引用本文:
范一强, 王玫, 高峰, 庄俭, 唐刚, 张亚军. 液滴微流控系统在数字聚合酶链式反应中的应用研究进展[J]. 分析化学,
2016, 44(8): 1300-1307.
doi:
10.11895/j.issn.0253-3820.160074

Citation: FAN Yi-Qiang, WANG Mei, GAO Feng, ZHUANG Jian, TANG Gang, ZHANG Ya-Jun. Recent Development of Droplet Microfluidics in Digital Polymerase Chain Reaction[J]. Chinese Journal of Analytical Chemistry, 2016, 44(8): 1300-1307. doi: 10.11895/j.issn.0253-3820.160074
Citation: FAN Yi-Qiang, WANG Mei, GAO Feng, ZHUANG Jian, TANG Gang, ZHANG Ya-Jun. Recent Development of Droplet Microfluidics in Digital Polymerase Chain Reaction[J]. Chinese Journal of Analytical Chemistry, 2016, 44(8): 1300-1307. doi: 10.11895/j.issn.0253-3820.160074
液滴微流控系统在数字聚合酶链式反应中的应用研究进展
摘要:
数字聚合酶链式反应(PCR)技术近年来发展迅速。与以实时荧光定量PCR为代表的传统PCR技术相比,数字PCR技术显著提高了定量分析的精确度和灵敏度。数字PCR的快速发展与近年来微流控技术在数字PCR技术中的广泛应用有着密切的联系。早期的研究和商业化产品使用的是大规模集成流路微流控芯片,加工过程复杂且价格高昂。近年来,液滴微流控芯片被应用到数字PCR技术中,它可以在短时间内产生102~107个微液滴,每一个微液滴都是最多只含有一个目的基因片段的PCR反应器。PCR扩增后,通过对单个微液滴的观察计数,就可以获得绝对定量的分析数据。本文综述了不同种类的液滴微流控系统在数字PCR技术中的应用,以及液滴数字PCR微流控芯片在生物、医药、环境等领域的应用。
English
Recent Development of Droplet Microfluidics in Digital Polymerase Chain Reaction
Abstract:
Digital polymerase chain reaction (PCR) has been experiencing a rapid development during the past few years. Comparing with the traditional real-time quantitative PCR (RT-qPCR), using the same primer and probe, the accuracy for the absolute quantification of target gene is significantly improved. The development of digital PCR is directly related to the development of microfluidics. The integrated fluid circuit is an early combination of the microfluidics and digital PCR, which has a complicated fabrication process with high cost. Recently, researchers are trying to apply the droplet microfluidics in digital PCR, and the droplet microfluidic chip is able to generate millions of droplets within a short time. Each of these droplets containing no more than one target gene is a reaction chamber during the amplification process. After amplification, each droplet is tested to achieve the absolute quantification of the target gene. This paper reviews the recent progresses of droplet digital PCR, and the applications of droplet digital PCR in biological, medical and environmental fields.
-
Key words:
- Digital polymerase chain reaction
- / Droplet microfluidics
- / Genetic testing
- / Gene sequencing
