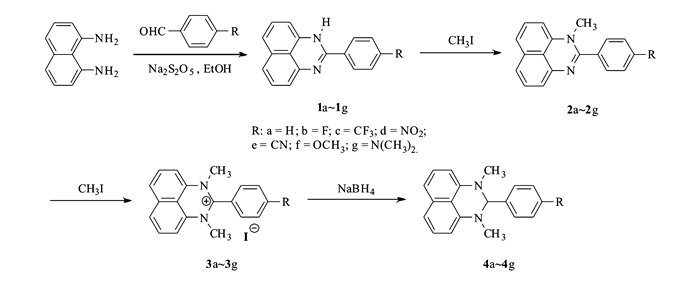
 Figure Scheme 1.
Synthesis of compounds 4a~4g
Figure Scheme 1.
Synthesis of compounds 4a~4g
Citation:
YUAN Lin, LI Zhongyan, JIA Guokai, ZHANG Min, YUAN Xianyou. Synthesis and Biological Activity of Perimidine Derivatives[J]. Chinese Journal of Applied Chemistry,
2017, 34(6): 685-692.
doi:
10.11944/j.issn.1000-0518.2017.06.160393

呸啶衍生物的合成及抑菌性
摘要:
呸啶是一种多核含氮杂环化合物,在工业和生物活性研究领域研究中发挥着重要作用长期以来,由于含氮杂环的存在,广大研究者们对呸啶化合物进行了大量的研究本文以1, 8-萘二胺和醛为原料,设计合成了一系列呸啶衍生物(4a~4g和6), 并通过红外光谱、核磁共振氢谱、核磁共振碳谱和高分辨质谱对其结构进行了表征其中化合物4a~4g和6的结构进行了单晶衍射测定单晶衍射结果显示,化合物4b和6的晶体均属于属于单斜晶系,P21/n空间群此外,还对目标化合物进行了抑菌试验,结果表明,化合物4b~4g和6对所测试的植物性病菌具有广谱的抑菌活性,说明呸啶衍生物在将来的抗菌生物领域具有很好应用前景4研究结果还表明,抑菌性并不具有明显的取代基效应。
English
Synthesis and Biological Activity of Perimidine Derivatives
Abstract:
Perimidine is a multi-nuclear N-heterocyclic compound, which plays an important role in industrial and biological fields. Perimidines have been drawn extensive examinations from many researchers for a long time due to the nitrogen heterocyclic ring. Here, a series of perimidine derivatives(4a~4g and 6) was synthesized through 1, 8-diaminonaphthalene and aldehydes. The products were characterized by infrared spectroscopy(IR), proton or carbon nuclear magnetic resonance(1H NMR, 13C NMR), high resolution mass spectrometry(HRMS). Compounds 4a and 6 were investigated by single-crystal X-ray diffraction. X-ray analysis reveals that both of crystals of 4a and 6 are made up of monoclinic unit cells with space group P21/n. In addition, the biological activity assay was carried out. The results show that perimidine derivatives have a diverse range of biological activities against the fungi, which indicates that perimidine derivatives have great potential applications in antibacterial fields in the future. And test data indicate that there is no obvious regulation between fungicidal activity and the different substituents on the benzene ring.
-
Key words:
- perimidine
- / synthesis
- / crystal structure
- / biological activity
-
Multi-nuclear N-heterocyclic compounds like perimidines have drawn extensive examinations from many researchers for a long time, because they exhibit a diverse range of biological activities. Perimidines also can be used as dyes and have a wide application in industrial field, and the famous product is reported as Solvent Black 3[1]. Perimidine derivatives were usually used as ligands with NCN pincers which have some advantages, such as an easy synthetic access to derived complexes, low cost, and endurance against oxygen under aerobic conditions. Thus, various NCN pincer complexes with diverse functional groups have been synthesized and reported[2]. In addition, perimidine derivatives can be obtained easily with several mature preparative methods[3-7] and the most common method for the preparation of perimidines is the condensation reaction of 1, 8-diaminonaphthalene with a carbonyl group[8-10]. Recently, perimidine derivatives have drawn more extensive examinations because of a diverse range of biological activities[11-14]. Because of the continuing interest in this field, investigation into new perimidine derivatives and their biological activities is of great importance. However, the biological activity of perimidine derivatives is mostly focused on application in bio-medical field, little attention has been paid to application in pesticide field[15-16]. In this paper, we report the synthesis, characterization and biological activity of a series of perimidine derivatives(Schemes 1 and 2).
