Table 1.
Crystallographic data and corresponding structural refinements of 3c, 3f, and 4d
Citation:
Fangxiang SUN, Qing ZHANG, Yifan ZHANG, Haoyi SUN, Akim V. Shmal′ko, Sergey A. Anufriev, Igor B. Sivaev, Deshuang TU, Hong YAN. Pd-catalyzed B—H bond activation and annulation of nido-carborane with terminal olefins: Facile construction of 2D-3D fused polycyclic compounds[J]. Chinese Journal of Inorganic Chemistry,
2026, 42(3): 467-478.
doi:
10.11862/CJIC.20250347

钯催化巢式碳硼烷与末端烯烃的B—H键活化环化反应——二维-三维稠合多环化合物的快速构建
English
Pd-catalyzed B—H bond activation and annulation of nido-carborane with terminal olefins: Facile construction of 2D-3D fused polycyclic compounds
Abstract:
To address the longstanding challenge in traditional carborane methodology of rapidly and efficiently constructing carboranyl-based polycyclic frameworks, Pd-catalyzed one-pot reactions between pyridyl-substituted nido-carboranes and alkynes directly afford two distinct types of 2D-3D fused carboranyl polycyclic compounds: 3a-3f, 4a-4d. The structures of this series of compounds were characterized by nuclear magnetic resonance spectroscopy, single-crystal X-ray diffraction, and high-resolution mass spectrometry, and a plausible reaction mechanism was proposed. Crystal structures reveal that the multiple rings in such 2D-3D fused carboranyl polycyclic compounds exhibit a certain degree of coplanarity. Furthermore, these compounds exhibited properties distinct from those of conventional 2D polycyclic systems.
-
Key words:
- carborane
- / B—H bond activation
- / polycyclic compounds
-
0. Introduction
Polycyclic compounds represent ubiquitous and pivotal molecular architectures in both fundamental research and practical applications[1-4]. The conventional paradigm for constructing high-performance polycyclics, as exemplified by graphene, relies on the extension of aromatic systems[5-7]. Inspired by these established structures, we are driven to explore novel synthetic strategies to access new polycyclic systems.
In contrast to 2D aromatic rings, boron clusters such as carboranes exhibit 3D aromaticity[8-12], featuring multiple B—H and/or C—H bonds. Their unique structures and delocalized multi-center bonding endow them with exceptional properties, including 3D aromaticity, high thermal and chemical stability, rigid geometry, low toxicity, bulky size, and hydrophobicity. These attributes make carboranes and their derivatives highly attractive for a wide range of applications, spanning materials science to medicine. Consequently, carboranes have been widely employed as building blocks for functional molecules[13-18] and as tunable pharmacophores, particularly in boron neutron capture therapy (BNCT) agents[19-21]. In most of these studies, however, the carborane cluster is typically utilized as an 'appended' or peripheral unit. A more intriguing and distinct approach would be to develop carborane‑ embedded polycyclic systems where the cluster plays an integral, central role[22-25]. In such architectures, the carborane and its extended framework would form a unified structural skeleton.
Herein, we report a novel 2D-3D fused molecular architecture, constructed through a facile Pd-catalyzed B—H activation and cascade heteroannulation between a nido-carborane cluster and a pyridine moiety with alkynes. This efficient cage-extension protocol, which proceeds under mild and additive-free conditions, afforded two distinct types of fused skeletons in high yields. The resulting 2D-3D fused systems not only expand the structural diversity of aromatic polycyclics but also demonstrate significant potential for applications in luminescent materials and pharmaceutical sciences.
1. Experimental
1.1 Materials and methods
All the commercially available reagents were purchased from commercial sources and used directly. The process for the synthesis of nido-carboranes has been optimized according to literature reports[4]. Analytical thin-layer chromatography (TLC) was performed on silica gel (GF 254) plates, while column chromatography was performed on Silica Gel (200-300 mesh or 300-400 mesh). Visualization of the developed chromatogram was performed by UV absorbance (254 nm).
1H, 11B, 11B{1H}, and 13C nuclear magnetic resonance (NMR) spectra were recorded using Bruker AVANCE Ⅲ 400 MHz spectrometers under ambient conditions unless otherwise stated. All chemical shifts (δ values) were reported in ppm with the residual solvent resonances of the deuterated solvents for proton and carbon chemical shifts. 11B chemical shifts were measured utilizing external Et2O·BF3 (δ=0) as reference. The high-resolution mass spectra (HRMS) were recorded on Agilent Micromass 6540 Q-Tof LC/MS. Elemental analyses were performed on a PerkinElmer 240 analyzer. Single-crystal X-ray crystallographic data were collected on a Bruker SMART Apex Ⅱ CCD diffractometer by means of graphite monochromated Mo Kα radiation (λ=0.071 07 nm). The structures were solved by direct method and refined by full-matrix least-squares method on F2 using the SHELXTL or Olex2 crystallographic software package.
1.2 Synthesis of 2D-3D fused carborane polycyclic compounds
The 1-Py-2-H-1, 2-C2B10H10 were synthesized according to literature procedures[26-28].
Deboronation of closo-carborane substrates: A 50 mL Schlenk tube was added with substituted 1-Py-2-H-1, 2-C2B10H10 (5.0 mmol), KOH (1.12 g, 20 mmol), and 25 mL EtOH. The mixture was refluxed for 6 h before cooling down to room temperature. CO2 was flowed to adjust the pH value to 7-8. After filtration and evaporation, the resulting white solid was dissolved in 50 mL of deionized water. Then, an aqueous solution of NMe3·HCl (525 mg, 5.5 mmol) was added dropwise. The resulting mixture was stirred for 30 min and filtered. The filter cake was washed with hexane and diethyl ether three times separately and then dried under vacuum to give the corresponding nido‑ carborane substrates 1.
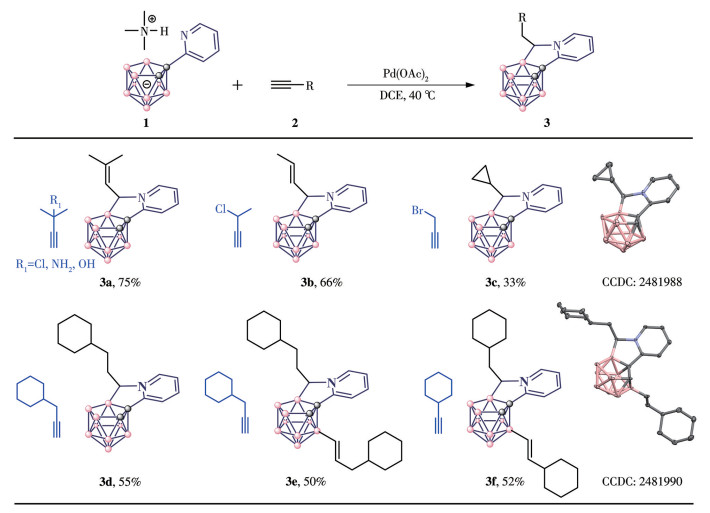
General procedure for carborane substrates: nido-Carborane (0.1 mmol), alkyne (0.15 mmol), and Pd(OAc)2 (0.005 mmol) were introduced into a 10 mL Schlenk tube under an argon atmosphere. Subsequently, anhydrous dichloroethane (DCE, 2.0 mL) was added via syringe under the same inert atmosphere. The reaction mixture was stirred at 40 ℃ for 3 h. Upon completion, as indicated by TLC analysis, the reaction was concentrated under reduced pressure. The resulting crude residue was purified by column chromatography on silica gel using a dichloromethane (DCM)/hexane mixture as the eluent to yield the desired products 3a-3f and 4a-4d.