- / Review
-
1. 引言
早在1992年,已有学者提出了数字聚合酶链式反应(Digital PCR, dPCR)的概念[1],并且指出其在DNA的定量分析方面具有显著优势。但是初期的dPCR技术发展十分缓慢,这是由于如果使用传统的96孔板或者384孔板进行PCR扩增,需要数个多孔板同时进行扩增,不仅操作复杂,且实验样品和试剂的消耗量非常大[2]。在当时的条件下,研究者很难开展数字PCR的研究。此外,在DNA定量研究方面,由于实时荧光定量PCR (Real-time quantitative PCR, RT-qPCR)[3, 4]等技术的发展和广泛应用,也使得dPCR技术的发展缓慢。
随着微流控技术的出现和近年来的高速发展,研究者尝试将微流控技术与数字PCR技术结合起来。与微流控芯片结合后的dPCR技术的灵敏度和精确度有了很大提高[5],在单细胞研究[6, 7]、基因测序[8]、医学诊断[9-11]、环境监测[12, 13]、食品安全[14-16]等领域得到了越来越多的应用,展现出了广泛的应用潜力。近年来,还涌现出一批商业化的dPCR检测设备。本文综述了近年来dPCR技术及液滴微流控芯片的发展状况,并介绍了dPCR技术在医学、生命科学等领域的实际应用情况。
2. dPCR原理
传统的实时定量PCR以指数形式体现定量信息,而dPCR技术所能提供的DNA量化信息是以线性的数字化形式体现的。dPCR的原理如图 1所示[17],将含有目标基因、引物、聚合酶等的溶液稀释后,根据实验需要被分成102~107份不等的溶液(反应单元),每份溶液中至多只含有一个目的基因拷贝。这些溶液单元经过与传统的PCR技术相同的热循环扩增后,对每份溶液进行检测。如果含有目的基因,则计为“1”(阳性反应);不含有目的基因,则计为“0”(阴性反应),这些代表阳性反应的数字“1”和代表阴性反应的数字“0”类似于数字电路中的通和断两种状态,这也是dPCR名称的由来。最后通过对阳性反应的单元计数可以获知扩增之前目的基因的绝对数量。考虑到有可能出现扩增前一个反应单元里有两个或以上目的基因的情况,使用泊松分布进行校正可以进一步增加对扩增前目的基因计量的准确度。
图 1
与传统的PCR技术相比,由于热循环和荧光探针标记等技术已经较为成熟,dPCR主要的技术难点体现在对稀释后的反应溶液进行大规模等分的技术上。在对稀释后的溶液进行等分方面,最初采用了96孔或384孔板的方法,但这种方法试剂消耗量巨大(每孔约5μL),且可实现的反应单元数量较少。后来尽管在反应单元的数量上有所增加,如Shen等[18]在玻璃基底加工了1280个反应单元,但仍存在溶液等分不能自动完成和反应单元数量较少的问题。直到微流控技术被应用到dPCR领域后,反应单元的数量才得到了爆发式的增长。关于微流控技术在dPCR领域的应用,本文后续部分会详细介绍。
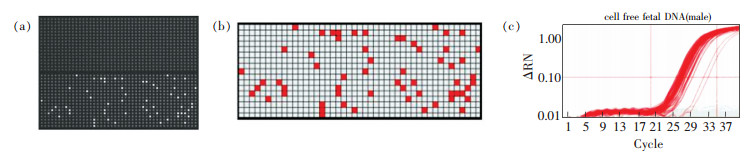
为了进一步说明dPCR技术的原理,图 2显示了dPCR技术在胎儿早期疾病诊断中的应用。图 2a是检测过程中一组12块多孔板中的1块,具有765个反应单元,每个反应单元的容积为6 nL,含有目标基因、引物、荧光探针等的反应溶液通过微流控技术被均匀送入各个反应单元中。经过PCR扩增后,由TaqMan探针标记的含有目标基因的反应单元发出荧光,可以很容易地观察到。经过计算机图像处理,转换为图 2b所示的热点图(Heat map), 每个红点代表一个含有目标基因的反应单元,通过热点图就可以准确分析出目标基因的数量。图 2c是作为参考的dPCR的扩增曲线,需要指出的是,dPCR对目标基因的测量并不需要扩增曲线的帮助,而在实时荧光定量PCR检测中,对目标基因扩增数量的检测直接依赖于与标准曲线的对比,由于标准曲线与实际样品扩增效率不同等原因,导致荧光实时定量PCR检测的准确度偏低。Hindson[19]和Whale[20]等对实时荧光PCR技术和dPCR技术进行了系统对比,结果表明,相比于实时荧光定量PCR,dPCR对目标基因定量检测的灵敏度、精度、检测时间等方面有着非常显著的优势。在检测精度方面,dPCR与实时荧光PCR相比,相对标准偏差(RSD)显著减小,测量精密度提高[19];在检测时间方面,如果保持与荧光PCR相同的精度,dPCR的日检测量增加了7倍[19]。
图 2
 图 2 采用dPCR技术在微流控阵列中对采自孕妇血浆中的胎儿DNA进行扩增[10]。(a)同一个芯片阵列在扩增前后的对比(b)阳性反应的热点图(c)扩增过程的指数图Figure 2. Digital PCR amplification of fetal DNA from maternal plasma analyzed in a digital array microfluidics chip[10]. (a) Same chip before and after PCR amplification process, (b) Heat map for the positive reactions, (c) Exponential plot for the amplification process