Figure Scheme 1.
Synthesis of compounds 4a~4g
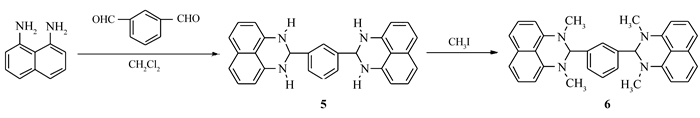 Figure Scheme 2.
Synthetic route of compoud 6
Figure Scheme 2.
Synthetic route of compoud 6
1 Experimental
1.1 Instruments and Reagents
AVANCE Ⅲ HD400 NMR spectrometer(Bruker, Germany); APEX Ⅱ Smart CCD with MoKα radiation(Bruker, Germany).
All chemicals and solvents were purchased from Aladdin Industrial Corporation and used without further purification.
1.2 Experimental Methods
1.3 Crystal Data and Structure Determination
The crystal of the tested compounds were cultivated from ethyl acetate, and the prisms were selected for X-ray diffraction analysis. All measurements were made on a Bruker Smart CCD area detector with Mo Kα radiation(λ=0.071073 nm). The data were collected at 293(2) K. The structures were solved by direct methods with SHELXS-97 program[18]. Refinements were done by full-matrix least-squares on F2 with SHELXL-97[19]. All of the non-H atoms were refined anisotropically by full-matrix least-squares. The hydrogen atoms were placed in the calculated positions and refined isotropically. The crystallographic data are summarized in Table 1.
 Table 1.
Crystal data and structure refinement for compounds 4a and 6
Table 1.
Crystal data and structure refinement for compounds 4a and 6

1.4 Biological Experiment
The biological activity of compounds 4b~4g and 6 were conducted by fungi growth inhibition method according to the reference using potato dextrose agar(PDA) as cultivation medium[20]. The assay was performed according to a previously described method[21]with some modifications. A stock solution of the tested compound was prepared at 500 mg/L using sterilized water containing 2 drops of N, N-dimethylformamide(DMF) as solvent. Subsequently, 1 mL of the stock solution was transferred into a 10 cm diameter of Petri dish, and 9 mL of PDA was added to prepare the plate containing 50 mg/L of the tested compound. Before the plate solidification, the PDA was thoroughly mixed by turning around the Petri dish in the sterilized operation desk 5 times to scatter the compound in PDA evenly. Then, 4 mm of diameter of fungi cake was inoculated on the plate and cultured in the tank at 24~26 ℃. The diameter of fungi spread was measured 2 days later. Growth inhibition was then calculated using the corresponding blank. Azoxystrobin was used as the positive control. Fungi tested in this study included Alternaria solani(AS), Botrytis cinerea(BC), Phytophthora infestans(Mont.) de Bary(PI), Cercospora arachidicola(CA), Physalospora piricola(PP), Gibberella zeae(GZ), Sclerotinia sclerotiorum(SS), Pellicularia sasakii(PS) and Rhizoctonia cerealis(RC).
1.2.6 Preparation of compound 6
Compound 5(0.01 mol) was dissolved in dry dimethylformamide(20 mL) and treated with potassium tert-butoxide(0.025 mol) under N2. After 30 min at room temperature, iodomethane(1.20 mol) was added to the reaction mixture and kept for 4 h under N2, giving a white solid. After the reaction, the solid was filtered, washed with moderate petroleum ether and dried under vacuum to yield the desired product. Colorless prisms of the product were obtained upon recrystallization from ethyl acetate.