3a: Yield 66% (inseparable isomers with a molar ratio of 3.2∶1 determined by NMR). White solid. 1H NMR (400 MHz, CDCl3): δ 8.27 (d, J=6.4 Hz, 1H), 8.21 (d, J=6.4 Hz, 1H), 8.07 (t, J=8.0 Hz, 1H), 7.51 (t, J=6.8 Hz, 1H), 7.35 (d, J=8.0 Hz, 1H), 5.52 (d, J=6.8 Hz, 1H), 5.17 (d, J=6.8 Hz, 1H, pyridyl and olefinic H), 2.27 (s, 1H, cage H), 1.92 (s, 3H), 1.88 (s, 3H), -2.83 (br, 1H, B—H—B). 13C NMR (101 MHz, CDCl3): δ 143.5, 142.1, 141.9, 137.6, 125.5, 123.3, 122.9, 120.3, 37.2, 25.9, 18.6. 11B{1H} NMR (128 MHz, CDCl3): δ 0.6 (1B), -6.5 (1B), -11.7 (1B), -14.7 (1B), -20.0 (1B), -20.5 (1B), -26.0 (1B), -31.2 (1B), -33.3 (1B). HRMS (m/z): Calcd. for C12H21B9N ([M-H]-): 277.254 2. Found: 277.255 4. Elemental Anal. Cacld.(%): C, 51.92; H, 7.99; B, 35.05; N, 5.05. Found(%): C, 51.51; H, 8.11; B, 35.35; N, 5.03.
3b: Yield 75% (inseparable isomers with a molar ratio of 3.3∶1). White solid. 1H NMR (400 MHz, CDCl3): δ 8.40 (d, J=6.4 Hz, 1H), 8.35 (d, J=6.4 Hz, 1H), 8.08 (t, J=8.0 Hz, 1H), 7.53 (t, J=6.8 Hz, 1H), 7.37 (d, J=8.0 Hz, 1H), 5.90-6.11 (m, 1H), 5.69-5.85 (m, 1H), 4.85 (d, J=6.4 Hz, 1H, pyridyl and olefinic H), 2.29 (s, 1H, cage H), 1.86 (d, J=6.8 Hz, 1H), 1.56 (s, 1H), -2.79 (br, 1H, B—H—B). 13C NMR (101 MHz, CDCl3): δ 143.7, 142.0, 133.5, 131.9, 131.8, 129.6, 122.8, 120.3, 18.0. 11B{1H} NMR (128 MHz, CDCl3): δ 0.1 (1B), -6.2 (1B), -11.8 (1B), -14.7 (1B), -19.5 (1B), -20.6 (1B), -26.1 (1B), -31.0 (1B), -33.4 (1B). HRMS (m/z): Calcd. for C11H19B9N ([M-H]-): 262.238 6. Found: 262.232 6. Elemental Anal. Cacld.(%): C, 50.13; H, 7.65; B, 36.91; N, 5.31. Found(%): C, 50.71; H, 7.42; B, 36.61; N, 5.26.
3c: Yield 33% (inseparable isomers with a molar ratio of 3.2∶1). Yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.76 (d, J=6.4 Hz, 1H), 8.72 (d, J=6.4 Hz, 1H), 8.09 (t, J=8.0 Hz, 1H), 7.54 (t, J=6.8 Hz, 1H), 7.37 (d, J=8.0 Hz, 1H, pyridyl H), 4.06 (d, J=9.6 Hz, 1H), 3.89 (d, J=9.6 Hz, 1H), 2.30 (s, 1H, cage H), 2.23 (s, 1H, cage H), 1.25 (s, 1H), 0.79-0.96 (m, 4H), -2.81 (br, 1H, B—H—B). 13C NMR (101 MHz, CDCl3): δ 141.2, 139.2, 120.4, 118.2, 27.4, 16.3, 13.1, 5.4, 5.0, 0, -0.7. 11B{1H} NMR (128 MHz, CDCl3): δ -0.3 (1B), -5.8 (1B), -11.8 (1B), -14.6 (1B), -19.5 (1B), -20.6 (1B), -26.1 (1B), -30.9 (1B), -33.4 (1B). HRMS (m/z): Calcd. for C11H19B9N ([M-H]-): 262.238 6. Found: 262.232 2. Elemental Anal. Cacld.(%): C, 50.13; H, 7.65; B, 36.91; N, 5.31. Found(%): C, 50.66; H, 7.21; B, 36.74; N, 5.39.
3d: Yield 55%. Yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.44 (d, J=6.4 Hz, 1H), 8.07 (t, J=8.0 Hz, 1H), 7.54 (t, J=6.8 Hz, 1H), 7.37 (d, J=8.0 Hz, 1H, pyridyl H), 4.31 (d, J=10.8 Hz, 1H), 2.30 (s, 1H, cage H), 1.98-2.12 (m, 2H), 1.67-1.79 (m, 6H), 1.59-1.66 (m, 3H), 1.16-1.28 (m, 4H), -2.81 (br, 1H, B—H—B). 13C NMR (101 MHz, CDCl3): δ 143.3, 141.2, 122.6, 120.5, 37.8, 36.0, 33.5, 33.4, 33.1, 26.6, 26.3, 26.3. 11B{1H} NMR (128 MHz, CDCl3): δ 0.4 (1B), -5.7 (1B), -12.0 (1B), -14.6 (1B), -19.7 (1B), -21.1 (1B), -26.2 (1B), -31.3 (1B), -33.7 (1B). HRMS (m/z): Calcd. for C16H29B9N ([M-H]-): 333.316 8. Found: 333.318 0. Elemental Anal. Cacld.(%): C, 57.59; H, 9.06; B, 29.15; N, 4.20. Found(%): C, 57.90; H, 9.01; B, 28.88; N, 4.21.