图 2 采用dPCR技术在微流控阵列中对采自孕妇血浆中的胎儿DNA进行扩增[10]。(a)同一个芯片阵列在扩增前后的对比(b)阳性反应的热点图(c)扩增过程的指数图Figure 2. Digital PCR amplification of fetal DNA from maternal plasma analyzed in a digital array microfluidics chip[10]. (a) Same chip before and after PCR amplification process, (b) Heat map for the positive reactions, (c) Exponential plot for the amplification process早期的微流控技术与dPCR的结合应用体现在以集成流路微流控芯片(Integrated fluid circuit, IFC)[21]为基础的dPCR技术。这种微流控芯片采用多层光刻技术加工而成,具有非常复杂的流路结构以及气动的微泵与微阀,通过微泵和微阀将溶液送入反应腔阵列,其加工过程复杂,成本非常高,且受到加工技术和成本的限制,IFC芯片可以实现的反应单元数量一般不超过10000个[22]。
近年来,液滴微流控技术的发展给dPCR带来了新的发展机遇。采用液滴微流控技术,经过高度稀释的含有模板DNA和引物等的反应溶液可以在微流控芯片中很容易地被分成102~107小液滴[23],每个体积只有纳升[24]甚至皮升[25]的小液滴都是只含有最多一个目的基因的微型反应器,经过PCR扩增后,通过荧光检测,可以很直观地定量分析出原始溶液中目的基因的拷贝数量。
与IFC芯片相比,液滴微流控芯片的优势不止体现在反应单元的数量,其最大优势在于成本方面:IFC芯片的加工采用接近纳米尺度的多层光刻技术,成本非常高;而液滴微流控芯片大多只有一层结构,且加工尺度在微米级,因此大大降低了芯片的加工成本(微流控芯片的加工成本占芯片总成本的大部分),这对于dPCR技术的发展至关重要。
3. 液滴微流控技术
3.1. 微流控系统中微液滴的产生方法
如前所述,微流控技术对于dPCR发展的贡献主要在于可以大规模增加反应单元的数量,通过微流控技术,高效精确地将样本溶液分割为102~107份。微流控技术对样本溶液的分割,最初是由气动微泵、微阀控制分割样本溶液[21]的IFC芯片,不仅结构较为复杂,加工难度也较大,可以分割的份数也非常有限,单块芯片的成本非常高[26]。近年来,研究者开始使用微流控系统产生大量微液滴的办法实现对样本溶液的分割[27, 28],每个产生的含有目的基因、探针和引物等的微液滴都可以作为一个独立的反应单元进行PCR扩增,而微滴产生的速度、数量、体积等均可以通过微流控系统精确控制。相比于IFC芯片,液滴微流控芯片的加工过程简单,成本低廉,代表了未来微流控与dPCR结合的发展方向。下面将结合微液滴在dPCR领域的应用,简要介绍使用微流控系统产生及操控微液滴的技术。
通过微流控系统产生大量应用于dPCR技术的微液滴,最常用的方法是乳化技术(Emulsion),即两种不相溶的液体混合时,一种液体会以微滴的形式分散在另一种溶液中。微流控芯片可以在微尺度上对一种溶液(分散相)在另外一种不相溶的溶液(连续相)中产生微液滴的过程进行精确控制,继而获得大量体积在纳升甚至皮升级,且大小均一的微液滴。
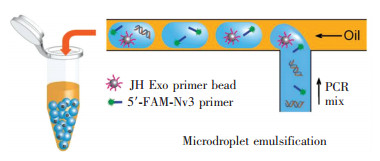
图 3显示了使用液滴dPCR技术对癌症基因进行筛选和测序的应用研究[28]。如图 3所示,在一个T形(T-junction)的微通道中,黄色代表油,蓝色代表包含引物、荧光探针以及目标基因等的水溶液。由于两种液体不相溶,在微通道的液体流动过程中,油在微通道内在向左侧流动的过程中对水溶液不断进行挤压,作为分散相的水溶液在连续相油的不断挤压中被最终剪断,形成了一个微液滴(本例中体积为2.5 nL)。分散后的微液滴含有至多一个目的基因以及引物、荧光探针等,分散后的液滴可以使用传统的PCR技术进行扩增,由于油的阻隔,在扩增过程中,每个微液滴都是一个相对独立的反应单位。
图 3
除了图 3所展示的T形微通道的液滴产生系统,类似的还有Y形[29]、十字形[30]等多种微通道结构,都可以用于产生微液滴。除了乳化法之外,还可以采用流动聚焦(Flow-focusing)等方法[31]产生单/双/多层包裹的微液滴(Single/double/multiple emulsion)。采用微流控技术产生微液滴的大小与微通道的尺寸及连续相和分散相的流速、粘度等都有直接关系[32]。现阶段使用微流控技术可以产生的微液滴的体积范围从0.05 pL到1 nL不等,对应的微液滴直径为5~120μm[33],实际应用中可以灵活选择。文献[29]中采用的是以油包水(W/O)方式产生的微液滴,根据实验需要,同样的微流控系统还可以产生水包油(O/W)的微液滴,甚至气体包水(W/G)的微液滴。
在连续相液体的选择方面,多采用矿物油(Mineral oil),对微液滴形状的保持和微液滴间的阻隔起到了重要作用,且对微液滴中参与PCR扩增的生物大分子、酶等的活性和结构没有影响[14]。为了增强液滴产生的稳定性和控制分散相微液滴的大小,有的研究者在矿物油中加入了0.5%~3.0%的表面活性剂[34, 35]。在水或油中添加表面活性剂对微液滴的稳定起到了重要作用,且未发现毒副作用。未来的研究中,可以通过改变微通道表面的物理化学性质等方法(物理/化学修饰),增加微流控系统产生微液滴的稳定性,从而摆脱对价格昂贵的表面活性剂的依赖,减少对溶液的污染。
微流控芯片材料的选择与其应用环境有直接关系。目前,在dPCR领域,无论是IFC芯片,还是液滴微流控芯片,通常使用聚二甲基硅氧烷[14, 36](PDMS)材质的微流控芯片。PDMS具有良好的生物相容性和透气性,且微加工性能良好[37],但在某些有机溶剂的作用下会发生溶解和变形。根据目前微流控技术发展的总体趋势,未来非常有可能产生基于热塑性塑料(如:PMMA、PS、PC等),甚至纸基等低成本微流控dPCR芯片。
3.2. 微液滴的高通量生成技术
液滴微流控芯片在dPCR领域的应用是利用了液滴微流控系统可以迅速产生大量的微液滴的优势。近年来,在产生高通量微液滴的微流控芯片方面进行了大量研究,目前,单一微液滴产生装置的液滴产生速度可以高达10 kHz[38],如果将多块微流控芯片并行使用,或者在一块微流控芯片上加工数十到数百个微液滴产生装置,还可以极大提升液滴的产生速度。Nisisako等在[39]一块圆形的玻璃基底上呈辐射状排布了128个Y形微液滴产生器,每小时可产生1 L粒径为96.4μm的微液滴。
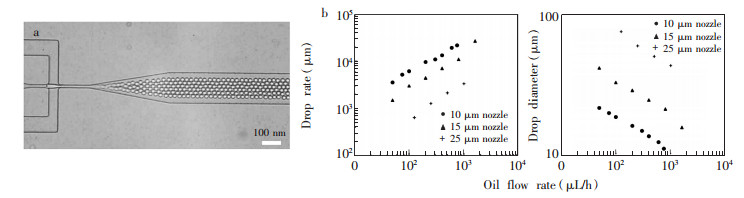
图 4展示了Holtze等[40]制作的高速微液滴发生微流控芯片,芯片采用了软光刻的方法,使用传统紫外光刻技术对SU-8进行曝光冲洗后,利用SU-8作为模具,对PDMS进行倒模,最后将PDMS键合在玻璃片上制成微流控芯片。他们不仅在连续相的油中添加了表面活性剂,还在分散相的水中也添加了具有良好生物相容性的表面活性剂(PFPE-PEG共聚物),从而极大提高了微液滴高速产生时的稳定性,液滴的产生速度可以达到30 kHz(图 4a)。从图 4b可见,随着微通道内油流量上升,微液滴的产生速度也迅速上升,产生的微液滴的直径随之变小。为了验证PFPE-PEG共聚物作为表面活性剂的生物兼容性,在微液滴中进行了质粒DNA体外编译β-Galactosidase酶的实验,获得了满意的效果。
图 4
 图 4 液滴产生过程中的稳定性[40](a)使用流动聚焦的方法从25μm的喷嘴中产生的微液滴,(b)在水流速不变的情况下,微液滴产生的频率和大小与油流速之间的关系Figure 4. Droplet stability during formation[39]. (a)Droplets are generated by flow-focusing from a 25-mm nozzle; (b) Droplet production frequency and drop diameter as a function of oil flow rate at constant flow rate of the water phase
图 4 液滴产生过程中的稳定性[40](a)使用流动聚焦的方法从25μm的喷嘴中产生的微液滴,(b)在水流速不变的情况下,微液滴产生的频率和大小与油流速之间的关系Figure 4. Droplet stability during formation[39]. (a)Droplets are generated by flow-focusing from a 25-mm nozzle; (b) Droplet production frequency and drop diameter as a function of oil flow rate at constant flow rate of the water phase值得注意的是,高通量的微液滴产生技术对微流控芯片系统以及扩增后的液滴荧光检测都提出了更高的要求。首先,产生高通量液滴的微流控芯片对反应液体进样的稳定性和连续性都提出了更高要求,这种情况下,反应液体进样常需要通过价格高昂的恒压泵实现。其次,高通量的液滴产生微流控系统对芯片的键合也提出了更高要求,内部的高压液体对芯片的键合强度要求更高。最后,对液滴的荧光检测的速度方面也提出了更高要求,检测系统不仅需要在极短时间内对微流控芯片中按顺序排布流动的微液滴进行荧光检测,还需要高速的数据处理和统计能力。
4. dPCR与液滴微流控芯片的结合和应用
目前,基于液滴微流控芯片的dPCR(Droplet digital PCR, ddPCR)已被广泛应用于疾病检测[41, 42]、基因诊断[43]、基因测序[44]、食品安全[45]等多个领域。
在疾病检测和基因诊断方面,Heredia等[46]采用液滴dPCR技术对乳腺癌肿瘤细胞中的表皮生长因子受体(HER2)的表达水平进行了研究。与之前研究者的结果相比,采用dPCR显著提高了对HER2表达水平的准确度,为研究HER2表达水平与乳腺癌发病之间的关系提供了准确数据。dPCR技术在疾病诊断领域的应用远不止于乳腺癌领域,在肺癌[47]、结肠癌[48]等疾病的检测领域,研究者也利用dPCR技术对其相应的标志物(如:EGFR、KRAS等)进行了检测,从而实现了在基因水平上对疾病诊断的目的。利用dPCR技术的高灵敏度,还可以对微量的游离在血液中的肿瘤导致的CNAs进行检测,对于癌症的早期诊断有着重要的意义。实验表明,利用dPCR技术,可以达到在106个野生型中检测出一个突变基因的灵敏度[49]。Contente-Cuomo等使用液滴dPCR对微量的游离在细胞外可作为癌症标志物的DNA片段进行了检测[50],验证了在血液的复杂背景中,利用dPCR技术对循环肿瘤标志物检测的可行性。
在环境工程和食品安全领域,Bian等[14]采用经过油饱和处理的基于PDMS材料的微流控芯片,对饮用水中E. coliO157和L. monocytogenes两种细菌进行了dPCR检测(图 5),分别用两种不同颜色的荧光探针标记两种细菌,PCR扩增后可以同时对两种颜色荧光微液滴进行检测计数,从而得到两种细菌在初始溶液中的绝对定量结果[14],用这种方法对饮用水的检测精度可以达到10 CFU/mL[15]。在食品安全领域,为了适应欧盟对转基因农作物的严格要求,Dobnik等[51]提出并验证了利用液滴dPCR技术对欧盟境内种植的12种转基因玉米进行快速检测的方法。Dany等[45]也使用dPCR—实时荧光PCR联用技术对食品、饲料、种子中的转基因成分进行了测定,在提高精度的同时也降低了检测成本。
图 5
 图 5 使用液滴dPCR对E. coliO157和L. monocytogenes同时进行检测[14](a)微流控芯片的加工(b)产生微液滴(c)在微流控芯片上进行扩增和使用通过荧光分析结果Figure 5. Droplet digital PCR workflow for simultaneous detection of E. coli O157 and L. monocytogenes[14] (a)Fabrication of microfluidic chip; (b) generation of droplets; (c) On-chip amplification followed by fluorescence read out
图 5 使用液滴dPCR对E. coliO157和L. monocytogenes同时进行检测[14](a)微流控芯片的加工(b)产生微液滴(c)在微流控芯片上进行扩增和使用通过荧光分析结果Figure 5. Droplet digital PCR workflow for simultaneous detection of E. coli O157 and L. monocytogenes[14] (a)Fabrication of microfluidic chip; (b) generation of droplets; (c) On-chip amplification followed by fluorescence read out在液滴微流控技术与dPCR的结合应用方面也有一些商业化产品,如Bio-Rad公司的QX系列液滴dPCR检测系统,其中QX100系统中的液滴微流控芯片可以在一次分析中将样品溶液分成11.2万~12.8万个小液滴,每块芯片的价格仅为24美元。在QX100系统中,使用了前文提到过的十字形的微流道,分散相溶液在连续相油的挤压中不断产生小液滴,通过数个液滴产生微流控芯片的并行协作,在短时间内产生了大量的小液滴,每个小液滴就是一个反应容器;PCR热循环扩增结束后,又使用微流控技术,使小液滴依次通过检测装置对其进行荧光检测,完成计数过程。与之相似,RainDance公司的Rraindrop系统附带的微流控芯片的8个进样通道同时工作时,最高可将样品溶液分为8000万个pL级的微液滴,每块微流控芯片的成本也只有80~240美元。相比之下,Fluidigm公司BioMark系统使用的基于IFC技术的微流控芯片中的12个反应单元阵列加起来也仅有9180个反应单元,每块芯片价格却高达400美元[17]。由此可见,从单个芯片的价格和对反应溶液分割的份数来看,液滴微流控芯片相比于IFC芯片都拥有巨大的优势。
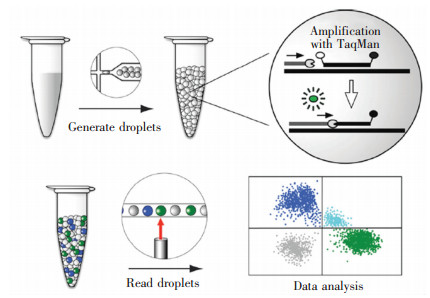
典型的商业化液滴dPCR系统的工作原理如图 6所示,含有目的基因、Taq聚合酶、引物、分子信标(如:TaqMan等)的溶液经过微流控芯片,被分为数百万个微液滴,分散的微液滴被收集在离心管中进行PCR热循环扩增,扩增的同时,通过荧光探针/分子信标标记目的基因,最后,微液滴被重新送入微流控芯片中,微液滴依次通过荧光传感器进行检测,最后得到数据分析图。值得一提的是,现阶段微液滴一般可以通过荧光探针标记两种颜色,如果对荧光探针的强度进行控制,基于两种颜色的探针最多可以达到12个标记的多重分析[53]。
图 6
5. 结语和展望
与传统的依靠标准曲线和循环阈值进行定量检测的实时荧光PCR技术相比,在目标基因的定量精度和灵敏度方面,dPCR具有非常明显优势:通过对溶液的高度稀释和分割,每份溶液中都至多含有一个目标基因,在扩增后,通过荧光检测,可以准确定量分析出原始溶液中的目标基因数量。
dPCR技术在基因工程、疾病检测以及环境工程等领域得到了较为广泛的应用,无疑是未来PCR技术的发展方向。液滴dPCR是近年来兴起的一项新dPCR技术,是微流控技术与dPCR结合产生的新成果。相比于基于IFC的dPCR芯片,液滴dPCR对反应溶液的分割数量增加了上千倍,可以容易地将反应溶液分割为数百万份含有至多一个目标基因的微液滴,大大提高了定量分析精密度,不仅推动了dPCR在更多领域的应用,且显著降低了dPCR技术的成本。
目前,包括液滴dPCR在内的整个dPCR技术还处于发展初期,与液滴dPCR相关的液滴微流控芯片技术、多重荧光探针、反应后微液滴的高速检测等相比还有很大发展的空间。在液滴微流控芯片技术方面,其技术难点在于大规模高通量的液滴产生技术,高通量往往是通过增加芯片中的液体流动速度和采用数块微流控芯片并行运作实现的,这对芯片的设计、微通道的加工、芯片的键合等都提出了更高的要求,是未来技术攻关的重要难点之一。在多重荧光探针的使用方面,多数实验室研究和商业化产品使用的都是1~2个荧光探针,可以实现对1~2个目标基因的同时检测。在未来的dPCR技术发展中,提高荧光探针数量必然是发展趋势之一,在一次dPCR检测中对多个目标基因进行定量分析,大大提高效率,降低成本,对医学检测等领域的应用有着重要意义。前面提到的高通量液滴产生技术和多重荧光探针技术也都对PCR扩增后的荧光检测系统提出了更高的要求,在非常短的时间内,不仅需要荧光检测系统对液滴是否具有荧光做出判定,还需要对荧光的颜色做出准确判断,所以,未来dPCR技术发展中高速荧光检测和信号处理也是亟待解决的难题。
此外,值得重点关注的是液滴dPCR技术的高成本问题。目前,为了保证内部微流道洁净,只能一次性使用的液滴微流控芯片的成本仍然过高,使得整个液滴dPCR技术的检测成本高昂,影响了液滴dPCR技术的普及。微流控芯片成本的绝大部分源自微加工技术和芯片材料,相信未来随着微流控技术的高速发展,各种低成本的微加工方法的不断涌现和各类低成本材料的使用,会在很大程度上降低液滴dPCR技术的成本。从降低成本方面考虑,研发应用于液滴dPCR技术中的高通量、低成本微流控芯片也必将是未来的发展方向之一。
-
-
[1]
Sykes P,Neoh S,Brisco M,Hughes E,Condon J,Morley A A.Biotechniques,1992,13(3):444-449
-
[2]
Vogelstein B,Kinzler K W.P.Natl.Acad.Sci.USA,1999,96(16):9236-9241
-
[3]
Chen C,Ridzon D A,Broomer A J,Zhou Z,Lee D H,Nguyen J T,Barbisin M,Xu N L,Mahuvakar V R,Andersen M R.Nucleic Acids Res.,2005,33(20):e179-e179
-
[4]
Bustin S,Benes V,Nolan T,Pfaffl M.J.Mol.Endocrinol.,2005,34(3):597-601
-
[5]
ZHU Qiang-Yuan,YANG Wen-Xiu,GAO Yi-Bo,YU Bing-Wen,QIU Lin,ZHOU Chao,JIN Wei,JIN Qin-Han,MU Ying.Chem.J.Chinese Universities,2013,34(3):538-544朱强远,杨文秀,高一博,于丙文,邱琳,周超,金伟,金钦汉,牟颖.高等学校化学学报,2013,34(3):538-544
-
[6]
Warren L,Bryder D,Weissman I L,Quake S R.P.Natl.Acad.Sci.USA,2006,103(47):17807-17812
-
[7]
Shen F,Du W,Kreutz J E,Fok A,Ismagilov R F.Lab Chip,2010,10(20):2666-2672
-
[8]
White R A,Blainey P C,Fan H C,Quake S R.BMC Genomics,2009,10(1):116
-
[9]
Fan H C,Blumenfeld Y J,El-Sayed Y Y,Chueh J,Quake S R.Am.J.Obstet.Gynecol.,2009,200(5):541-547
-
[10]
Zimmermann B G,Grill S,Holzgreve W,Zhong X Y,Jackson L G,Hahn S.Prenat.Diagn.,2008,28(12):1087-1093
-
[11]
Malapelle U,De Luca C,Vigliar E,Ambrosio F,Rocco D,Pisapia P,Bellevicine C,Troncone G.J.Clin.Pathol.,2016,69(5):454-457
-
[12]
Ottesen E A,Hong J W,Quake S R,Leadbetter J R.Science,2006,314(5804):1464-1467
-
[13]
Tadmor A D,Ottesen E A,Leadbetter J R,Phillips R.Science,2011,333(6038):58-62
-
[14]
Bian X,Jing F,Li G,Fan X,Jia C,Zhou H,Jin Q,Zhao J.Biosens.Bioelectron.,2015,74:770-777
-
[15]
Coudray-Meunier C,Fraisse A,Martin-Latil S,Guillier L,Delannoy S,Fach P,Perelle S.Int.J.Food Microbiol.,2015,201:17-26
-
[16]
DONG Lian-Hua,ZHANG Ling,JIANG Jun,WANG Jiang-Nan,WANG Jing,CHEN Wei-Jun.Chinese J.Anal.Chem.,2015,43(3):319-324董莲华,张玲,姜君,王江南,王晶,陈唯军.分析化学,2015,43(3):319-324
-
[17]
Baker M.Nat Methods,2012,9(6):541-544
-
[18]
Shen F,Du W,Kreutz J E,Fok A,Ismagilov R F.Lab Chip,2010,10(20):2666-2672
-
[19]
Hindson C M,Chevillet J R,Briggs H A,Gallichotte E N,Ruf I K,Hindson B J,Vessella R L,Tewari M.Nat.Methods,2013,10(10):1003-1005
-
[20]
Whale A S,Huggett J F,Cowen S,Speirs V,Shaw J,Foy C A,Scott D J.Nucleic Acids Res.,2012,40(11):e82
-
[21]
Ramakrishnan R,Jian Q,Jones R C,Weaver L S.Method Mol.Biol.,2013,949:423-431
-
[22]
Devonshire A S,Sanders R,Wilkes T M,Taylor M S,Foy C A,Huggett J F.Methods,2013,59(1):89-100
-
[23]
Heyries K A,Tropini C,VanInsberghe M,Doolin C,Petriv I,Singhal A,Leung K,Hughesman C B,Hansen C L.Nat.Methods,2011,8(8):649-651
-
[24]
Hindson B J,Ness K D,Masquelier D A,Belgrader P,Heredia N J,Makarewicz A J,Bright I J,Lucero M Y,Hiddessen A L,Legler T C,Kitano T K,Hodel M R,Petersen J F,Wyatt P W,Steenblock E R,Shah P H,Bousse L J,Troup C B,Mellen J C,Wittmann D K,Erndt N G,Cauley T H,Koehler R T,So A P,Dube S,Rose K A,Montesclaros L,Wang S,Stumbo D P,Hodges S P,Romine S,Milanovich F P,White H E,Regan J F,Karlin-Neumann G A,Hindson C M,Saxonov S,Colston B W.Anal.Chem.,2011,83(22):8604-8610
-
[25]
Beer N R,Wheeler E K,Lee-Houghton L,Watkins N,Nasarabadi S,Hebert N,Leung P,Arnold D W,Bailey C G,Colston B W.Anal.Chem.,2008,80(6):1854-1858
-
[26]
Blow N.Nat.Methods,2009,6(9):683-686
-
[27]
Thompson A M,Gansen A,Paguirigan A L,Kreutz J E,Radich J P,Chiu D T.Anal.Chem.,2014,86(24):12308-12314
-
[28]
Shuga J,Zeng Y,Novak R,Lan Q,Tang X,Rothman N,Vermeulen R,Li L,Hubbard A,Zhang L,Mathies R A,Smith M T.Nucleic Acids Res.,2013,41(16):e159
-
[29]
Giuffrida M C,Zanoli L M,D'Agata R,Finotti A,Gambari R,Spoto G.Anal.Bioanal.Chem.,2015,407(6):1533-1543
-
[30]
Shih S C,Gach P C,Sustarich J,Simmons B A,Adams P D,Singh S,Singh A K.Lab Chip,2015,15(1):225-236
-
[31]
Woodward A,Cosgrove T,Espidel J,Jenkins P,Shaw N.Soft Matter,2007,3(5):627-633
-
[32]
Yobas L,Martens S,Ong W L,Ranganathan N.Lab Chip,2006,6(8):1073-1079
-
[33]
Guo M T,Rotem A,Heyman J A,Weitz D A.Lab Chip,2012,12(12):2146-2155
-
[34]
Choi C H,Jung J H,Hwang T S,Lee C S.Macromol.Res.,2009,17(3):163-167
-
[35]
Song H,Chen D L,Ismagilov R F.Angew.Chem.Int.Ed.,2007,45(44):7336-7356
-
[36]
Zhu Q,Qiu L,Yu B,Xu Y,Gao Y,Pan T,Tian Q,Song Q,Jin W,Jin Q,Mu Y.Lab Chip,2014,14(6):1176-1185
-
[37]
Whitesides G M.Nature,2006,442(442):368-373
-
[38]
Joensson H N,Andersson Svahn H.Angew.Chem.Int.Ed.Eng.,2012,51(49):12176-12192
-
[39]
Nisisako T,Torii T.Lab Chip,2008,8(2):287-293
-
[40]
Holtze C,Rowat A C,Agresti J J,Hutchison J B,Angilè F E,Schmitz C H,K ster S,Duan H,Humphry K J,Scanga R A.Lab Chip,2008,8(10):1632-1639
-
[41]
Roberts C H,Anna L,Sandra M G,Eunice C,Robert B,Meno N,Elizabeth M C,Burr S E,Mabey D C,Bailey R L.J.Clin.Microbiol.,2013,51(7)::2195-2203
-
[42]
Maharaj A,Rampersad S N.Mol.Biotechnol.,2011,50(3):237-249
-
[43]
Phillip B,Tanner S C,Regan J F,Ryan K,Hindson B J,Brown A S.Clin.Chem., 2013,59(6):1155-1156
-
[44]
Thaitrong N,Kim H,Renzi R F,Bartsch M S,Meagher R J,Patel K D.Electrophoresis,2012,33(23):3506-3513
-
[45]
Dany M,Dejan T,Mojca M,Kristina G,Jana E.Plos One,2013,8(5):e62583-e62583
-
[46]
Heredia N J,Belgrader P,Wang S,Koehler R,Regan J,Cosman A M,Saxonov S,Hindson B,Tanner S C,Brown A S.Methods,2013,59(1):S20-S23
-
[47]
Yung T K F,Chan K C A,Mok T S K,Joanna T,Ka-Fai T,Lo Y M D.Clin.Cancer Res.,2009,15(6):2076-2084
-
[48]
Fearon E R,Vogelstein B.Cell,1990,61(5):759-767
-
[49]
Heyries K A,Tropini C,Vaninsberghe M,Doolin C,Petriv O I,Singhal A,Leung K,Hughesman C B,Hansen C L.Nat.Methods,2011,8(8):649-651
-
[50]
Contente-Cuomo T,Murtaza M.Cancer Res.,2015,75(15 Supplement):2404
-
[51]
Dobnik D,Spilsberg B,Bogožalec Košir A,Holst-Jensen A,Žel J.Anal.Chem.,2015,87(16):8218-8226
-
[52]
Mazaika E,Homsy J.Curr.Protoc.Hum.Genet.,2014,82:7.24.1-13
-
[53]
Zhong Q,Bhattacharya S,Kotsopoulos S,Olson J,Taly V,Griffiths A D,Link D R,Larson J W.Lab Chip,2011,11(13):2167-2174
-
[1]
-
Figure 2 Digital PCR amplification of fetal DNA from maternal plasma analyzed in a digital array microfluidics chip[10]. (a) Same chip before and after PCR amplification process, (b) Heat map for the positive reactions, (c) Exponential plot for the amplification process
Figure 4 Droplet stability during formation[39]. (a)Droplets are generated by flow-focusing from a 25-mm nozzle; (b) Droplet production frequency and drop diameter as a function of oil flow rate at constant flow rate of the water phase
Figure 5 Droplet digital PCR workflow for simultaneous detection of E. coli O157 and L. monocytogenes[14] (a)Fabrication of microfluidic chip; (b) generation of droplets; (c) On-chip amplification followed by fluorescence read out
-

 下载:
下载:


 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 28
- 文章访问数: 1812
- HTML全文浏览量: 440