Compound 4a:light yellow crystal, yield 70%, mp 164~165 ℃; IR(KBr), σ/cm-1:3094.0, 3049.2, 2921.3, 2878.7, 1649.8, 1588.7, 1558.0, 1443.3, 1418.4, 1275.2, 1198.8, 1103.6, 1040.1, 874.9, 818.0, 755.8, 705.0, 660.1; 1H NMR(400 MHz, DMSO-d6), δ:7.286(t, J=7.7 Hz, 2H), 7.214(s, 3H), 7.098(d, J=7.9 Hz, 2H), 7.018(s, 2H), 6.414(d, J=7.4 Hz, 2H), 5.513(s, 1H), 2.847(s, 6H); 13C NMR(400 MHz, DMSO-d6), δ:141.84, 138.48, 134.02, 128.82, 128.49, 127.85, 126.94, 116.36, 112.99, 102.88, 78.11, 36.21; HRMS(ESI) m/z:calcd. for C19H18N2[M+H]+:275.1543[M+H]+; found:275.1363.
Compound 4b:light yellow crystal, yield 68%, mp 255~257 ℃; IR(KBr), σ/cm-1:3058.1, 2941.5, 2914.6, 2869.8, 1584.1, 1503.3, 1473.4, 1374.8, 1326.9, 1207.3, 1177.4, 1120.6, 1081.7, 962.1, 839.5, 755.8, 705.0; 1H NMR (400 M, DMSO-d6), δ:7.292(t, J=7.6 Hz, 2H), 7.060(d, J=8.0 Hz, 2H), 7.043(d, J=7.1 Hz, 4H), 6.424(d, J=7.1 Hz, 2H), 5.550(s, 1H), 2.840(s, 6H); 13C NMR(400 MHz, DMSO-d6), δ:163.45, 161.03, 141.62, 134.81, 134.78, 134.01, 128.95, 128.87, 127.89, 116.49, 115.79, 115.58, 112.34, 103.04, 77.25, 36.12; HRMS(ESI) m/z:calcd. for C19H17N2F[M+H]+:293.1449[M+H]+; found:293.1264.
Compound 4c:white crystal, yield 69%, mp 162~164 ℃; IR(KBr), σ/cm-1:3054.6, 2922.2, 2884.1, 2820.3, 1614.8, 1590.8, 1478.8, 1419.1, 1378.5, 1326.0, 1221.5, 1128.5, 1087.7, 1014.0, 848.3, 809.8, 790.5; 1H NMR (400 MHz, DMSO-d6), δ:7.638(d, J=7.8 Hz, 2H), 7.294(t, J=7.8 Hz, 2H), 7.115(d, J=7.7 Hz, 2H), 6.963(d, J=8.0 Hz, 2H), 6.440(d, J=7.5 Hz, 2H), 5.645(s, 1H), 2.846(s, 6H); 13C NMR(400 MHz, DMSO-d6), δ:143.05, 141.37, 134.01, 129.09, 128.77, 127.91, 127.65, 125.89, 123.17, 116.72, 112.84, 103.22, 77.21, 36.16; HRMS(ESI) m/z:calcd. for C20H17N2F3[M+H]+:343.1417[M+H]+; found:343.1237.
