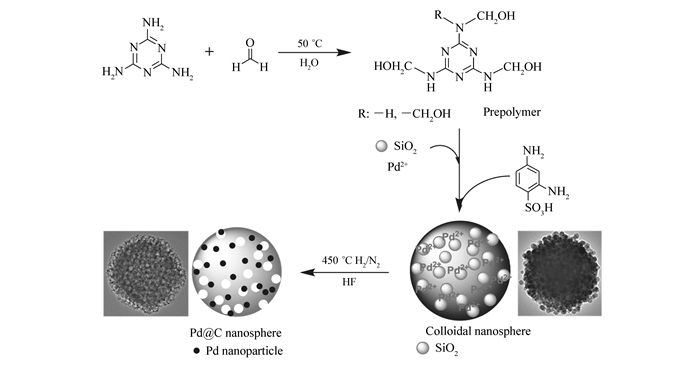
图 1.
Pd@C形成过程示意图
Figure 1.
Schematic illustration for the formation mechanism of Pd@C

钯纳米粒子由于粒径小、比表面积大以及具有量子限域效应[1-3],在催化领域受到广泛关注[4-6]。但正由于钯粒子粒径小,使其具有高的表面能,钯粒子极易发生团聚。针对这一问题,人们通过将钯纳米粒子分散在载体材料上来解决[7-9]。碳材料作为载体负载钯纳米粒子,相比于二氧化硅[10-12]、金属氧化物[13-14]等载体,具有高温稳定性好、化学惰性以及金属易回收等优点,被认为是优异的载体材料[15-16]。负载型纳米催化剂的制备通常采用浸渍法[17]、沉积法[18]、表层反应法[19]等,但这些方法在后续还原、焙烧等阶段难以实现金属纳米粒子尺寸的有效控制、使其保持在较小的尺寸范围。在碳载体形成的同时将钯纳米粒子原位分散,使钯粒子与载体紧密结合,是解决上述问题的有效途径。
基于此,本文在水溶液中合成三聚氰胺甲醛预聚物,利用预聚物中N原子与Pd2+相互作用,化学锚定Pd2+,预聚物进一步反应生成胶体纳米微球,并将Pd2+原位分散于胶体微球中。将胶体微球在H2和N2混合气体(物质的量比5:95)下焙烧,得到炭载钯纳米粒子(Pd@C)纳米微球材料。该制备过程条件温和、操作简单可控。由于Pd2+被原位锚定在胶体微球中以及碳载体对Pd纳米粒子的稳定作用,Pd@C纳米材料中Pd粒子具有高分散性。同时碳载体高孔隙率和化学稳定性,为Pd@C催化剂高催化活性提供可能。在Suzuki模型催化反应中,摩尔分数1%Pd@C在5 min内获得99.3%收率。
三聚氰胺、氯亚钯酸铵、碘苯、苯硼酸、碳酸钾和乙醇均为分析纯试剂,2, 4-二氨基苯磺酸(质量分数为98%)、二氧化硅溶胶水溶液(粒径30 nm,质量分数为50%)、氢氟酸(质量分数为40%)和甲醛(质量分数为37%~41%),以上试剂均由国药集团化学试剂有限公司提供,试剂均直接使用。实验用水均为电阻18.2兆欧的去离子水。
XD-3A型X射线衍射仪(XRD,日本岛津公司);Nova Nano-SEM 450型场发射扫描电子显微镜(SEM,美国FEI公司),加速电压5 kV;JEOL-200CX型透射电子显微镜(TEM,日本电子公司),工作电压300 kV;3H-2000PM型物理吸附仪(贝士德仪器科技(北京)有限公司);6130MSD型液质联用仪器(HPLC-MS,美国Agilent公司);AR 2130型电子精密天平(0.0001 g,奥豪斯(上海)公司);Agilent 1100型高效液相色谱仪(HPLC,美国Agilent公司);RCT basic型恒温加热磁力搅拌器(德国IKA集团);H-1650型台式高速离心机(湖南湘仪仪器有限公司);OTL-1200型管式炉(南京大学仪器厂);Escalab 250型X射线光电子能谱仪(XPS,美国Thermo-VG公司),工作电压15 kV,线源为Al-KαX,使用284.6 eV的C1s峰校准结合能及软件XPS PEAK 4.1进行曲线拟合;Optima 2000DV型电感耦合等离子发射光谱仪(ICP,美国PerkinElmer Precisely公司),其工作参数为:射频功率1300 W;辅助气流量0.20 L/min;冷却气流量15 L/min;样品提升量1.5 mL/min;积分时间10 s;观察高度15 mm。
向100 mL单口烧瓶中加入0.19 g三聚氰胺(1.5 mmol)、0.5 mL甲醛溶液和40 mL去离子水,在50 ℃、500 r/min条件下搅拌至反应体系均一透明。加入0.5 mL二氧化硅水凝胶,搅拌30 min后,准确量取0.75 mL (NH4)2PdCl4(0.5 mol/L)水溶液,加入到反应体系中,反应液呈浅黄色,继续搅拌60 min后,加入0.038 g 2, 4-二氨基苯磺酸(0.2 mmol),继续搅拌反应2 h。将反应液倒入离心管中,6000 r/min,离心10 min,20%乙醇溶液清洗后再离心,此过程重复3次,采用ICP-AES确认离心后溶液中Pd2+质量分数降低至0.01%以下,也证明Pd2+被锚定在胶体纳米微球。将离心得到的产物在105 ℃烘干10 h,然后在摩尔分数5%氢气及95%氮气气氛下450 ℃焙烧2 h,焙烧后产物用5%氢氟酸浸泡1 h除去二氧化硅,得到催化剂Pd@C。
量取已配制的0.05 mol/L碘苯和0.05 mol/L苯硼酸乙醇溶液各10 mL,加入到50 mL圆底烧瓶中,再称量0.039 g Pd@C催化剂(Pd与碘苯的物质的量比为1:100)加入烧瓶中,搅拌下加热至体系回流后,加入0.2 g碳酸钾固体继续搅拌,并采用HPLC监测反应进程。HPLC检测条件:流动相A为水相(含0.6%乙酸、0.3%三乙胺);流动相B为甲醇。流动相比例为V(A):V(B)=4:1,流速为0.8 mL/min,检测波长为(250±4) nm,进样量为5 μL。
三聚氰胺与甲醛在中性条件下、50 ℃水溶液中反应,形成三聚氰胺羟甲基化合物,常被称为预聚物;向预聚物中加入二氧化硅水凝胶及氯亚钯酸铵水溶液,预聚物中N原子与Pd2+发生相互作用[20-22],化学锚定Pd2+。加入2, 4-二氨基苯磺酸后,酸催化下—NHCH2OH可形成—NHCH2+[23],该碳正离子可与2, 4-二氨基苯磺酸的氨基发生亲电取代反应,形成—NHCH2NH—桥连,并借助2, 4-二氨基苯磺酸两个氨基的连接作用实现预聚物分子间的交联;同时,酸催化下—NHCH2OH的羟基也可脱水成醚,形成—NHCH2OCH2NH—桥连[24],使锚定Pd2+的预聚物形成胶体纳米微球。由于Pd2+与预聚物中N原子的相互作用,使其被原位锚定在预聚物中,因此可以均匀地分布在胶体纳米微球中;二氧化硅由于硅羟基与亚氨基或羟甲基的氢键作用,也可以均匀地分布在胶体纳米微球中。将得到的胶体纳米微球在H2/N2标准气体氛围下、450 ℃焙烧,胶体微球碳化的同时Pd2+被还原为Pd0。经氢氟酸除去二氧化硅,即可得到Pd@C纳米材料。Pd@C的形成过程如图 1所示。
对反应形成预聚物及Pd@C纳米材料采用HPLC-MS、SEM、TEM、XRD、XPS及BET等手段进行了表征,证明了Pd@C的形成机理和组成。
三聚氰胺分子中含有3个氨基,每个氨基上的活泼氢原子均可与甲醛发生亲核加成反应,在甲醛量足够的情况下,可生成六羟甲基三聚氰胺。实际反应中,三聚氰胺和甲醛反应的产物是含1~6个羟甲基的三聚氰胺预聚物的混合物,产物的比例受反应物的比例、反应温度、反应时间、pH值等条件的影响。本实验中,控制三聚氰胺和甲醛的物质的量比为1:4.5,50 ℃下搅拌1 h,得到预聚物。对该预聚物进行HPLC-MS联机分析,相对分子质量为216的三羟甲基三聚氰胺HPLC相对质量分数为56.8%,相对分子质量为246的四羟甲基三聚氰胺HPLC相对质量分数为37.1%,预聚物中三羟甲基三聚氰胺和四羟甲基三聚氰胺的含量达93.9%。预聚物中大量的—NHCH2OH及—NH—基团,为化学锚定Pd2+及均匀分散二氧化硅水凝胶提供了保障。
酸催化条件下,预聚物分子结构中的—NHCH2OH既可生成—NHCH2+对氨基进行亲电取代反应,形成—NHCH2NH—桥连,也可以发生脱水成醚的反应,形成—NHCH2OCH2NH—链接,实现预聚物分子间的聚合,得到胶体微球。该胶体微球在H2/N2标准气体下焙烧时Pd2+被还原,得到Pd@C纳米材料。图 2为所制备的Pd@C材料的SEM及TEM照片,其中图 2A是碳化胶体微球的SEM照片,其表面布满颗粒状凸起,经HF溶液处理后,凸起消失,微球表面呈蜂窝状(见图 2B),说明这些凸起是二氧化硅,蜂窝孔是HF刻蚀除去二氧化硅形成的,孔径为30 nm,与二氧化硅水凝胶的粒径一致,蜂窝孔提高材料高空隙率,为催化反应提供通道[25-27];蜂窝孔的分布也较均匀,说明含羟甲基或亚氨基的预聚物与含硅羟基的二氧化硅水凝胶间存在氢键相互作用,使二氧化硅能够在预聚物聚合得到的微球中均匀分散;图 2C是除去二氧化硅后微球的TEM照片,进一步显示出蜂窝孔在整个微球中均匀分布;图 2D是单个微球的放大TEM图,图中可以清晰看到高分散的小黑点,这些小黑点即是Pd纳米粒子,平均粒径为(2.4±0.87) nm,说明Pd2+被原位锚定在聚合物胶体微球中,且分布均匀,经焙烧处理后,还原得到的Pd纳米粒子仍均匀分布在碳球中。

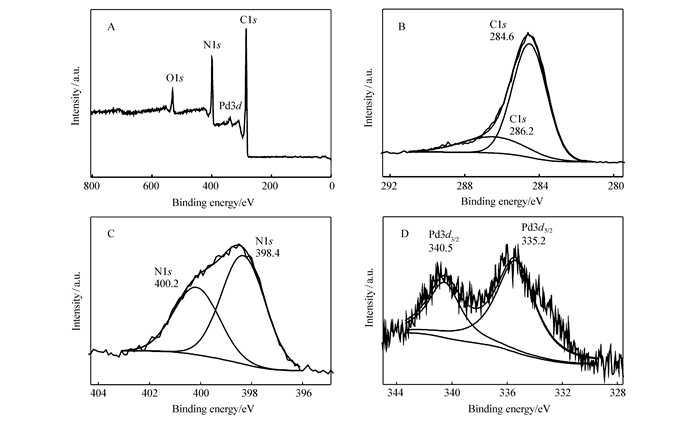
各种原子、分子不同轨道的电子结合能是一定的,具有标识性。由于元素所处的化学环境不同,其内层电子的轨道结合能会发生变化。因此,测定电子结合能即可进行元素的定性分析。图 3A是Pd@C的XPS宽扫描谱图,显示出C1s、N1s、Pd3d和O1s的4个典型特征峰,分别对应位于285、348、399和532 eV处的峰。图 3B的C1s结合能284.6和286.2 eV可归属为C—C和C—N;图 3C的N1s的也是两个峰,它们分别对应吡啶N(398.4 eV)、吡咯N(400.2 eV)[23];图 3D中Pd3d的两个峰为335.2和340.5 eV,分别对应Pd3d5/2和Pd3d3/2,证明钯是Pd0形式存在[28],材料制备过程在氢气条件下还原制得,所以Pd是以零价钯存在。即所制备的Pd@C主要含有C、N、Pd和O元素,其中Pd是零价钯。
Pd@C纳米材料的元素图像分析(图 4)可进一步确定Pd@C纳米材料中含有C、N和Pd 3种元素,这与XPS光谱分析结果相吻合。同时,Pd元素在分析图像上分散均匀,也表明Pd粒子在微球上分布均匀。

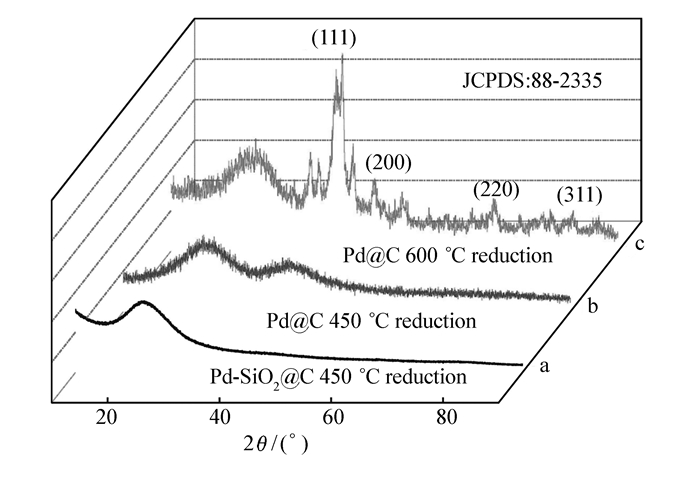
图 5为纳米材料Pd@C的XRD谱图。图 5谱线a和b为450 ℃还原后Pd-SiO2@C和Pd@C纳米材料的XRD谱图,未发现Pd颗粒的标准特征峰,这是由于此时Pd纳米粒子粒径比较小,衍射峰特征峰不明显,分别被二氧化硅和碳衍射峰所覆盖。为了证明Pd@C纳米材料中Pd粒子的存在,将Pd@C纳米材料的还原温度提高到600 ℃,高温下小粒径的Pd纳米粒子将进一步聚集,形成Pd大颗粒。图 5谱线c为600 ℃还原后Pd@C纳米材料的XRD,此时可以清晰的看到出现在40°、46°、68°和82°的Pd颗粒的特征衍射峰,分别对应Pd的(111)、(200)、(220)和(311)晶面,参照标准卡片(JCPDS:88-2335),表明纳米材料中Pd纳米粒子粒径较小,这是由于Pd2+被化学锚定在胶体微球中及碳对Pd纳米粒子的稳定作用。
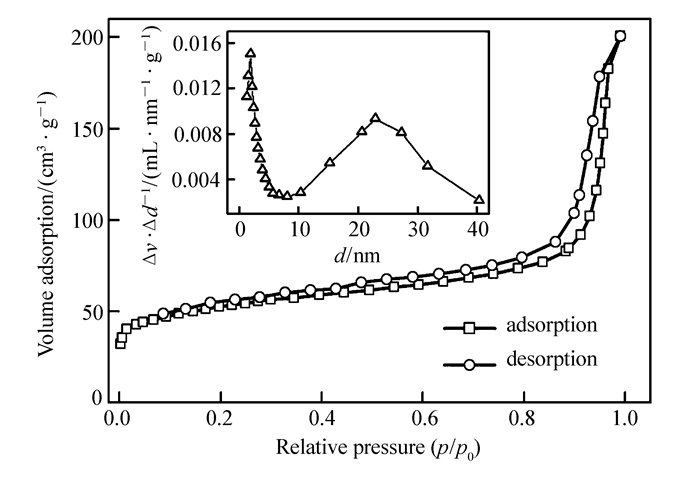
Pd@C纳米材料的氮气吸附-脱附等温曲线及孔径分布如图 6所示。根据BET计算结果,Pd@C材料的比表面积为169.3 m2/g,总孔体积为3.2 m3/g,平均孔径为24.74 nm,这与TEM图像中观察到刻蚀二氧化硅后形成的孔径基本相符,且为多孔结构。这种孔结构的存在有利于Pd纳米粒子与反应物接触,有利于Pd纳米粒子的催化性能。
以Suzuki偶联反应为模型反应,测试Pd@C纳米材料的催化性能[29],研究了反应时间、催化剂用量及催化剂循环使用对产物收率的影响,Scheme 1为Suzuki偶联反应的条件及反应方程式。
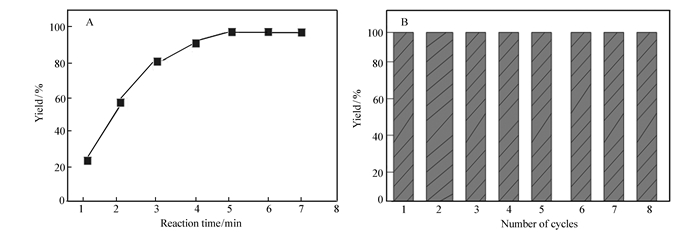
Pd@C催化剂用量选用与碘苯物质的量比为1:100,用已配制的碘苯与苯硼酸乙醇溶液进行Suzuki反应。分别在1、2、3、4、5、6和7 min时取样,进行液相色谱分析。结果表明(如图 7A),随着时间的增加,收率逐渐增大,反应1 min时,收率为22.7%;反应5 min时,收率可达到99.3%,反应时间进行到6和7 min时,收率增大不明显。表明Pd@C纳米材料在5 min即可催化Suzuki反应完成。
在其它条件不变的情况下,通过控制催化剂的用量来探讨其对反应速率的影响,Pd@C催化剂用量分别取用与碘苯物质的量比0.3:100、0.5:100、1:100、1.5:100和2:100进行Suzuki反应,均在5 min时取样。液相检测分析结果表明:Pd@C催化剂用量摩尔分数在0.3%、0.5%、1%时,随着催化剂的用量的增加,收率增大;而催化剂与碘苯物质的量比在1:100、1.5:100和2:100时,收率基本相同,即与碘苯物质的量比1:100是催化剂的合适用量。
以产物收率为考察指标,Pd@C催化剂用量与碘苯物质的量比为1:100时,反应5 min,收率为99.3%。催化剂循环使用8次,反应5 min时,收率未见明显下降,保持在98%以上(图 7B),说明所制备的Pd@C催化材料对碘苯与苯硼酸的Suzuki偶联反应具有较好的循环使用稳定性。
本文合成了一种碳载高分散Pd纳米粒子催化剂Pd@C。利用三聚氰胺甲醛预聚物中N原子与Pd2+配合作用,将Pd2+原位锚定在预聚物中,加入二氧化硅水溶胶造孔,通过2, 4-二氨基苯磺酸促进预聚物聚合,形成Pd2+均匀分布的胶体纳米球;再经过焙烧还原及HF刻蚀除去SiO2,得到Pd@C纳米材料。基于N原子对Pd2+的化学锚定及碳载体对Pd纳米粒子的稳定作用,Pd纳米粒子均匀分布在碳球中,平均粒径为(2.4±0.87) nm。在Suzuki模型催化反应中,与反应物物质的量比为1:100的Pd@C在5 min内获得99.3%的收率,证明Pd@C纳米材料在催化方面具有较好的应用前景。
Tsuji J. Palladium Reagents and Catalysts[M]. New Perspectives for the 21st Century; Wiley: West Sussex, U.K., 2004.
Malleron J L, Fiaud J C, Legros J Y, Handbook of Palladium-catalyzed Organic Reactions[M]. Academic Press:London, 2000.
Burda C, Chen X B, Narayanan R. Chemistry and Properties of Nanocrystals of Different Shapes[J]. Chem Rev, 2005, 105(4): 1025-1102.
Yuan B Z, Pan Y Y, Li Y W. A Highly Active Heterogeneous Palladium Catalyst for the Suzuki Miyaur and Ullmann Coupling Reactions of Aryl Chlorides in Aqueous Media[J]. Angew Chem Int Ed, 2010, 49(24): 4054-4058. doi: 10.1002/anie.201000576
Reetz M T, Westermann E. Phosphane-free Palladium-Catalyzed Coupling Reactions:The Decisive Role of Pd Nanoparticles[J]. Angew Chem Int Ed, 2000, 39(1): 165-168. doi: 10.1002/(SICI)1521-3773(20000103)39:1<165::AID-ANIE165>3.0.CO;2-B
Jadhav S N, Kumbhar A S, Rodeb C V. Ligand-free Pd Catalyzed Cross-coupling Reactions in an Aqueous Hydrotropic Medium[J]. Green Chem, 2016, 18(7): 1898-1911. doi: 10.1039/C5GC02314A
Wan X K, Guan Z J, Wang Q M. Homoleptic Alkynyl-Protected Gold Nanoclusters:Au44(PhC/C)28 and Au36(PhC/C)24[J]. Angew Chem Int Ed, 2017, 56(38): 11494-11497. doi: 10.1002/anie.201706021
Mitsudome T, Yamamoto M, Maeno Z. One-Step Synthesis of Core-Gold/Shell-Ceria Nanomaterial and Its Catalysis for Highly Selective Semihydrogenation of Alkynes[J]. J Am Chem Soc, 2015, 137(42): 13452-13455. doi: 10.1021/jacs.5b07521
Wang T, Yuan X, Li S R. CeO2-Modified Au@SBA-15 Nanocatalysts for Liquid-Phase Selective Oxidation of Benzyl Alcohol[J]. Nanoscale, 2015, 7(7): 7593-7602.
Lin X J, Zhong A Z, Sun Y B. In Situ Encapsulation of Pd Inside the MCM-41 Channel[J]. Chem Commun, 2015, 51(35): 7482-7485. doi: 10.1039/C5CC00300H
Zhong A Z, Zou W, Mao W X. A Continuous Etching Process for Highly-active Pd Nanoclusters and Their in Situ Stabilization[J]. RSC Adv, 2014, 4(45): 23637-23641. doi: 10.1039/c4ra02047b
Li C L, Zhang Q H, Wang Y. Preparation, Characterization and Catalytic Activity of Palladium Nanoparticles Encapsulated in SBA-15[J]. Catal Lett, 2008, 120(1/2): 126-136.
Cargnello M, Delgado Jaen J J, Hernandez Garrido J C. Exceptional Activity for Methane Combustion over Modular Pd@CeO2 Subunits on Functionalized Al2O3[J]. Science, 2012, 337(6095): 713-717. doi: 10.1126/science.1222887
Jin Z, Xiao M D, Bao Z H. A General Approach to Mesoporous Metal Oxide Microspheres Loaded with Noble Metal Nanoparticles[J]. Angew Chem Int Ed, 2012, 51(26): 6406-6410. doi: 10.1002/anie.201106948
Liu R, Priestley R D. Rational Design and Fabrication of Core shell Nanoparticles Through a One-Step/Pot Strategy[J]. J Mater Chem A, 2016, 4(18): 6680-6692. doi: 10.1039/C5TA09607C
Zhu C Z, Li H, Fu S F. Highly Efficient Nonprecious Metal Catalysts Towards Oxygen Reduction Reaction Based on Three-Dimensional Porous Carbon Nanostructures[J]. Chem Soc Rev, 2016, 45(3): 517-531.
White R J, Luque R, Budarin V L. Supported Metal Nanoparticles on Porous Materials, Methods and Applications[J]. Chem Soc Rev, 2009, 38(2): 481-494.
Haruta M, Tsubota S, Kobayashi T. Low-Temperature Oxidation of CO over Gold Supported on TiO2, α-Fe2O3, and Co3O4[J]. J Catal, 1993, 144(1): 175-192.
Jeong U, Joo J B, Kim Y. Au Nanoparticle-Embedded SiO2-Au@SiO2 Catalysts with Improved Catalytic Activity, Enhanced Stability to Metal Sintering and Excellent Recyclability[J]. RSC Adv, 2015, 5(69): 55608-55618. doi: 10.1039/C5RA07175E
Haas K L, Franz K J. Application of Metal Coordination Chemistry to Explore and Manipulate Cell Biology[J]. Chem Rev, 2009, 109(10): 4921-4960. doi: 10.1021/cr900134a
Yang J, Sargent E H, Kelley S O. A General Phase-Transfer Protocol for Metal Ions and Its Application in Nanocrystal Synthesis[J]. Nat Mater, 2009, 8: 683-689. doi: 10.1038/nmat2490
Ogasawara S, Kato S. Palladium Nanoparticles Captured in Microporous Polymers:A Tailor-Made Catalyst for Heterogeneous Carbon Cross-Coupling Reactions[J]. J Am Chem Soc, 2010, 132(13): 4608-4613. doi: 10.1021/ja9062053
Liu M H, Liu Y C, Gao Z M. Nitrogen and Sulfur Co-Doped Carbon Nanospheres for Highly Efficient Oxidation of Ethylbenzene[J]. New J Chem, 2018, 42(19): 15962-15967. doi: 10.1039/C8NJ02948B
Wu Y S, Li Y, Qin L. Monodispersed or Narrow-Dispersed Melamine Formaldehyde Resin Polymer Colloidal Spheres:Preparation, Size-Control, Modification, Bioconjugation and Particle Formation Mechanism[J]. J Mater Chem B, 2013, 1(2): 204-212. doi: 10.1039/C2TB00043A
Zhang H W, Noonan O, Huang X D. Surfactant-Free Assembly of Mesoporous Carbon Hollow Spheres with Large Tunable Pore Sizes[J]. ACS Nano, 2016, 10(4): 4579-4586. doi: 10.1021/acsnano.6b00723
Wang G, Sun Y H, Li D B. Controlled Synthesis of N-Doped Carbon Nanospheres with Tailored Mesopores Through Self-assembly of Colloidal Silica[J]. Angew Chem Int Ed, 2015, 54(50): 15191-15196. doi: 10.1002/anie.201507735
Kim S Y, Suh W H, Choi J H. Template-Free Synthesis of High Surface Area Nitrogen-Rich Carbon Microporous Spheres and Their Hydrogen Uptake Capacity[J]. J Mater Chem A, 2014, 2(7): 2227-2232. doi: 10.1039/C3TA14030J
Zhang D L, Zhaorigetu B, Bao Y S. Supported Palladium Nanoparticles Catalyzed Ortho-directed C-C Coupling Reaction via a Pd0/PdII/PdIV Catalytic Cycle[J]. J Phys Chem C, 2015, 119(35): 20426-20432. doi: 10.1021/acs.jpcc.5b04735
Budarin V L, Clark J H, Luque R. Palladium Nanoparticles on Polysaccharide-Derived Mesoporous Materials and Their Catalytic Performance in C-C Coupling Reactions[J]. Green Chem, 2008, 10(4): 382-387. doi: 10.1039/B715508E

图 2 Pd-SiO2@C (A)和Pd@C(B)材料SEM照片;Pd@C材料TEM照片(C, D)(D中插图为Pd纳米粒子粒径分布图)
Figure 2 SEM images of Pd-SiO2@C(A) and Pd@C(B); TEM images of Pd@C(C, D) (The inset of D shows the particle size distribution of Pd nanoparticles)

图 3 Pd@C材料的XPS光谱宽扫描光谱(A)及C 1s (B)、N1s (C)和Pd 3d (D)的元素高分辨率光谱
Figure 3 XPS survey spectra of Pd@C(A) and high resolution spectra for the elements of C1s(B), N1s(C), and Pd3d(D)

图 4 Pd@C材料的元素分布图像: Pd@C材料的暗场扫描透射电子显微镜照片(STEM-DF)(A)、C元素(B)、N元素(C)和Pd元素(D)
Figure 4 Element mapping images of Pd@C: STEM-DF of Pd@C(A), C(B), N(C)and Pd(D)

图 6 Pd@C材料的氮气吸附-脱附曲线及孔径分布图
Figure 6 Nitrogen adsorption-desorption isotherms of Pd@C(The inset shows the pore size distribution of Pd@C)

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:


