10.1002/pola.21107
O'Reilly R K. Using Controlled Radical Polymerisation Techniques for the Synthesis of Functional Polymers Containing Amino Acid Moieties[J]. Polym Int, 2010, 59: 568-573.
Lee Y, Sampson N S. Polymeric ADAM Protein Mimics Interrogate Mammalian Sperm Egg Binding[J]. ChemBioChem, 2009, 10: 929-937. doi: 10.1002/cbic.200800791
Bauri K, De P, Shah P N. Polyisobutylene-Based Helical Block Copolymers with pH-Responsive Cationic Side-Chain Amino Acid Moieties by Tandem Living Polymerizations[J]. Macromolecules, 2013, 46(15): 5861-5870. doi: 10.1021/ma401395f
Mori H, Takahashi E, Ishizuki A. Tryptophan-Containing Block Copolymers Prepared by RAFT Polymerization:Synthesis, Self-Assembly, and Chiroptical and Sensing Properties[J]. Macromolecules, 2013, 46(16): 6451-6465. doi: 10.1021/ma400596r
Bielawski C W, Hillmyer M A. Telechelic Polymers from Olefin Metathesis Methodologies[B]. Handbook of Metathesis; Grubbs, R. H., ED.; Wiley-VCH: Weinheim, Germany, 2003: 255-282.
Penczek S, Cypryk M, Duda A. Living Ring-opening Polymerizations of Heterocyclic Monomers[J]. Prog Polym Sci, 2007, 32: 247-282. doi: 10.1016/j.progpolymsci.2007.01.002
Wu J, Yu T L, Chen C T. Recent Developments in Main Group Metal Complexes Catalyzed/Initiated Polymerization of Lactides and Related Cyclic Esters[J]. Coord Chem Rev, 2006, 250: 602-626. doi: 10.1016/j.ccr.2005.07.010
Maynard H D, Okada S Y, Grubbs R H. Inhibition of Cell Adhesion to Fibronectin by Oligopeptide-Substituted Polynorbornenes[J]. J Am Chem Soc, 2001, 123(7): 1275-1279. doi: 10.1021/ja003305m
Biagini SC G, Gareth Davies R, Gibson V C. Ruthenium Initiated Ring Opening Metathesis Polymerisation of Amino-acid and -Ester Functionalised Norbornenes and a Highly Selective Chain-End Functionalisation Reaction Using Molecular Oxygen[J]. Polymer, 2001, 42: 6669-6671. doi: 10.1016/S0032-3861(01)00146-X
Sutthasupa S, Shiotsuki M, Masuda T. Alternating Ring-Opening Metathesis Copolymerization of Amino Acid Derived Norbornene Monomers Carrying Nonprotected Carboxy and Amino Groups Based on Acid? Base Interaction[J]. J Am Chem Soc, 2009, 131(30): 10546-10551. doi: 10.1021/ja903248c
Osawa K, Kobayashi S, Tanaka M. Synthesis of Sequence-Specific Polymers with Amide Side Chains via Regio-/Stereoselective Ring-Opening Metathesis Polymerization of 3-Substituted cis-Cyclooctene[J]. Macromolecules, 2016, 49(21): 8154-8161. doi: 10.1021/acs.macromol.6b01829
Kobayashi S, Fukuda K, Kataoka M. Regioselective Ring-Opening Metathesis Polymerization of 3-Substituted Cyclooctenes with Ether Side Chains[J]. Macromolecules, 2016, 49(7): 2493-2501. doi: 10.1021/acs.macromol.6b00273
Zhang J, Matta M E, Martinez H. Precision Vinyl Acetate/Ethylene(VAE) Copolymers by ROMP of Acetoxy-Substituted Cyclic Alkenes[J]. Macromolecules, 2013, 46(7): 2535-2543. doi: 10.1021/ma400092z
Zhang J, Matta M E, Hillmyer M A. Synthesis of Sequence-Specific Vinyl Copolymers by Regioselective ROMP of Multiply Substituted Cyclooctenes[J]. ACS Macro Lett, 2012, 1(12): 1383-1387. doi: 10.1021/mz300535r
Martinez H, Mir P, Charbonneau P. Selectivity in Ring-Opening Metathesis Polymerization of Z-Cyclooctenes Catalyzed by a Second-generation Grubbs Catalyst[J]. ACS Catal, 2012, 2(12): 2547-2556. doi: 10.1021/cs300549u
Kobayashi S, Pitet L M, Hillmyer M A. Regio- and Stereoselective Ring-Opening Metathesis Polymerization of 3-Substituted Cyclooctenes[J]. J Am Chem Soc, 2011, 133(15): 5794-5797. doi: 10.1021/ja201644v
Martinez H, Ren N, Matta M E. Ring-Opening Metathesis Polymerization of 8-Membered Cyclic Olefins[J]. Polym Chem, 2014, 5: 3507-3532. doi: 10.1039/c3py01787g
Li M, Cui F, Li Y. Crystalline Regio-/Stereoregular Glycine-Bearing Polymers from ROMP:Effect of Microstructures on Materials Performances[J]. Macromolecules, 2016, 49(24): 9415-9424. doi: 10.1021/acs.macromol.6b02244
Bates C M, Bates F S. 50th Anniversary Perspective:Block Polymers-Pure Potential[J]. Macromolecules, 2017, 50(1): 3-22.
Ahmed S R, Bullock S E, Cresce A V. Polydispersity Control in Ring Opening Metathesis Polymerization of Amphiphilic Norbornene Diblock Copolymers[J]. Polymer, 2003, 44: 4943-4948. doi: 10.1016/S0032-3861(03)00487-7
Gill G B, Idris M S, Kirollos K S. Ene Reactions of Indane-1, 2, 3-trione(A Super-Enophile) and Related Vicinal Tricarbonyl Systems[J]. J Chem Soc, Perkin Transactions 1, 1992, 18: 2355-2365.
Gill G B, Idris M S, Kirollos K S. Oxidative Cleavage of Indane-1, 2, 3-Trione-Ene Adducts; A Convenient Synthesis of Allyl and Allenyl Carboxylic Acids[J]. J Chem Soc, Perkin Trans 1, 1992, 18: 2367-2369.
Te Velde G, Bickelhaupt F M, Baerends E J. Chemistry with ADF[J]. J Comput Chem, 2001, 22: 931-967. doi: 10.1002/jcc.1056
Baerends E J, Ellis D E, Ros P. Self-consistent Molecular Hartree-Fock-Slater Calculations Ⅰ.The Computational Procedure[J]. Chem Phys, 1973, 2: 41-51. doi: 10.1016/0301-0104(73)80059-X
Baerends E J and Ros P. Self-Consistent Molecular Hartree-Fock-Slater Calculations Ⅱ.The Effect of Exchange Scaling in Some Small Molecules[J]. Chem Phys, 1973, 2: 52-59. doi: 10.1016/0301-0104(73)80060-6
Becke A D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior[J]. Phys Rev A, 1988, 38: 3098-3100. doi: 10.1103/PhysRevA.38.3098
Perdew J P. Accurate and Simple Density Functional for the Electronic Exchange Energy:Generalized Gradient Approximation[J]. Phys Rev B, 1986, 33: 8822-8824. doi: 10.1103/PhysRevB.33.8822
Perdew J P. Erratum:Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas[J]. Phys Rev B, 1986, 34: 7406-7406.
Yamamoto Y, Yatagai H, Maruyama K. Reaction of Allylic Boron and Aluminum "ate" Complexes with Organic Halides and Carbonyl Compounds. Trialkylboranes as Regio-, Stereo-, and Chemoselective Control Elements[J]. J Am Chem Soc, 1981, 103(8): 1969-1975. doi: 10.1021/ja00398a016
Rabotyagova O S, Cebe P, Kaplan D L. Protein-Based Block Copolymers[J]. Biomacromolecules, 2011, 12(2): 269-289. doi: 10.1021/bm100928x
Broyer R M, Grover G N, Maynard H D. Emerging Synthetic Approaches for Protein-Polymer Conjugations[J]. Chem Commun, 2011, 47: 2212-2226. doi: 10.1039/c0cc04062b
Sutthasupa S, Shiotsuki M, Matsuoka H. ing-Opening Metathesis Block Copolymerization of Amino Acid Functionalized Norbornene Monomers. Effects of Solvent and pH on Micelle Formation[J]. Macromolecules, 2010, 43(4): 1815-1822. doi: 10.1021/ma902405g
Förster S, Antonietti M. Amphiphilic Block Copolymers in Structure-Controlled Nanomaterial Hybrids[J]. Adv Mater, 1998, 10: 195-217. doi: 10.1002/(SICI)1521-4095(199802)10:3<195::AID-ADMA195>3.0.CO;2-V
Rodríguez-Hernández J, Chécot F, Gnanou Y. Toward Smart' Nano-objects by Self-assembly of Block Copolymers in Solution[J]. Prog Polym Sci, 2005, 30: 691-724. doi: 10.1016/j.progpolymsci.2005.04.002

Figure 2 Mn, Mw/Mn(A) and GPC(B) curves of the poly(1) obtained with the ROMP using Grubbs second generation catalyst in THF at room temperature

Figure 4 Optimized M06-L E TS2 structures for the proximal and distal reactions of monomer 2. Hydrogens were omitted for simplicity

Figure 5 (A) 1H NMR spectra of poly(1)50-block-poly(2)50 in DMSO-d6. (B)1H NMR spectra of poly(1)50-block-poly(2)50 in acetone-d6; (C)13C NMR spectra of poly(1)50-block-poly(2)50 in CDCl3

Figure 6 (A) Transmittance of poly(1)50-block-poly(2)50 solution(ρ=1%, mass fraction) in THF and acetone. (B)Difference between the transmittance(T) of THF and acetone solutions of poly(1)50-block-poly(2)50 at different concentration at 435 nm

Figure 7 (A) Size distribution(Rh:hydrodynamic radius) of poly(1)50-block-poly(2)50 solution in acetone(mass fraction 1%) measured by dynamic light scattering. (B)TEM micrographs of poly(1)50-block-poly(2)50 micelle in acetone solution(ρ=0.5%, mass fraction)
Table 1. Homo- and copolymerization of monomers 1 and 2a
| Feed ratio n(1):n(2) |
Yieldb/% | Mnc | Mw/Mnc | Unit ratiod n(1):n(2) |
| 100:0 | 66 | 48 600 | 1.40 | 100:0 |
| 75:25 | 82 | 21 700 | 1.39 | 72:28 |
| 50:50 | 73 | 14 200 | 1.45 | 58:42 |
| 25:75 | 77 | 33 100 | 1.60 | 24:76 |
| 0:100 | 70 | 37 500 | 1.51 | 0:100 |
| a.Conditions: [M]total=0.35 mol/L in THF; Grubbs second generation(G2) as catalyst, [M]total/[G2]=100:1; 30 ℃; 4 h; b.diethyl ether-isoluble part; c.determined by GPC(DMF, PMMA calibration); d.detemined by 1H NMR(DMSO-d6). | ||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Block copolymerization of monomers 1 and 2a
| Feed ratio n(1):n(2) |
Unit ratiod n(1):n(2) |
Yieldb/% | Mnc | Mw/Mnc |
| 75:25 | 64 | 17 300 | 1.59 | 74:26 |
| 60:40 | 52 | 14 500 | 1.40 | 62:38 |
| 50:50 | 57 | 17 800 | 1.46 | 53:47 |
| 40:60 | 79 | 13 500 | 1.49 | 44:56 |
| 25:75 | 75 | 17 800 | 1.51 | 34:66 |
| a.Conditions: [M]total=0.35 mol/L in THF; Grubbs second generation(G2) as catalyst, [M]total/[G2]=100:1; 30 ℃; 4 h; b.diethyl ether-isoluble part; c.determined by GPC(DMF, PMMA calibration); d.detemined by 1H NMR(DMSO-d6). | ||||
下载: 导出CSV
Table 3. Solubility of polymersa
| Polymer | PE | Et2O | DCM | CHCl3 | THF | Acetone | DMF | MeOH | DMSO |
| poly(1) | - | - | + | + | + | + | + | + | + |
| poly(2) | - | - | - | - | + | - | + | + | + |
| poly(1-co-2) | |||||||||
| 75:25 | - | - | + | + | + | ± | + | + | + |
| 50:50 | - | - | ± | + | + | ± | + | + | + |
| 25:75 | - | - | - | ± | + | - | + | + | + |
| poly(1)-block-poly(2) | |||||||||
| 75:25 | - | - | + | + | + | + | + | + | + |
| 60:40 | - | - | + | + | + | + | + | + | + |
| 50:50 | - | - | ± | + | + | + | + | + | + |
| 40:60 | - | - | - | + | + | ± | + | + | + |
| 25:75 | - | - | - | ± | + | ± | + | + | + |
| a.(-)insoluble; (±)partly soluble; (+)soluble. | |||||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们