3e: Yield 50%. Yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.43 (d, J=6.4 Hz, 1H), 7.98 (t, J=8.0 Hz, 1H), 7.49 (t, J=6.8 Hz, 1H), 7.24 (s, 1H), 5.86 (dd, J=6.4, 17.6 Hz, 1H), 5.48 (d, J=17.6 Hz, 1H), 4.39 (t, J=8.0 Hz, 1H, pyridyl and olefinic H), 2.19 (s, 1H, cage H), 1.89-2.13 (m, 7H), 1.63-1.75 (m, 10H), 1.22-1.30 (m, 8H), 0.98-1.04 (m, 3H), -2.60 (br, 1H, B—H—B). 13C NMR (101 MHz, CDCl3): δ 151.6, 142.0, 141.4, 122.2, 121.6, 47.0, 43.2, 35.2, 34.60, 32.8, 32.7, 32.6, 29.7, 26.5, 26.3, 26.2, 26.1, 26.0, 22.7, 14.1. 11B{1H} NMR (128 MHz, CDCl3): δ 0.9 (1B), -6.3 (2B), -12.9 (1B), -18.7 (1B), -20.8 (1B), -25.6 (1B), -33.3 (2B). HRMS (m/z): Cacld. for C25H43B9N ([M-H]-): 455.426 4. Found: 455.427 8. Elemental Anal. Cacld.(%): C, 65.86; H, 9.73; B, 21.34; N, 3.07. Found(%): C, 66.13; H, 9.62; B, 21.22; N, 3.03.
3f: Yield 52%. Yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.44 (d, J=6.0 Hz, 1H), 7.98 (t, J=8.0 Hz, 1H), 7.50 (t, J=6.8 Hz, 1H), 7.28 (s, 1H), 5.95 (m, 1H), 5.51 (d, J=17.2 Hz, 1H), 4.31 (d, J=11.2 Hz, 1H, pyridyl and olefinic H), 2.27 (s, 1H) (cage H), 2.21 (s, 1H), 1.91 (t, J=7.2 Hz, 2H), 1.62-1.78 (m, 12H), 1.21-1.30 (m, 9H), -2.66 (br, 1H, B—H—B). 13C NMR (101 MHz, CDCl3): δ 162.0, 144.8, 142.1, 141.4, 122.2, 121.5, 43.9, 40.2, 37.9, 37.9, 36.0, 33.5, 33.3, 33.2, 33.1, 29.7, 26.6, 26.3; 11B{1H} NMR (128 MHz, CDCl3): δ 0.3 (1B), -6.2 (1B), -13.2 (1B), -18.7 (1B), -20.8 (1B), -25.6 (2B), -33.4 (2B). HRMS (m/z): Cacld. for C23H39B9N ([M-H]-): 427.395 1. Found: 427.396 3. Elemental Anal. Cacld.(%): C, 64.56; H, 9.42; B, 22.74; N, 3.27. Found(%): C, 64.92; H, 9.33; B, 22.45; N, 3.30.
4a: Yield 58% (inseparable isomers with a molar ratio of 3.4∶1). White solid. 1H NMR (400 MHz, CDCl3): δ 8.40 (d, J=6.4 Hz, 1H), 8.05 (t, J=8.0 Hz, 1H), 7.54 (t, J=6.8 Hz, 1H), 7.35 (d, J=8.0 Hz, 1H), 5.04 (d, J=12.8 Hz, 1H), 4.63 (d, J=12.8 Hz, 1H, pyridyl and olefinic H), 2.29 (s, 1H, cage H), 2.22 (s, 1H, cage H), 0.22 (s, 9H, CH3), -2.79 (br, 1H, B—H—B). 13C NMR (101 MHz, CDCl3): δ 143.6, 141.4, 123.5, 120.8, 37.1, 30.2, 29.3, 25.4, 0.1, 0. 11B {1H} NMR (128 MHz, CDCl3): δ 1.2 (1B), -5.6 (1B), -11.9 (1B), -14.5 (1B), -19.5 (1B), -20.8 (1B), -26.2 (1B), -30.9 (1B), -33.5 (1B). HRMS (m/z): Cacld. for C12H23B9NSi ([M-H]-): 307.246 8. Found: 307.248 6. Elemental Anal. Cacld.(%): C, 46.84; H, 7.86; B, 31.62; N, 4.55; Si, 9.13. Found(%): C, 47.42; H, 7.69; B, 31.40; N, 4.35; Si, 9.14.
4b: Yield 56% (inseparable isomers with a molar ratio of 3.0∶1). White solid. 1H NMR (400 MHz, CDCl3): δ 8.40 (t, J=6.4 Hz, 1H), 8.07 (t, J=8.0 Hz, 1H), 7.58 (t, J=6.8 Hz, 1H), 7.34 (d, J=8.0 Hz, 1H), 4.83 (d, J=12.8 Hz, 1H), 4.42 (d, J=12.8 Hz, 1H, pyridyl and olefinic H), 2.28 (s, 1H, cage H), 2.22 (s, 1H, cage H), 0.94-1.06 (m, 9H), 0.66-0.80 (m, 6H), -2.81 (br, 1H, B—H—B). 13C NMR (101 MHz, CDCl3): δ 161.7, 143.2, 140.9, 123.2, 123.1, 120.2, 36.7, 29.7, 24.1, 20.1, 7.7, 4.0, 3.9. 11B {1H} NMR (128 MHz, CDCl3): δ 1.3 (1B), -5.6 (1B), -11.9 (1B), -14.4 (1B), -19.6 (1B), -20.6 (1B), -26.2 (1B), -30.9 (1B), -33.5 (1B). HRMS (m/z): Cacld. for C15H29B9NSi ([M-H]-): 349.293 8. Found: 349.295 1. Elemental Anal. Cacld.(%): C, 51.51; H, 8.65; B, 27.81; N, 4.00; Si, 8.03. Found(%): C, 51.98; H, 8.55; B, 27.52; N, 4.06; Si, 7.89.
4c: Yield 47%. White solid. 1H NMR (400 MHz, CDCl3): δ 7.91 (t, J=8.0 Hz, 1H), 7.68 (d, J=6.8 Hz, 6H), 7.39-7.50 (m, 8H), 7.22-7.33 (m, 3H), 7.13 (t, J=7.2 Hz, 1H), 4.58 (s, 1H, phenyl, pyridyl, and olefinic H), 2.23 (s, 1H, cage H), -2.76 (br, 1H, B—H—B). 13C NMR (101 MHz, CDCl3): δ 142.9, 141.5, 135.9, 133.5, 130.2, 128.4, 123.0, 120.1, 25.7. 11B{1H} NMR (128 MHz, CDCl3): δ 2.2 (1B), -5.0 (1B), -11.5 (1B), -14.1 (1B), -19.4 (1B), -20.8 (1B), -26.2 (1B), -30.8 (1B), -33.3 (1B). HRMS (m/z): Cacld. for C27H29B9NSi ([M-H]-): 493.293 8. Found: 493.294 9. Elemental Anal. Cacld.(%): C, 65.66; H, 6.12; B, 19.70; N, 2.84; Si, 5.69. Found(%): C, 66.15; H, 6.01; B, 19.39; N, 2.67; Si, 5.78.
4d: Yield 55% (inseparable isomers with a molar ratio of 2.2∶1). White solid. 1H NMR (400 MHz, CDCl3): δ 8.45 (d, J=6.0 Hz, 1H), 8.42 (d, J=6.0 Hz, 1H), 8.08 (t, J=8.0 Hz, 1H), 7.60 (t, J=7.2 Hz, 1H), 7.34 (d, J=8.0 Hz, 1H), 5.04 (d, J=12.8 Hz, 1H), 4.63 (d, J=12.8 Hz, 1H, pyridyl and olefinic H), 2.28 (s, 1H, cage H), 2.20 (s, 1H)(cage H), 1.24-1.36 (m, 3H) (CH), 1.15 (s, 18H) (CH3), -2.78 (br, 1H, B—H—B). 13C NMR (101 MHz, CDCl3): δ 161.8, 143.2, 140.7, 123.4, 123.2, 120.2, 36.7, 22.6, 22.3, 19.2, 19.1, 19.1, 18.5, 18.0, 17.7, 11.9, 11.6, 11.5. 11B {1H} NMR (128 MHz, CDCl3): δ 1.3 (1B), -5.5 (1B), -11.9 (1B), -14.4 (1B), -19.5 (1B), -20.7 (1B), -26.2 (1B), -30.8 (1B), -33.5 (1B). HRMS (m/z): Cacld. for C18H35B9NSi ([M-H]-): 391.340 7. Found: 391.343 1. Elemental Anal. Cacld.(%): C, 55.17; H, 9.26; B, 24.83; N, 3.57; Si, 7.17. Found(%): C, 54.68; H, 9.43; B, 25.14; N, 3.53; Si, 7.22.
1.3 X-ray crystallography
The regular block single crystal was selected to be wrapped with vaseline and encapsulated in a fine loop of appropriate size. Crystal data were obtained using a Bruker Smart-CCD diffractometer with monochromated Mo Kα radiation (λ=0.071 07 nm) at 173 K. The structures were solved by direct methods using SHELXS-2014, implemented in the Olex2 interface, and refined with full-matrix least-squares techniques[29]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms attached to carbon and boron atoms were placed in idealized positions. Crystallographic data and refinement details for compounds 3c, 3f, and 4d are summarized in Table 1.
Table 1

 下载:
导出CSV
下载:
导出CSV
Parameter 3c 3f 4d Empirical formula C11H20B9N C23H40B9N C18H36B9NSi Formula weight 263.57 430.25 391.86 Temperature/K 296.15 296.15 296.15 Crystal system Monoclinic Monoclinic Triclinic Space group P21/n P21/n P1 a/nm 1.072 6(2) 0.950 99(5) 0.781 78(7) b/nm 0.998 8(3) 1.537 36(10) 1.503 90(14) c/nm 1.495 8(4) 1.795 07(11) 2.143 40(19) α/(°) 73.108(3) β/(°) 105.320(8) 95.099(2) 80.767(3) γ/(°) 84.063(3) Volume/nm3 1.545 5(7) 2.614 0(3) 2.375 8(4) Z 4 4 1 Dc/(g·cm-3) 1.133 1.093 1.096 μ/mm-1 0.056 0.058 0.104 F(000) 552.0 928.0 840.0 Crystal size/mm 0.12×0.11×0.1 0.13×0.11×0.1 0.13×0.12×0.11 2θ range/(°) 4.96-55.358 4.556-55.052 4.01-55.034 Reflection collected 10 018 24 053 21 109 Data, number of restraints, number of parameters 3 544, 0, 194 6 023, 3, 414 10 772, 0, 567 Goodness-of-fit on F2 1.115 0.966 1.024 R1, wR2 [I≥2σ(I)] 0.120 2, 0.274 0 0.077 2, 0.178 6 0.079 5, 0.168 3 R1, wR2 (all data) 0.239 7, 0.312 4 0.132 6, 0.208 1 0.165 1, 0.210 7 2. Results and discussion
2.1 Condition optimization
Based on previous research, we initiated our investigation into the cyclization reaction between (NMe3H)[7-(2-pyridyl)-nido-C2B9H10] (1a) and alkyne (2d) under various reaction conditions. Upon completion, the reaction mixture was purified by chromatography on silica gel eluting with DCM/petroleum ether to afford the desired product 3a. The screening of catalysts, solvents, temperature, and reaction time was listed in Table 2. Consequently, Pd(OAc)2 facilitated a rapid and quantitative conversion. While no product was observed with other metal catalysts, even when the reaction temperature was increased to 60 ℃ and the reaction time was extended to 12 h (Table 2, entries 2-7). Subsequently, the effect of temperature and solvents (Table 2, entries 8-13) was examined. To our delight, the isolated yield reached up to 70% in DCE. Furthermore, the presence of air and moisture exerted only a minimal influence on the reaction yield (Table 2, entry 14). In the absence of a metal catalyst, no reaction was observed (Table 2, entry 15). When phenyl was used in place of the directing pyridyl group in 1a, no product was detected (Table 2, entry 16). The experimental results demonstrate that both a Pd(Ⅱ) catalyst and an N-directing group are essential for the reaction, likely playing key roles in facilitating the formation of intermediates. Furthermore, increasing the amount of 2d to three equivalents under standard reaction conditions resulted in the formation of the disubstituted product 3e with a 50% isolated yield.
Table 2
Table 2. Screening of catalysts, solvents, temperature, and reaction timea下载:
导出CSV
Entry Catalyst Solvent Time/h T/℃ Yieldb/% 1 Pd(OAc)2 DCE 3 40 70 2 Pd(PPh3)4Cl2 DCE 12 40 Trace 3 Cu(OAc)2 DCE 12 40 — 4 [Cp*RhCl2]2 DCE 12 40 — 5 [(p-cymene)RuCl2]2 DCE 12 40 — 6 Ni(OAc)2 DCE 12 40 — 7 Co(OAc)2 DCE 12 40 — 8 Pd(OAc)2 DCE 3 RT 50 9 Pd(OAc)2 DCE 3 60 65 10 Pd(OAc)2 DCE 3 80 61 11 Pd(OAc)2 DCM 3 40 60 12 Pd(OAc)2 CH3CN 3 40 — 13 Pd(OAc)2 THF 3 40 — 14c Pd(OAc)2 DCE 3 40 60 15 None DCE 3 40 — 16d Pd(OAc)2 DCE 3 40 — a Reactions were conducted at 0.1 mmol scale in 2.0 mL of solvent in a Schlenk tube; b Yield of isolated product; c In air; d Phenyl instead of pyridyl in compound 1a. 2.2 Substrate scope
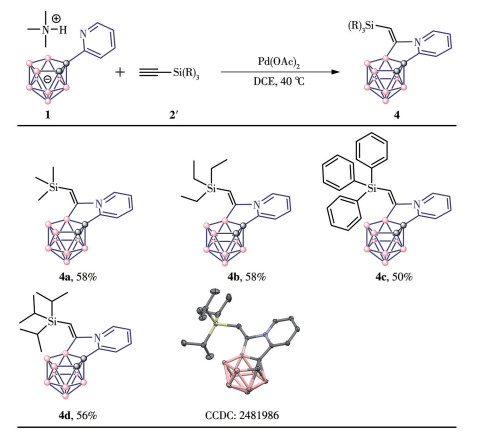
With the optimal reaction conditions in hand, the substrate scope of alkynes was examined. We were pleased to find that many alkynes could react with 1a smoothly to generate carborane-fused tricyclic compounds in good isolated yields. As shown in Fig.1, through B—H activation and subsequent annulation with terminal aryl alkynes, carborane-fused products were formed. These products feature a new five-membered ring containing a chiral center (3a-3f). Furthermore, the reaction tolerated substrates bearing an alkyne moiety with a leaving group, affording products containing carbon-carbon double bonds or a tricyclic ring system (3a-3c). When the amount of alkene was scaled to three equivalents, disubstituted products 3e and 3f were obtained. Using silyl acetylene as the starting material afforded new five-membered ring products 4a-4d, which incorporated cis-trans isomers (Fig.2). In contrast to the previous products, this new five-membered ring incorporates a sp2-hybridized carbon. Based on further mechanistic studies, this result is due to the significant steric hindrance of the silicon substituent.
Figure 1
 Figure 1. Reaction scope Ⅰ
Figure 1. Reaction scope ⅠReaction conditions for 3a-3d: 1 (0.1 mmol), 2 (0.15 mmol), Pd(OAc)2 (x=5%), DCE (4.0 mL), 40 ℃, 3 h; Reaction conditions for 3e and 3f: 1 (0.1 mmol), 2 (0.3 mmol), Pd(OAc)2 (x=5%), DCE (4.0 mL), 40 ℃, 3 h; The B—H—B bridging proton of 3a-3f is omitted for clarity.
Figure 2
 Figure 2. Reaction scope Ⅱ
Figure 2. Reaction scope ⅡReaction conditions: 1 (0.1 mmol), 2 (0.15 mmol), Pd(OAc)2 (5%), DCE (4.0 mL), 40 ℃, 3 h; The B—H—B bridging proton of 4a-4d is omitted for clarity.
The structures of all the new carborane derivatives were characterized by 1H, 13C, and 11B NMR and HRMS, among which 3c, 3f, and 4b were unambiguously identified by single-crystal X-ray diffraction (Table 1). The ratio of the inseparable mixture was determined by NMR analysis. Overall, two types of boron cluster-cored tricyclics are obtained in a one-pot reaction. Here, the synthetic methodology of boron cage extension represents a brand-new strategy for the construction of different carborane-cored tricyclics through control of the type of alkynes used.
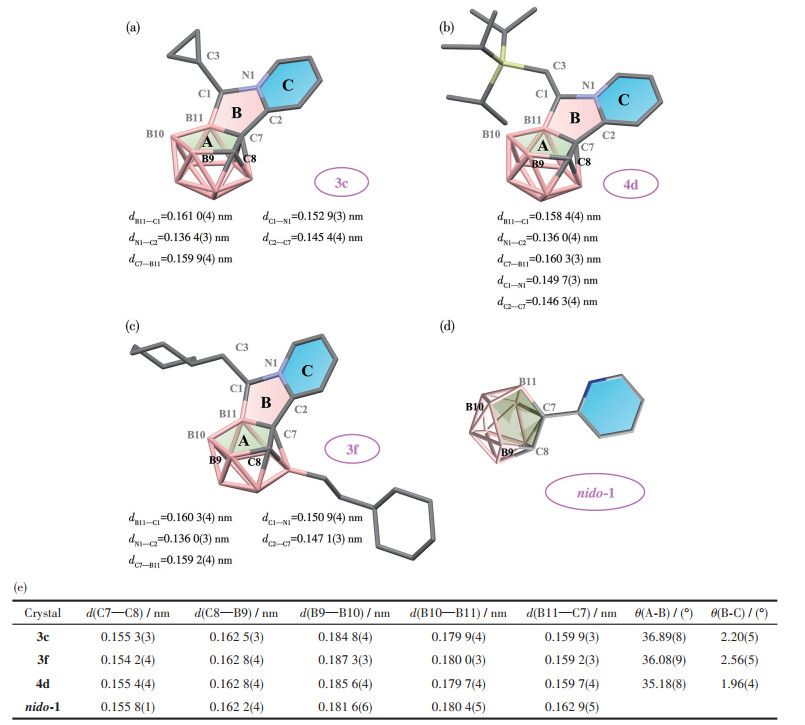
2.3 Crystal structure analysis
To gain structural insights into the above new fused structures, the selected crystal structures of 3c, 3f, 4d, and the non-fused control crystal structures of nido-1 were studied (Fig.3). The carborane cage is indeed fused with the pyridyl ring by sharing the middle five-membered ring to generate completely new polycyclic compounds. But they present a non-planar geometry, which is distinct from the 2D fused rings like planar phenanthrene. The dihedral angles between the C2B3 face (plane A) in carborane and the fused ring (plane B) are 36.89(8)°, 36.08(9)°, and 35.18(8)°, in comparison to smaller dihedral angles of 2.20(5)°, 2.56(5)° and 1.96(4)° between planes B and C for 3c, 3f, and 4d, respectively. Moreover, a certain degree of deformation of the nido-carborane unit was observed in 3c, 3f, and 4d as the B11—C7 bonds [0.159 2(3)-0.159 9(3) nm] are shortened in comparison with those in nido-1 [0.162 9(5) nm, Fig.3]. In the mid-rings (plane B), some bonds are also shortened [e.g. dB11—C1=0.158 4(4) nm for 4d vs dB—C=0.160 6 nm for the classical B—C single bond[30]; dC2—C7=0.146 3(3) nm for 4d vs dC—C=0.153 6 nm for the classical C—C bond]. These data indicate certain electronic delocalization for the fused systems.
Figure 3
 Figure 3. Crystal structures of carborane-based tricyclic compounds (a) 3c, (b) 4d, (c) 3f, and (d) control compound nido-1; (e) Bond lengths and dihedral angles for the selected crystal structures
Figure 3. Crystal structures of carborane-based tricyclic compounds (a) 3c, (b) 4d, (c) 3f, and (d) control compound nido-1; (e) Bond lengths and dihedral angles for the selected crystal structures2.4 Reaction mechanism
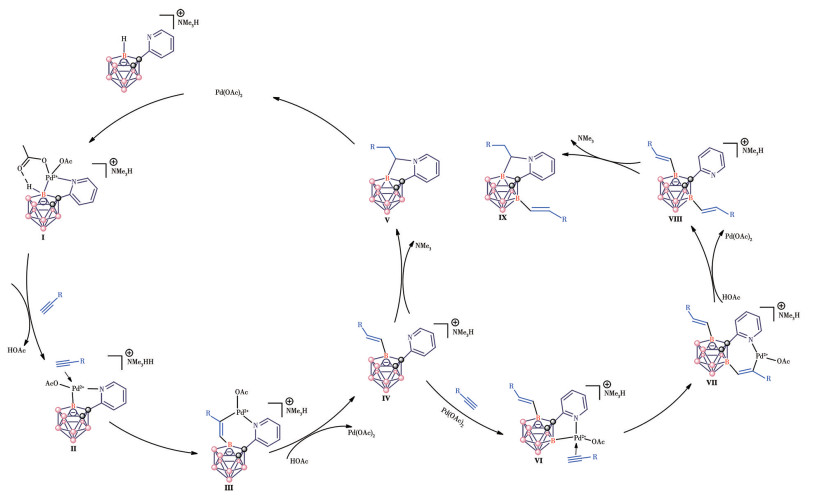
Based on our previous research and relevant literature, plausible mechanisms were put forward[31-37] (Fig.4). Regarding the cyclization products, following the coordination of pyridyl to the metal [i.e., Pd(Ⅱ)], selective B—H activation and regioselective alkyne insertion into the Pd—B bond occur in sequence, leading to intermediate Ⅲ. This intermediate then further undergoes protodemetalation to yield intermediate Ⅳ. In the final step, the nitrogen atom on the pyridyl group performs a nucleophilic attack on both sites of the double bond, followed by the elimination of one molecule of trimethylamine, yielding the target five-membered ring and compounds as the final products. If we use intermediate Ⅳ as the starting material, disubstituted product Ⅸ can be obtained via the same mechanism.
Figure 4
 Figure 4. Proposed reaction mechanism for five-membered ring product 3
Figure 4. Proposed reaction mechanism for five-membered ring product 3The B—H—B bridging proton is omitted for clarity.
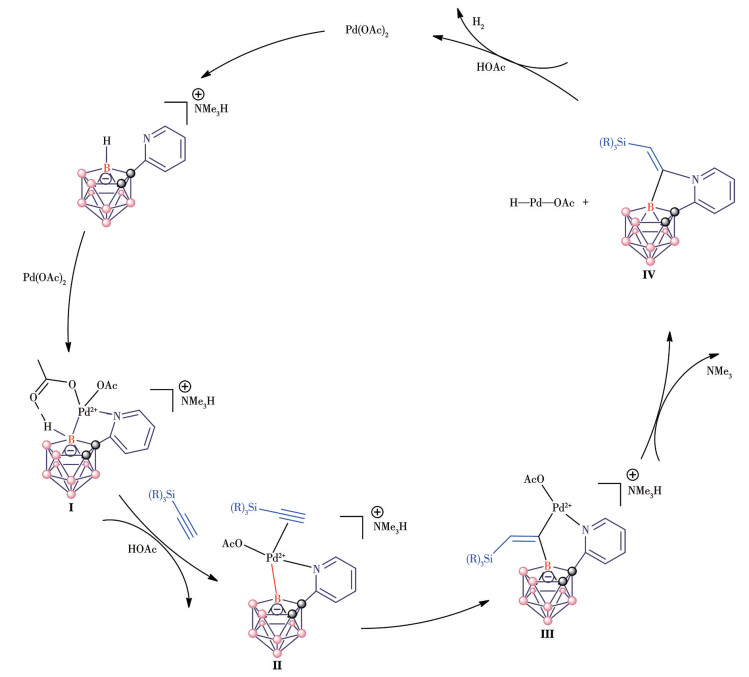
For another five-membered ring product 4, after the coordination of pyridyl to metal [i.e., Pd(Ⅱ)], intermediate Ⅰ is formed. Then, the regioselective B—H metalation in the B11-position occurs via a metalation process to afford a five-membered ring intermediate Ⅱ. Due to the relatively large steric hindrance of silicon atoms, insertion of alkyne then leads to the key six-membered ring intermediate Ⅲ, which undergoes reductive elimination to give rise to the five-membered ring products Ⅳ. The metal Pd(0) reacted with acetic acid to generate the H—Pd(Ⅱ)—L species. Then the H—Pd(Ⅱ)—L species reacted with acetic acid and finally recycled the catalyst by the release of hydrogen (Fig.5).
Figure 5
 Figure 5. Proposed reaction mechanism for five-membered ring product 4
Figure 5. Proposed reaction mechanism for five-membered ring product 4The B—H—B bridging proton is omitted for clarity.
3. Conclusions
We report a new synthetic method for 2D-3D fused polycyclic compounds. We developed a one-pot Pd-catalyzed B—H activation and cascade annulation reaction of nido-carboranes with N-heterocycles and alkynes, enabling the rapid construction of two types of boron cluster-cored tricyclic compounds. Based on our previous research and relevant literature, plausible mechanisms have been proposed. Based on the analysis of the crystal structure, these polycyclic compounds exhibit a structure different from the ordinary 2D polycyclic compounds. This study establishes a rational design strategy for boron cluster‑cored polycyclic molecules, showcasing their potential for various applications.
Acknowledgments: This work was supported by Zhejiang Provincial Natural Science Foundation (Grant No.LQ24B020001), Huzhou Municipal Natural Science Foundation of China (Grant No.2023YZ09), National Natural Science Foundation of China (Grants No.92261202, 92461308, W2412072, 22401141), the Natural Science Foundation of Jiangsu Province (Grant No.BK20241229), and the Fundamental Research Funds for the Central Universities (Grant No.2024300362). Syntheses of compounds 3a-3c were supported by the Ministry of Science and Higher Education of the Russian Federation (Contract No.075-00276-25-00). -
-
[1]
ITO H, OZAKI K, ITAMI K. Annulative π-extension (APEX): Rapid access to fused arenes, heteroarenes, and nanographenes[J]. Angew. Chem. ‒Int. Edit., 2017, 56(37): 11144-11164. doi: 10.1002/anie.201701058
-
[2]
VON GROTTHUSS E, JOHN A, KAESE T, WAGNER M. Doping polycyclic aromatics with boron for superior performance in materials science and catalysis[J]. Asian J. Org. Chem., 2017, 7(1): 37-53.
-
[3]
WANG Z S, JIANG L F, JI J W, ZHOU F L, LAN J B, YOU J S. Construction of cationic azahelicenes: Regioselective three-component annulation using in situ activation strategy[J]. Angew. Chem. ‒Int. Edit., 2020, 59(52): 23532-23536. doi: 10.1002/anie.202010051
-
[4]
BORISSOV A, MAURYA Y K, MOSHNIAHA L, WONG WS, ŻYŁA-KARWOWSKA M, STĘPIEŃ M. Recent advances in heterocyclic nanographenes and other polycyclic heteroaromatic compounds[J]. Chem. Rev., 2021, 122(1): 565-788.
-
[5]
ALZAHRANI A Z. First-principles study on the structural and electronic properties of graphene upon benzene and naphthalene adsorption[J]. Appl. Surf. Sci., 2010, 257(3): 807-810. doi: 10.1016/j.apsusc.2010.07.069
-
[6]
CHEN F, TAO N J. Electron transport in single molecules: From benzene to graphene[J]. Accounts Chem. Res., 2009, 42(3): 429-438. doi: 10.1021/ar800199a
-
[7]
DOMENICO J, SCHNEIDER A M, SOHLBERG K. From benzene to graphene: Exploring the electronic structure of single-layer and bilayer graphene using polycyclic aromatic hydrocarbons[J]. J. Chem. Educ., 2019, 96(10): 2225-2237. doi: 10.1021/acs.jchemed.9b00331
-
[8]
CHI X, ZHANG Z X, LI M Q, JIAO Y, LI X M, MENG F C, XUE B, WU D Q, ZHANG F. Vinylene-linking of polycyclic aromatic hydrocarbons to π-extended two-dimensional covalent organic framework photocatalyst for H2O2 synthesis[J]. Angew. Chem. ‒Int. Edit., 2024, 64(7): e202418895.
-
[9]
GRIMES R N. Carboranes[M]. [S. l. ]: Academic Press, 2016: 7-17
-
[10]
SOLA M. Aromaticity rules[J]. Nat. Chem., 2022, 14(6): 585-590. doi: 10.1038/s41557-022-00961-w
-
[11]
POATER J, VIÑAS C, OLID D, SOLÀ M, TEIXIDOR F. Aromaticity and extrusion of benzenoids linked to [o-COSAN]-: Clar has the answer[J]. Angew. Chem. ‒Int. Edit., 2022, 61(22): e202200672. doi: 10.1002/anie.202200672
-
[12]
GAO Y H, SZATHMÁRI B, BUZSÁKI D, KELEMEN Z. Reassessing the possibility of π-σ-π full electron delocalization through 3D aromatic carboranes[J]. Chem. Eur. J., 2025, 31(41): e202501806. doi: 10.1002/chem.202501806
-
[13]
MUKHERJEE S, THILAGAR P. Boron clusters in luminescent materials[J]. Chem. Commun., 2016, 52(6): 1070-1093. doi: 10.1039/C5CC08213G
-
[14]
OCHI J, TANAKA K, CHUJO Y. Recent progress in the development of solid-state luminescent o-carboranes with stimuli responsivity[J]. Angew. Chem. ‒Int. Edit., 2020, 59(25): 9841-9855. doi: 10.1002/anie.201916666
-
[15]
FISHER S P, TOMICH A W, LOVERA S O, KLEINSASSER J F, GUO J, ASAY M J, NELSON H M, LAVALLO V. Nonclassical applications of closo-carborane anions: From main group chemistry and catalysis to energy storage[J]. Chem. Rev., 2019, 119(14): 8262-8290. doi: 10.1021/acs.chemrev.8b00551
-
[16]
WANG Q Y, WANG J, WANG S, WANG Z Y, CAO M, HE C L, YANG J Q, ZANG S Q, MAK T C W. o-Carborane-based and atomically precise metal clusters as hypergolic materials[J]. J. Am. Chem. Soc., 2020, 142(28): 12010-12014. doi: 10.1021/jacs.0c04638
-
[17]
WANG Z L, GOU X Y, SHI Q Y, LIU K, CHANG X M, WANG G, XU W J, LIN S M, LIU T H, FANG Y. Through-space charge transfer: A new way to develop a high-performance fluorescence sensing film towards opto-electronically inert alkanes[J]. Angew. Chem. ‒Int. Edit., 2022, 61(35): e202207619. doi: 10.1002/anie.202207619
-
[18]
CHEN M, SUN Z F, WANG L Y, ZONG J B, WEI G F, LU C S, TANG S, TU D S, YAN H. Direct B—H bond activation polymerization of boron clusters[J]. J. Am. Chem. Soc., 2025, 147(47): 43946-43956. doi: 10.1021/jacs.5c16451
-
[19]
ZHOU Y T, CHENG K, LIU B, CAO Y C, FAN J X, LIU Z G, ZHAO Y D. Recent progress of nano-drugs in neutron capture therapy[J]. Theranostics, 2024, 14(8): 3193-3212. doi: 10.7150/thno.95034
-
[20]
MA W L, WANG Y Y, XUE Y L, WANG M M, LU C S, GUO W H, LIU Y H, SHU D Y, SHAO G Q, XU Q F, TU D S, YAN H. Molecular engineering of AIE-active boron clustoluminogens for enhanced boron neutron capture therapy[J]. Chem. Sci., 2024, 15(11): 4019-4030. doi: 10.1039/D3SC06222H
-
[21]
MA W L, ZHANG J Y, ZONG J B, REN H Y, TU D S, XU Q F, ZHONG TANG B, YAN H. Luminescence modulation in boron-cluster-based luminogens via boron isotope effects[J]. Angew. Chem. ‒Int. Edit., 2024, 63(52): e202410430. doi: 10.1002/anie.202410430
-
[22]
KREBS J, HÄFNER A, FUCHS S, GUO X, RAUCH F, EICHHORN A, KRUMMENACHER I, FRIEDRICH A, JI L, FINZE M, LIN Z, BRAUNSCHWEIG H, MARDER T B. Backbone-controlled LUMO energy induces intramolecular C—H activation in ortho-bis-9-borafluorene-substituted phenyl and o-carboranyl compounds leading to novel 9, 10-diboraanthracene derivatives[J]. Chem. Sci., 2022, 13(47): 14165-14178. doi: 10.1039/D2SC06057D
-
[23]
JI L, RIESE S, SCHMIEDEL A, HOLZAPFEL M, FEST M, NITSCH J, CURCHOD B F E, FRIEDRICH A, WU L, AL MAMARI H H, HAMMER S, PFLAUM J, FOX M A, TOZER D J, FINZE M, LAMBERT C, MARDER T B. Thermodynamic equilibrium between locally excited and charge-transfer states through thermally activated charge transfer in 1-(pyren-2′-yl)-o-carborane[J]. Chem. Sci., 2022, 13(18): 5205-5219. doi: 10.1039/D1SC06867A
-
[24]
ZHANG C H, LIU X C, WANG J Y, YE Q. A three-dimensional inorganic analogue of 9, 10-diazido-9, 10-diboraanthracene: A Lewis superacidic azido borane with reactivity and stability[J]. Angew. Chem. ‒Int. Edit., 2022, 61(36): e202205506. doi: 10.1002/anie.202205506
-
[25]
YRUEGAS S, AXTELL J C, KIRLIKOVALI K O, SPOKOYNY A M, MARTIN C D. Synthesis of 9-borafluorene analogues featuring a three‑dimensional 1, 1′‑bis(o‑carborane) backbone[J]. Chem. Commun., 2019, 55(20): 2892-2895. doi: 10.1039/C8CC10087J
-
[26]
AXTELL J C, KIRLIKOVALI K O, DJUROVICH P I, JUNG D, NGUYEN V T, MUNEKIYO B, ROYAPPA A T, RHEINGOLD A L, SPOKOYNY A M. Blue phosphorescent zwitterionic iridium complexes featuring weakly coordinating nido-carborane-based ligands[J]. J. Am. Chem. Soc., 2016, 138(48): 15758-15765. doi: 10.1021/jacs.6b10232
-
[27]
PARK K, BAE G, MOON J, CHOE J, SONG K H, LEE S. Synthesis of symmetrical and unsymmetrical diarylalkynes from propiolic acid using palladium-catalyzed decarboxylative coupling[J]. Org. Chem., 2010, 75(18): 6244-6251. doi: 10.1021/jo101398a
-
[28]
DOUCET H, HIERSO J C. Palladium-based catalytic systems for the synthesis of conjugated enynes by Sonogashira reactions and related alkynylations[J]. Angew. Chem. ‒Int. Edit., 2007, 46(6): 834-871. doi: 10.1002/anie.200602761
-
[29]
DOLOMANOV O V, BOURHIS L J, GILDEA R J, HOWARD J A K, PUSCHMANN H. OLEX2: A complete structure solution, refinement and analysis program[J]. J. Appl. Crystallogr., 2009, 42(2): 339-341. doi: 10.1107/S0021889808042726
-
[30]
DOLOMANOV O V, BOURHIS L J, GILDEA R J, HOWARD J A K, PUSCHMANN H. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds[J]. J. Chem. Soc. Perkin Trans. 2, 1987, 12: S1-S19.
-
[31]
SUN F X, TAN S M, CAO H J, XU J K, BREGADZE V I, TU D S, LU C S, YAN H. Palladium-catalyzed hydroboration of alkynes with carboranes: Facile construction of a library of boron cluster-based AIE-active luminogens[J]. Angew. Chem. ‒Int. Edit., 2022, 61(33): e202207125. doi: 10.1002/anie.202207125
-
[32]
SUN F X, TAN S M, CAO H J, LU C S, TU D S, POATER J, SOLÀ M, YAN H. Facile construction of new hybrid conjugation via boron cage extension[J]. J. Am. Chem. Soc., 2023, 145(6): 3577-3587. doi: 10.1021/jacs.2c12526
-
[33]
AL MASUM M, MEGURO M, YAMAMOTO Y. The two component palladium catalyst system for intermolecular hydroamination of allenes[J]. Tetrahedron Lett., 1997, 38(34): 6071-6074. doi: 10.1016/S0040-4039(97)01370-1
-
[34]
WARRATZ S, KORNHAASS C, CAJARAVILLE A, NIEPÖTTER B, STALKE D, ACKERMANN L. Ruthenium(Ⅱ)-catalyzed C—H activation/alkyne annulation by weak coordination with O2 as the sole oxidant[J]. Angew. Chem. ‒Int. Edit., 2015, 54(18): 5513-5517. doi: 10.1002/anie.201500600
-
[35]
MATHER B D, VISWANATHAN K, MILLER K M, LONG T E. Michael addition reactions in macromolecular design for emerging technologies[J]. Prog. Polym. Sci., 2006, 31(5): 487-531. doi: 10.1016/j.progpolymsci.2006.03.001
-
[36]
TEJEDOR D, MÉNDEZ-ABT G, COTOS L, GARCÍA-TELLADO F. Propargyl Claisen rearrangement: Allene synthesis and beyond[J]. Chem. Soc. Rev., 2013, 42(2): 458-471. doi: 10.1039/C2CS35311C
-
[37]
MURAI M, NISHIMURA K, TAKAI K. Palladium-catalyzed double-bond migration of unsaturated hydrocarbons accelerated by tantalum chloride[J]. Chem. Commun., 2019, 55(19): 2769-2772. doi: 10.1039/C9CC00223E
-
[1]
-
Figure 1 Reaction scope Ⅰ
Reaction conditions for 3a-3d: 1 (0.1 mmol), 2 (0.15 mmol), Pd(OAc)2 (x=5%), DCE (4.0 mL), 40 ℃, 3 h; Reaction conditions for 3e and 3f: 1 (0.1 mmol), 2 (0.3 mmol), Pd(OAc)2 (x=5%), DCE (4.0 mL), 40 ℃, 3 h; The B—H—B bridging proton of 3a-3f is omitted for clarity.
Figure 2 Reaction scope Ⅱ
Reaction conditions: 1 (0.1 mmol), 2 (0.15 mmol), Pd(OAc)2 (5%), DCE (4.0 mL), 40 ℃, 3 h; The B—H—B bridging proton of 4a-4d is omitted for clarity.
Figure 3 Crystal structures of carborane-based tricyclic compounds (a) 3c, (b) 4d, (c) 3f, and (d) control compound nido-1; (e) Bond lengths and dihedral angles for the selected crystal structures
Figure 4 Proposed reaction mechanism for five-membered ring product 3
The B—H—B bridging proton is omitted for clarity.
Figure 5 Proposed reaction mechanism for five-membered ring product 4
The B—H—B bridging proton is omitted for clarity.
Table 1. Crystallographic data and corresponding structural refinements of 3c, 3f, and 4d
Parameter 3c 3f 4d Empirical formula C11H20B9N C23H40B9N C18H36B9NSi Formula weight 263.57 430.25 391.86 Temperature/K 296.15 296.15 296.15 Crystal system Monoclinic Monoclinic Triclinic Space group P21/n P21/n P1 a/nm 1.072 6(2) 0.950 99(5) 0.781 78(7) b/nm 0.998 8(3) 1.537 36(10) 1.503 90(14) c/nm 1.495 8(4) 1.795 07(11) 2.143 40(19) α/(°) 73.108(3) β/(°) 105.320(8) 95.099(2) 80.767(3) γ/(°) 84.063(3) Volume/nm3 1.545 5(7) 2.614 0(3) 2.375 8(4) Z 4 4 1 Dc/(g·cm-3) 1.133 1.093 1.096 μ/mm-1 0.056 0.058 0.104 F(000) 552.0 928.0 840.0 Crystal size/mm 0.12×0.11×0.1 0.13×0.11×0.1 0.13×0.12×0.11 2θ range/(°) 4.96-55.358 4.556-55.052 4.01-55.034 Reflection collected 10 018 24 053 21 109 Data, number of restraints, number of parameters 3 544, 0, 194 6 023, 3, 414 10 772, 0, 567 Goodness-of-fit on F2 1.115 0.966 1.024 R1, wR2 [I≥2σ(I)] 0.120 2, 0.274 0 0.077 2, 0.178 6 0.079 5, 0.168 3 R1, wR2 (all data) 0.239 7, 0.312 4 0.132 6, 0.208 1 0.165 1, 0.210 7  下载: 导出CSV
下载: 导出CSV
Table 2. Screening of catalysts, solvents, temperature, and reaction timea
Entry Catalyst Solvent Time/h T/℃ Yieldb/% 1 Pd(OAc)2 DCE 3 40 70 2 Pd(PPh3)4Cl2 DCE 12 40 Trace 3 Cu(OAc)2 DCE 12 40 — 4 [Cp*RhCl2]2 DCE 12 40 — 5 [(p-cymene)RuCl2]2 DCE 12 40 — 6 Ni(OAc)2 DCE 12 40 — 7 Co(OAc)2 DCE 12 40 — 8 Pd(OAc)2 DCE 3 RT 50 9 Pd(OAc)2 DCE 3 60 65 10 Pd(OAc)2 DCE 3 80 61 11 Pd(OAc)2 DCM 3 40 60 12 Pd(OAc)2 CH3CN 3 40 — 13 Pd(OAc)2 THF 3 40 — 14c Pd(OAc)2 DCE 3 40 60 15 None DCE 3 40 — 16d Pd(OAc)2 DCE 3 40 — a Reactions were conducted at 0.1 mmol scale in 2.0 mL of solvent in a Schlenk tube; b Yield of isolated product; c In air; d Phenyl instead of pyridyl in compound 1a.
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 5
- 文章访问数: 620
- HTML全文浏览量: 80