Compound 4d:red crystal, yield 65%, mp 175~177 ℃; IR(KBr), σ/cm-1:3094.0, 3049.2, 2921.3, 2878.7, 1649.8, 1588.7, 1558.0, 1476.0, 1418.4, 1350.6, 1275.2, 1198.8, 874.9, 859.6, 755.8, 705.0, 660.1; 1H NMR (400 M, DMSO-d6), δ:8.098(d, J=8.3 Hz, 2H), 7.319(t, J=7.9 Hz, 2H), 7.268(d, J=8.7 Hz, 2H), 7.151(d, J=7.8 Hz, 2H), 6.472(d, J=7.1 Hz, 2H), 5.750(s, 1H), 2.894(s, 6H); 13C NMR(400 MHz, DMSO-d6), δ:147.48, 145.93, 141.04, 133.52, 127.98, 124.03, 116.91, 112.60, 103.39, 76.86, 36.06; HRMS(ESI) m/z:calcd. for C19H17N3O2[M+H]+:320.1394, found:320.1213.
Compound 4e:white crystal, yield 65%, mp 240~241 ℃; IR(KBr), σ/cm-1:3094.0, 2922.7, 2875.7, 2818.3, 1587.3, 1541.7, 1419.7, 1376.8, 1272.7, 1202.1, 1143.7, 1086.1, 1041.4, 808.7, 694.8, 603.4; 1H NMR(400 M, DMSO-d6), δ:7.694(d, J=8.2 Hz, 2H), 7.298(t, J=7.9 Hz, 2H), 7.173(d, J=8.3 Hz, 2H), 7.126(d, J=8.1 Hz, 2H), 6.446(d, J=7.6 Hz, 2H), 5.652(s, 1H), 2.858(s, 6H); 13C NMR(400 MHz, DMSO-d6), δ:143.88, 141.23, 133.98, 132.92, 127.93, 127.76, 119.02, 116.83, 112.77, 111.24, 103.35, 77.14, 36.14; HRMS(ESI) m/z:calcd. for C20H17N3[M+H]+:300.1495, [M+H]+; found:300.1312.
Compound 4f:white crystal, yield 77%. mp 101~103 ℃; IR(KBr), σ/cm-1:3051.8, 2924.7, 2853.5, 2812.2, 1650.3, 1590.8, 1541.3, 1489.3, 1474.0, 1372.9, 1250.6, 1171.8, 1058.1, 811.0, 759.3, 660.8; 1H NMR(400 M, DMSO-d6), δ:7.279(t, J=7.4 Hz, 2H), 7.085(d, J=7.5 Hz, 2H), 6.925(d, J=7.4 Hz, 2H), 6.768(d, J=7.7 Hz, 2H), 6.399(d, J=7.1 Hz, 2H), 5.462(s, 1H), 3.660(s, 3H), 2.822(s, 6H); 13C NMR(400 MHz, DMSO-d6), δ:148.19, 140.12, 136.80, 126.69, 118.07, 116.37, 106.09, 78.46, 52.76, 32.32; HRMS(ESI) m/z:calcd. for C20H20N2O [M+H]+:300.1649, [M+H]+; found:305.1474.
Compound 4g:white crystal, yield 75%, mp 168~170 ℃; IR(KBr), σ/cm-1:3047.8, 2922.5, 2922.5, 2815.3, 1612.1, 1592.9, 1521.4, 1475.3, 1446.0, 1343.4, 1201.5, 1084.8, 807.0, 756.8; 1H NMR(400 MHz, DMSO-d6), δ:7.260(t, J=7.5 Hz, 2H), 7.059(d, J=8.0 Hz, 2H), 6.828(d, J=7.3 Hz, 2H), 6.518(d, J=7.6 Hz, 2H), 6.372(d, J=7.0 Hz, 2H), 5.344(s, 1H), 2.800(s, 12H); 13C NMR(400 MHz, DMSO-d6), δ:150.49, 142.27, 134.04, 127.79, 127.74, 125.67, 116.04, 113.17, 112.34, 102.65, 78.14, 36.17; HRMS(ESI) m/z:calcd. for C21H23N3[M+H]+:318.1965, [M+H]+; found:318.1785.
Compound 6[15]:white crystal, yield 61%, mp 187~190 ℃.
1.2.4 Preparation of compound 4
Sodium borohydride(0.57 g, 0.015 mol) was added batch-wisely to a solution of compound 3(0.01 mol) in methanol(25 mL) under nitrogen atmosphere. The mixture was stirred at room temperature for 30 min, giving a white precipitate. After the completion of the reaction, the white solid was filtered, washed with water and dried under vacuum to yield the desired product.
1.2.1 Preparation of compound 1
Substituted benzaldehyde(0.01 mol) was added to a solution of 1, 8-naphthalenediamine(1.86 g, 0.01 mol) and sodium pyrosulfite(1.9 g, 0.01mol) in 150 mL of ethanol, followed by 50 mL of water. The mixture was refluxed with stirring and monitored by thin layer chromatography(TLC), giving a white precipitate. After cooling, the white solid was filtered, washed with ethanol and dried under vacuum to give the desired product. The product was used in the next reaction without purification.
1.2.3 Preparation of compound 3
Compound 3 was prepared in a manner similar to that of Pozharskii[17]. Iodomethane(3.02 g, 0.021 mol) was added in one portion to a solution of compound 2(0.01 mol) in dimethylformamide(15 mL) at 100 ℃. The mixture was heated for 12 h, giving a yellow precipitate. After cooling, the yellow solid was filtered and dried under vacuum to give the desired salt, which was used in the next reaction without purification.
1.2.2 Preparation of compound 2
Compound 2 was prepared by a modified procedure of Pozharskii[17]. Iodomethane(3.02 g, 0.021 mol) was added dropwisely over a period of 10 min to a solution of compound 1(0.01 mol) in 10 mL of dimethylformamide at 100 ℃. The mixture was heated for 12 h, giving a yellow precipitate. After cooling, the yellow solid was filtered and suspended in 120 mL of water. Aqueous ammonium hydroxide was added slowly with vigorous stirring till the solution was slightly basic to litmus. During the course of addition, a yellow-green solid appeared. The solid was filtered, washed with water, and dried under vacuum to give the desired product. The product was used in the next reaction without purification.
1.2.5 Preparation of compound 5
1, 8-Naphthalenediamine(3.72 g, 0.02 mol) and m-phthalaldehyde(0.01 mol) were added to 100 mL dichloromethane, followed by moderate molecular sieves. The mixture was stirred at room temperature for 36 h and monitored by TLC, giving a large amount of white precipitate. After the reaction, the solid was filtered, washed with dichloromethane and dried under vacuum to give the desired product. The product was used in the next reaction without purification.
2 Results and Discussion
2.1 Crystal data
The molecular structures of compounds 4a and 6 are shown in Fig. 1. The selected bond lengths and bond angles are listed in Table 2.
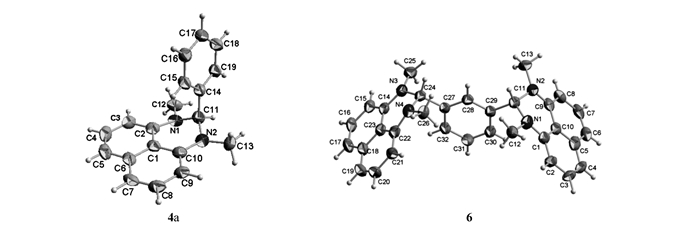 Figure 1.
Molecular structures of compounds 4a and 6
Table 2.
Selected bond lengths(nm) and bond angles(°) for compounds 4a and 6
Figure 1.
Molecular structures of compounds 4a and 6
Table 2.
Selected bond lengths(nm) and bond angles(°) for compounds 4a and 6

As shown in Table 2, in the structure of compound 4a, the bond angles of N(1)—C(11)—N(2), N(1)—C(11)—C(14) and N(2)—C(11)—C(14) are 109.27(14)°, 112.21(14)° and 114.90(14)° in turn, which indicates the sp3 hybridization state of C(11) atom. The new-formed three-membered heterocyclic ring is not coplanar obviously. Naphthalene and the plane through C(11), N(1), N(2) atoms are inclined 32.181° to each another and the atom C(11) deviates from the naphthalene molecular plane by 0.03120 nm. As a dimer of compound 4a, the compound 6 looks like a letter "M" and C(11) and C(24) atoms in the structure of compound 6 belong to the sp3 hybridization state.
2.2 Biological activity
The biological activities of the samples were tested and its fungicidal activities were listed in Table 3. The fungicidal activity against the typical fungi commonly occurring in the Chinese agro-ecosystem was detected at 50 mg/L in vitro according to the reported fungi growth inhibition method [22]. The results showed that all the tested compounds 4b~4g and 6 had broad spectrum of fungicide against the tested fungi. Especially for GZ, BC, PP and SS, perimidine derivatives showed better antibacterial activity. And the compounds 4b, 4c, 4d, 4e, 4f and 6 had obvious fungicidal activity against PP with growth inhibition of 74.72%, 76.74%, 74.42%, 72.05%, 69.74% and 67.44%, respectively. The compounds 4b, 4d and 6 had fungicidal activity against SS with growth inhibition of 70.27%, 67.57% and 72.97%, respectively. In addition, test data indicated that there was no obvious regulation between fungicidal activity and the different substituents on the benzene ring.
Table 3.
Fungicidal activity of the perimidine derivatives(%)

3 Conclusions
In summary, we have successfully prepared compounds 4a~4g and 6, which were characterized by 1H NMR. Compounds 4a and 6 were investigated by single-crystal X-ray diffraction. The biological activities of the target compounds were tested and the results showed that 4b~4g and 6 had broad spectrum of fungicide against the tested fungi. Especially for GZ, BC, PP and SS, perimidine derivatives showed very good antibacterial activity, which reveals that perimidine derivatives have great potential applications in antibacterial fields in the future.
-
-
[1]
Hashim J A, Kezhal M S. Synthesis, Characterization and Biological Activity of 2-Aryl-2, 3-dihydro-1H-Perimidine[J]. Res Pharm Biotechnol, 2014, (1): 1-6.
-
[2]
Jung I G, Son S U, Park K H. Synthesis of Novel Pd-NCN Pincer Complexes Having Additional Nitrogen Coordination Sites and Their Application as Catalysts for the Heck Reaction[J]. Organometallics, 2003, (23): 4715-4720.
-
[3]
Vanden E J J, Delfosse F, Mayence A. Old Reagents, New Results-aromatization of Hantzsch 1, 4-Dihydropyridines with Manganese-dioxide and 2, 3-Dichloro-5, 6-dicyano-1, 4-Benzoquinone[J]. Tetrahedron, 1995, (20): 5813-5818.
-
[4]
Hendrickson J B, Hussoin M S. Seeking the Ideal Dehydrating Reagent[J]. J Org Chem, 1987, (18): 4137-4139.
-
[5]
Deady L W, Rodemann T J. Synthesis of Perimidine and Fused Perimidine Derivatives from Reaction of 1, 8-Naphthalenediamine with an Iminoisocoumarin[J]. Heterocycl Chem, 1998, (6): 1417-1419.
-
[6]
Mueller-Westerhoff U T, Vance B, Dong I Y. The Synthesis of Dithiolene Dyes with Strong Near-IR Absorption[J]. Tetrahedron, 1991, (6): 909-932.
-
[7]
Starshikov M, Pozharskii F T. Synthesis of 2-(5-Halogeno-2-furyl)-2, 3-Dihydroperimidines[J]. Chem Heterocycl Compd, 1973, (7): 922-924.
-
[8]
Przemyslaw M, Anu C, Christopher R M. Optical Properties of Spiroconjugated Charge-transfer Dyes[J]. J Am Chem Soc, 1996, (6): 1471-1481.
-
[9]
Starshikov N M, Pozharskii A F. Heterocyclic Analogs of Pleiadiene[J]. Chem Heterocycl Compd, 1978, (10): 1413-1417.
-
[10]
Sokolov V I, Pozharskii A F, Kashparov I S. Heterocyclic Analogs of Pleiadiene[J]. Chem Heterocycl Compd, 1974, (4): 558-561.
-
[11]
Bu X, Deady L W, Finlay G. J. Synthesis and Cytotoxic Activity of 7-Oxo-7H-dibenz[f, ij]isoquinoline and 7-Oxo-7H-benzo[e]perimidine Derivatives[J]. J Med Chem, 2001, (12): 2004-2014.
-
[12]
Gong X W, Li X, Li W L. Synthesis and Crystal Structure of (E)-4-(Benzyloxy)-2-(cinnamoyl-oxy)-N, N, N-trimethyl-4-oxobutan-1-Aminium Chloride as a Double-prodrug[J]. Chinese J Struct Chem, 2008, (2): 177-182.
-
[13]
Ye J, Sun X X, Qiu S Y. Synthesis, Crystal Structure and Fungicidal Activity of N-(4-tert-buty)-5-(1, 2, 4-triazol-1-yl)thiaz-ol-2-yl) Propionamide[J]. Chinese J Struct Chem, 2014, (3): 429-433.
-
[14]
Fu C R, Pei J, Ning Y. Synthesis and Insecticidal Activities of Novel Pyrazole Oxime Ether Derivatives with Different Substituted Pyridyl Rings[J]. Pest Manage Sci, 2013, (8): 1207-1214.
-
[15]
Mobinikhaledi A, Sasani F, Hamta A. Molecular Iodine Catalyzed Synthesis of Some Biologically Active Dihydroperimidines[J]. Bulg Chem Commun, 2013, (3): 353-356.
-
Yuan L, Li Z Y, Zhang M. Synthesis, Crystal Structure and Anti-fungal Activity of 2-(4-Chlor-ophenyl)-1, 3-dimethyl-[JP]2, 3-dihydro-1H-Perimidine[J]. Chinese J Struct Chem, 2016, (8): 1181-1185.
-
[17]
Sokolov V I, Ardashev B I, Kashparov I S. Heterocyclic Analogs of Pleiadiene X.Reaction of 2-Substituted Quaternary Perimidinium Salts with Alkali[J]. Khim Geterosikl Soedin, 1973, (6): 782-784.
-
[18]
Sheldrick G M. SHELXS-97. Program for Solution of Crystal Structures[CP]. University of Gottingen, Germany, 1997.
-
[19]
Sheldrick G M. SHELXL-97. Program for Crystal Structure Refinement[CP]. University of Gottingen, Germany, 1997.
-
[20]
Fan Z J, Yang Z K, Zhang H K. Synthesis, Crystal Structure, and Biological Activity of 4-Methyl-1, 2, 3-thiadiazole Containing 1, 2, 4-Triazolo(3, 4-b)(1, 3, 4) thiadiazoles[J]. J Agric Food Chem, 2010, (5): 2630-2636.
-
[21]
Li F Y, Zhu Y J, Fan Z J. Synthesis, Crystal Structure and Biological Activity of 2-(1-(3-Bromo-1-(3-chloropyridin-2-yl)-1H-pyrazole-5-carbonyl)piperidin-4-yl)-N-isopropyl-1, 3-thiazole-4-Carboxamide[J]. Chinese J Struct Chem, 2015, (5): 659-666.
-
[22]
Fan Z J, Shi Z G, Zhang H K. Synthesis and Biological Activity Evaluation of 1, 2, 3-Thiadiazole Derivatives as Potential Elicitors with Highly Systemic Acquired Resistance[J]. J Agric Food Chem, 2009, (10): 4279-4286.
-
[1]
-


 下载:
下载:


 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 1
- 文章访问数: 454
- HTML全文浏览量: 58
 下载:
下载:
