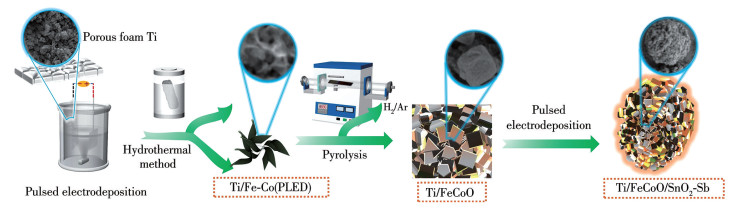
图 1.
Ti/FeCoO/SnO2-Sb复合电极的制备路线示意图
Figure 1.
Schematic diagram of the route of Ti/FeCoO/SnO2-Sb composite electrode

在传统高级氧化技术中,过氧化氢(H2O2)常用于在芬顿或类芬顿反应中生成羟基自由基(·OH),以降解水体中的有机污染物。而基于过氧单硫酸盐(PMS)活化的高级氧化技术所使用的原料在储存与运输方面的成本低于H2O2,被视为替代传统H2O2类高级氧化技术的有力方案[1]。PMS活化产生的硫酸根自由基(SO4·-)具有较高的氧化还原电位(2.50~3.10 V)、更好的选择性和较长的半衰期(30~40 μs)[2-4]。然而,PMS的本征活性较低,且其生成自由基的反应动力学过程缓慢,严重限制了其在污染物降解中的实际应用[5-6]。电催化氧化作为一种高效的高级氧化技术,能够在外加电场作用下,通过电极界面的直接电子转移或间接氧化反应,将有机污染物深度矿化为无机离子、CO2和H2O[7-12]。为进一步突破单一电化学氧化体系的效率瓶颈,研究者致力于将其与其他高级氧化技术耦合。其中,电化学协同活化PMS体系因其显著的协同增效潜力而备受关注。在该耦合体系的构建中,阳极材料起着决定性作用。目前常见的阳极主要包括掺硼金刚石(BDD)[13-14]、碳基材料(如石墨、碳纳米管)[15-16],以及金属氧化物电极(如SnO2-Sb[17-19]、PbO2[20-21]、RuO2[22-23]、MnO2[24-25]和IrO2[26-27]等)。尽管已有研究表明,BDD和碳基材料等阳极能有效活化PMS产生活性氧物种(ROS)以去除污染物[28-30],但BDD高昂的成本限制了其规模化应用,而碳基材料在高电位下易发生电化学腐蚀而导致失活,严重影响了系统的稳定性。相比之下,SnO2-Sb凭借其高析氧电位、优良的导电性及适中的成本,被视为极具潜力的候选材料之一。
然而,SnO2-Sb电极在实际应用中仍面临着提升材料稳定性与界面反应动力学的双重挑战。一方面,该电极服役寿命较短,其活性层在高电流密度下易发生机械剥落与钝化,导致催化性能迅速衰减,这是制约其工业化应用的最突出瓶颈。另一方面,传统SnO2-Sb涂层有限的比表面积限制了有效活性位点的暴露数量,导致其本征电催化效率及PMS活化速率难以进一步提升。更为关键的是,尽管SnO2-Sb与PMS的结合展现出降解持久性污染物的潜力,但该体系核心的界面化学机制仍不明晰:PMS分子在SnO2-Sb表面的具体吸附构型与电子转移路径尚待揭示;且在电场辅助下,污染物降解究竟是由自由基主导还是非自由基路径驱动,其深层反应机理亦亟待深入阐明。
因此,我们提出以三维泡沫钛(Ti)为基底,构建泡沫Ti/FeCo-Fe2O3-CoFe2O4/SnO2-Sb(简称为Ti/ FeCoO/SnO2-Sb)复合阳极。该设计策略旨在通过引入FeCoO多相物质(即FeCo合金、Fe2O3、CoFe2O4)作为关键中间层,利用其与SnO2-Sb活性层的结构耦合与功能互补,突破单一体系的性能局限。相较于传统SnO2-Sb电极或粉末态催化体系,该复合电极不仅保留了SnO2-Sb优异的析氧电位与导电骨架的稳定性,更借助Fe/Co双金属活性位点的协同效应,重构了电极界面微环境,显著增强了PMS的吸附与活化效能。通过强化界面电子转移并诱导多种ROS的高效生成,该体系实现了对有机污染物的多路径协同降解,为解决电化学/PMS耦合体系中活性组分利用率低这一关键难题提供了创新的设计思路与理论依据。
实验试剂及材料表征与测试信息详见Supporting information。将等物质的量的Fe(NO3)3·9H2O与Co(NO3)2·6H2O一并溶解于100 mL去离子水中,配制二者浓度均为0.1 mol·L-1的电沉积溶液。电沉积过程在电化学工作站的两电极体系中进行,其中工作电极为泡沫Ti(70 mm×10 mm×2 mm),对电极为铂网。电沉积反应通过多电位阶跃法(multi-potential steps,MPS)进行控制,具体参数:施加电位在-0.6~0 V之间交替变化,对应的保持时间分别为100和50 s,整个沉积过程共进行20个循环。电沉积完成后,将电极从溶液中取出,用去离子水反复清洗以去除表面残留电解质,随后置于60 ℃真空干燥箱中干燥2 h,得到Ti/Fe-Co基层状金属化合物脉冲电沉积(PLED)电极,命名为Ti/Fe-Co(PLED)。该PLED层主要充当与Ti基底强耦合的导电过渡和成核“种子”缓冲内层,可显著增强界面结合强度,降低界面电荷传递阻抗,并为后续Fe-Co基纳米结构的均匀生长提供成核位点。
接着,将Fe(NO3)3·9H2O(0.04 mol·L-1)和Co(NO3)2·6H2O(0.02 mol·L-1)共同溶解于100 mL去离子水中,得到溶液A;同时,将NaOH(0.096 mol·L-1)和Na2CO3(0.08 mol·L-1)共同溶解于100 mL去离子水中,得到溶液B。随后,在持续搅拌条件下将溶液B滴加至溶液A中,并维持体系充分混合直至形成均一悬浊液;继而使用H2SO4和NaOH调节pH至10。将上述反应混合液转移至含有聚四氟乙烯内衬的不锈钢高压釜中,加入上述制备好的Ti/Fe-Co(PLED)电极,于100 ℃下水热反应24 h。反应结束后冷却至室温,将所得产物分别用去离子水与乙醇各洗涤10次以去除表面残留盐与吸附物,随后置于60 ℃的真空干燥箱中干燥,得到负载Fe-Co基前驱体的Ti基底。以上步骤是通过在含CO32-、OH-的碱性体系中,在Ti/Fe-Co(PLED)表面异相生长高比表面积的Fe-Co基层状前驱体,从而构建2层缓冲结构。干燥后的前驱体在H2/Ar(体积比为5∶95)混合气体中,于200 ℃恒温处理180 min,以促进前驱体的相转化与结构定型,最终获得Ti/FeCoO电极。
采用电化学共沉积法制备Ti/FeCoO/SnO2-Sb电极材料。首先称取C4H6O6、Na4P2O7·10H2O、SnCl2·2H2O、SbCl3,并加入0.8 g·L-1明胶,共同溶解于100 mL去离子水中,配制得到各组分浓度分别为0.047、0.42、0.013和0.02 mol·L-1的电沉积溶液。电沉积过程采用两电极体系进行,其中铂片作为对电极,Ti/FeCoO(H,Ar)电极作为工作电极。沉积过程由脉冲直流电源进行控制。在电沉积过程中施加脉冲直流电流,通电阶段的电流密度设定为-50 mA·cm-2,单次脉冲持续时间为0.1 s。整个沉积过程采用间歇式操作模式,每个循环包括0.1 s通电和0.1 s断电,共进行24 000个循环。电沉积完成后,样品用去离子水反复清洗以去除表面残留电解质,随后置于马弗炉中500 ℃恒温煅烧2 h,最终获得Ti/FeCoO/SnO2-Sb复合电极材料。另外,将预处理后的泡沫Ti作为工作电极,采用与Ti/FeCoO/SnO2-Sb制备过程中相同的电沉积液和脉冲电沉积条件进行Sn-Sb共沉积,最终获得Ti/SnO2-Sb电极。
在带有旋转搅拌功能的100 mL圆柱形电解池中进行了相关污染物的电催化降解测试,如甲基橙(MO)、亚甲蓝(MB)、罗丹明B(RhB)等。以上文所述自制电极(70 mm×10 mm×2 mm)作为阳极,尺寸相同的铜片作为阴极。2个电极平行放置,相距1 cm,并与直流电源(DC,IT6874A,ITECH)相连。相关实验的具体操作条件如下:电流密度为50 mA·cm-2,初始溶液pH为4,初始染料浓度为20 mol·L-1,PMS浓度为0.100 0 mmol·L-1,电解质为Na2SO4(0.025 0 mol·L-1)。实验过程中,每30 min从电解池中取样一次,分析污染物的去除率、脱色效率和化学需氧量(COD)。所有实验均在40 ℃下进行。
图 1为Ti/FeCoO/SnO2-Sb复合电极的制备路线示意图。图 2a和2b展示了电沉积的Fe-Co基层状金属化合物的SEM图像,可以观察到,Fe-Co基层状金属化合物以均匀致密的纳米片形式覆盖于Ti基底表面,纳米片边界清晰,排列较为规整,呈现典型的层状结构特征。统计结果表明,Fe-Co基层状金属化合物纳米片的平均厚度为9.3 nm,横向长度约为83.5 nm。经水热处理后,Ti/FeCoO电极表面形貌发生明显演变。如图 2c和2d所示,原有纳米片结构部分转化为Fe/Fe2O3基不规则纳米立方体颗粒,其平均尺寸约为160 nm,尺寸分布较为集中,颗粒边缘清晰,表面较为平整,且未观察到明显团聚现象,表明其在后续热处理过程中具有较好的抗团聚和抗烧结能力,可较好地保持颗粒形貌与分散状态。同时,颗粒之间保留一定数量的孔隙结构,为后续活性层的嵌入提供了空间。为进一步解析其微观结构,采用HRTEM对Ti/FeCoO进行表征(图 2e和2f)。结果显示,该样品内部呈现多相共存特征:晶面间距为0.211、0.164和0.143 nm,分别对应Fe的(111)晶面、Fe2O3的(211)晶面以及FeCo合金的(200)晶面,说明水热与还原处理诱导了FeCoO的局部晶相重构。对应的扫描透射电子显微镜-能谱(STEM-EDS)元素映射结果(图 2g)表明,Fe、Co和O元素在整个结构中分布均匀,进一步证实了FeCoO中间层的成功构筑。在此基础上,通过PLED在FeCoO表面负载SnO2-Sb活性层。图 2h和2i显示,沉积后电极表面形成由SnO2-Sb纳米颗粒堆叠构成的粗糙包覆层,颗粒平均尺寸约为5 nm。该纳米颗粒层能够有效填充FeCoO表面的孔隙结构,有利于构建紧密的复合界面。在HRTEM图像(图 2j)中观察到0.264 nm的晶面间距,对应SnO2的(101)晶面;同时,STEM-EDS映射结果(图 2k)清晰显示Sn和Sb元素均匀分布,证实SnO2-Sb活性层已成功负载于FeCoO中间层之上。上述结果表明,所构筑的Ti/FeCoO/SnO2-Sb复合电极具有清晰的层级结构和良好的界面结合特征,为后续电化学性能与反应机理研究奠定了结构基础。

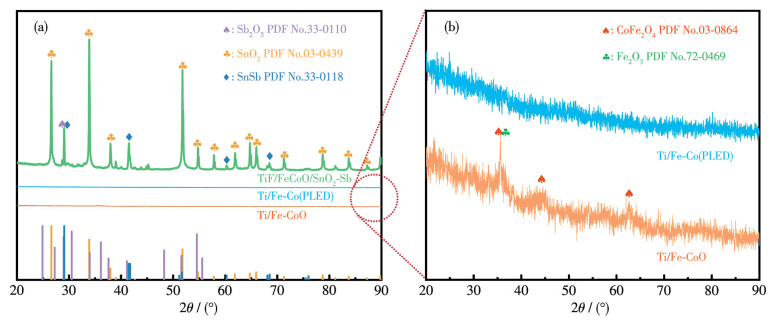
图 3展示了不同电极的XRD图。如图 3a所示,仅Ti/FeCoO/SnO2-Sb电极呈现出清晰可辨的衍射峰,这些峰主要对应SnO2与Sb2O5,并出现SnSb的特征信号,表明SnO2-Sb催化层已成功负载并形成于电极表面[33]。从图 3b中可以清晰观察到Ti/Fe-Co(PLED)和Ti/FeCoO电极的晶相信息。其中Ti/Fe-Co(PLED)以弥散背景为主,未出现明显晶相衍射峰,说明其主要呈无定形态。而Ti/FeCoO出现可分辨的特征衍射峰,其中2θ=35.6°处的衍射峰归属于Fe2O3的(110)晶面,而2θ=35.4°、43.1°和62.5°处的衍射峰分别对应CoFe2O4的(113)、(024)和(208)晶面,表明经水热与H2/Ar混合气氛煅烧处理后,FeCo合金与复合氧化物(Fe2O3-CoFe2O4)的协同晶化赋予了电极稳定的晶体框架。
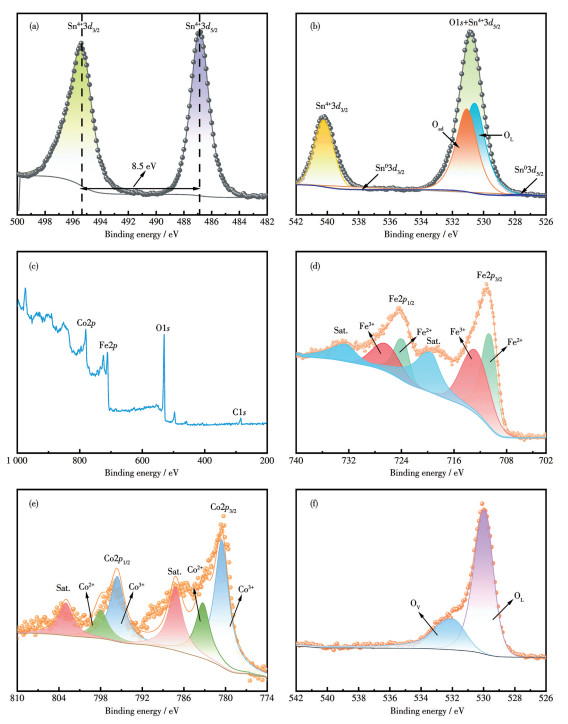
为进一步揭示Ti/FeCoO/SnO2-Sb电极表面元素组成及化学价态,对其进行了X射线光电子能谱(XPS)分析。图 4a为Ti/FeCoO/SnO2-Sb电极的高分辨Sn3d XPS谱图。该谱图可解卷积为位于487.3和495.8 eV的2个特征峰,分别对应于Sn4+3d5/2和Sn4+3d3/2,自旋-轨道分裂能为8.5 eV,与文献报道中SnO2的特征峰一致[34],表明Sn主要以Sn4+价态存在。进一步对Sb3d高分辨率XPS谱图进行分析(图 4b),其可解卷积为2个特征峰,分别位于531.0 eV(Sb4+3d5/2)和540.3 eV(Sb4+3d3/2)。XPS分析结果证实了电沉积的SnO2-Sb中Sb4+的存在。此外,在528.3 eV(Sb03d5/2)和537.7 eV(Sb03d3/2)处检测到的峰进一步证实了金属Sb的存在(二者之间的结合能差为9.4 eV)[35-36]。值得注意的是,Sb核心能级的自旋轨道分裂成分(特别是Sb3d5/2)与O1s峰存在重叠现象。O1s谱图可以分成2个峰(531.8和530.3 eV),分别归属于吸附氧(Oad)和晶格氧(OL),其中531.8 eV处的峰实际可以归属于氧空位[37],这是由于在表面氧空位上存在解离吸附的分子氧。在Ti/FeCoO电极中清晰检测到Fe、Co和O元素(图 4c)。图 4d为Ti/FeCoO电极中Fe2p的高分辨XPS谱图。位于711.0和724.1 eV的峰分别归属于Fe2+2p3/2和Fe2+2p1/2,而713.4和726.6 eV的峰分别对应于Fe3+2p3/2和Fe3+2p1/2。此外,在718.6和732.5 eV处出现的卫星峰进一步证实了Fe3+的存在[38],说明Fe2+与Fe3+在电极表面共存。相应地,Co2p高分辨XPS谱图(图 4e)则证实了Co3+与Co2+的共存[38](Co3+2p3/2:780.0 eV,Co3+2p1/2:795.3 eV,Co2+2p3/2:781.7 eV,Co2+2p1/2:797.2 eV,卫星峰:786.6和803.4 eV)。综上所述,Ti/FeCoO/SnO2-Sb电极表面同时存在Fe2+/Fe3+与Co2+/Co3+可逆氧化还原对,并伴随丰富的氧空位结构。多价态金属位点之间的协同耦合及其动态电子转移过程,为PMS活化和活性物种持续生成提供了有效的电子循环通道,从而构建了高效稳定的催化反应体系。此外,在O1s谱图中529 eV附近的峰归属于Fe—O键(OL),而更高结合能处的特征峰通常与氧空位(Ov)相关(图 4f)。
为了研究电催化活性,对Ti/SnO2-Sb和Ti/ FeCoO/SnO2-Sb电极进行了线性扫描伏安法(LSV)、循环伏安法(CV)和电化学阻抗谱(EIS)测试。LSV测试在室温下进行,使用50 mmol·L-1 Na2SO4溶液作为电解质,以50 mV·s-1的扫描速率测定不同电极的开路电位(OEP)。如图 5a所示,Ti/SnO2-Sb电极的OEP为2.16 V,而Ti/FeCoO/SnO2-Sb电极的OEP降低至1.76 V。OEP的降低表明引入FeCoO中间层后,电极表面电子结构发生调变,从而降低了反应启动所需的能垒,有利于后续氧化反应的进行[39]。
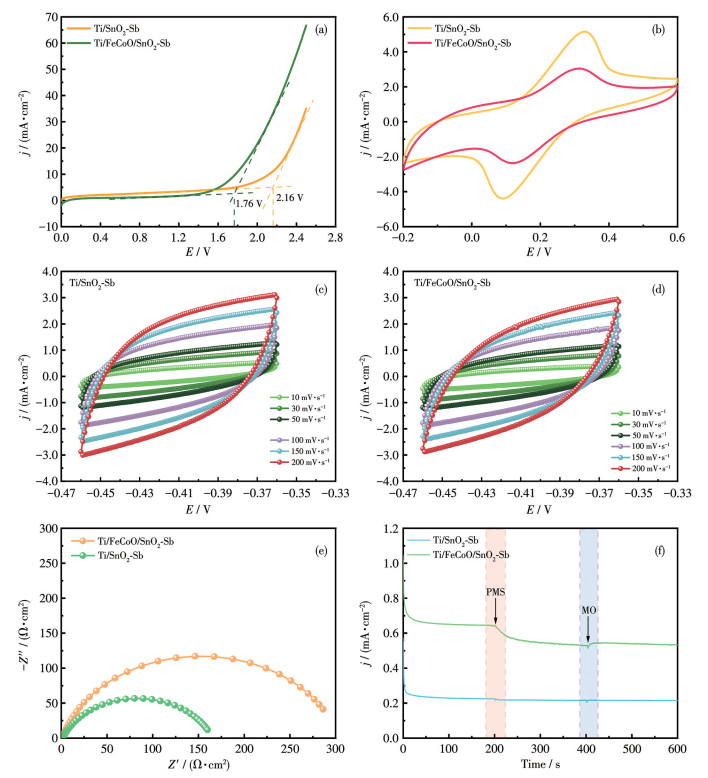
图 5b展示了2种电极在50 mV·s-1下、5 mmol·L-1 K3[Fe(CN)6]/K2[Fe(CN)4]溶液中以0.1 mol·L-1 KCl为电解质的CV曲线。可以观察到,二者均呈现一对可逆的氧化还原峰,表明电极表面存在赝电容行为。与Ti/SnO2-Sb相比,Ti/FeCoO/SnO2-Sb的峰电流略有降低,这主要归因于FeCoO层自身导电性较低,在一定程度上增加了电荷传输阻力。然而,该中间层的引入并未破坏电极的可逆氧化还原特性。为定量比较2种电极的电化学活性表面积(ECSA),在非法拉第电位区间(-0.46~-0.36 V)内,以20~200 mV·s-1的扫描速率记录CV曲线(图 5c和5d)。在选定电位处记录正向与反向扫描过程的电流密度(分别为jf和jr),并计算Δj/2(Δj=jf-jr)。如图S1所示,Δj/2与扫描速率(v)呈线性关系,其斜率对应电极的双电层电容CdL。结果表明,Ti/SnO2-Sb电极的CdL为19.37 mF·cm-2,而Ti/FeCoO/SnO2-Sb电极的CdL为16.76 mF·cm-2。相较Ti/SnO2-Sb,Ti/Fe-Co/SnO2-Sb电极的CdL略有降低,这主要归因于FeCoO中间层本征导电性相对较低,在一定程度上削弱了双电层电荷积累能力。然而,尽管CdL略有下降,FeCoO的引入有效构建了多价态Fe/Co活性位点与SnO2-Sb之间的协同界面,为后续电化学反应中的电子转移与反应动力学调控提供了有利条件。进一步采用EIS对电极界面电荷传输特性进行分析。如图 5e所示,随着FeCoO中间层的引入,Nyquist曲线中高频区半圆直径明显增大,说明电荷转移电阻有所增加,该结果与CV测试中观察到的峰电流变化趋势一致,表明FeCoO层成功构筑并稳定附着于电极表面。
为进一步研究氧化反应及污染物降解过程中的电子转移行为,采用计时安培法研究了Ti/FeCoO/SnO2-Sb电极在PMS与MO体系中的电子转移过程(图 5f)。实验中,在固定电位条件下依次向体系中加入100 μmol·L-1 PMS和100 μmol·L-1 MO。当PMS加入后,电流出现明显的瞬时下降,表明PMS在电极表面发生吸附并接受电子,形成高反应活性的中间体*PMS[40-41]。随后引入MO后,电流出现部分回升,说明*PMS与污染物之间发生电子转移反应并被持续消耗。相比之下,以Ti/SnO2-Sb作为阳极时,在加入PMS和MO后电流变化不明显,表明其直接电子转移能力有限。上述结果表明,FeCoO中间层的引入显著增强了电极在PMS体系中的直接电子转移(DET)能力,从而有利于污染物的高效降解。
鉴于有机染料对生态环境的潜在危害,选取了具有代表性的4种有机染料作为目标污染物,包括阳离子型RhB、阴离子型MO以及酸性红73,用于评估Ti/FeCoO/SnO2-Sb/PMS和Ti/SnO2-Sb/PMS两种体系的电催化降解性能。以COD去除率作为评价指标,根据式S1和S2计算COD去除率和COD去除过程中的表观反应速率常数(kobs),实验结果如图 6a和6d所示。结果表明,在Ti/FeCoO/SnO2-Sb+PMS体系中,COD去除率在150 min内即可达到100%,对应的kobs为0.016 6 min-1,显著高于Ti/SnO2-Sb+PMS体系的0.013 5 min-1。这表明引入FeCoO中间层后,电极对有机染料的电催化氧化能力得到明显提升。为进一步客观评价本工作中电极体系在处理印染废水方面的性能水平,将其与现有文献中报道的几种典型电极体系进行了对比分析(表S1)。结果显示,Ti/FeCoO/SnO2-Sb+PMS体系的COD去除率均优于多数已报道体系[42-43],充分体现了电催化过程与PMS活化之间的协同作用在提升污染物去除率方面的显著优势。
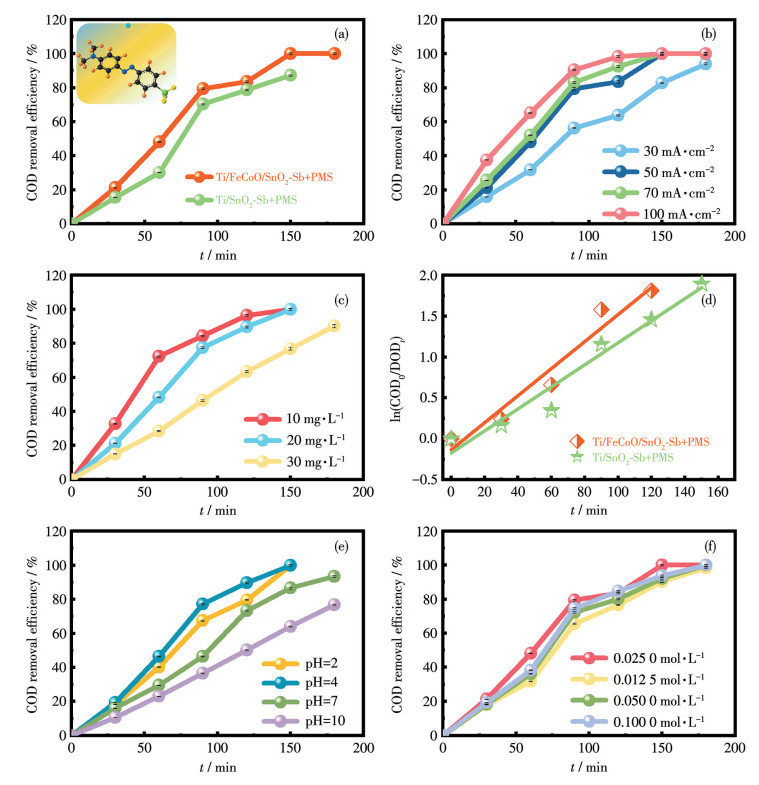
Inset: the molecular structure of MO; Reaction conditions in a and d: pollutant concentration: 20 mg·L-1, j=50 mA·cm-2, and pH=4, cPMS=0.100 0 mmol·L-1.
在电化学污染物降解过程中,电流密度是影响反应效率的关键工艺参数之一。如图 6b所示,在其他实验条件保持不变(初始MO质量浓度为20 mg· L⁻1,初始溶液pH=4,PMS浓度为0.100 0 mmol·L-1,电解质为0.025 0 mol·L-1 Na2SO4,下文工艺参数探索中,均保持其他最佳条件不变的情况下改变相应参数)的条件下,系统考察了不同电流密度对MO降解行为的影响。结果表明,随着电流密度由30 mA·cm-2提高至100 mA·cm-2,表观反应速率常数由0.014 6 min-1显著提升至0.033 7 min-1(图S2a)。该趋势表明,较高的电流密度有利于提高电极表面的ROS(如·OH)的生成速率,从而加快有机污染物的氧化降解过程。然而,过高的电流密度可能引发副反应并增加能耗。基于对COD去除率与能耗的综合考量,50 mA·cm-2被确定为本体系最优的电流密度条件。该电流密度既可保证较高的有机污染物COD的去除率,又能够有效避免不必要的能量损耗。
进一步系统考察了初始MO质量浓度(10、20和30 mg·L-1)对电催化降解性能的影响(图 6c)。结果表明,随着初始MO质量浓度的升高,其COD去除率呈现逐渐下降的趋势。这一现象主要归因于反应过程中中间产物的累积效应:一方面,中间产物会与MO竞争·OH,降低有效氧化能力;另一方面,其在Ti/ FeCoO/SnO2-Sb/PMS体系中占据部分电极表面活性位点,加速阳极钝化过程。而随着初始质量浓度的进一步升高,降解过程逐渐转变为由电流强度所决定的ROS生成速率控制。综合分析可知,当MO初始质量浓度为20 mg·L-1时,体系表现出最佳的降解性能,为后续工艺参数优化提供了重要实验依据。
溶液初始pH是影响电催化降解过程的重要因素之一。为系统评估pH对反应性能的影响,采用0.1 mol·L-1 H2SO4和0.1 mol·L-1 NaOH溶液对反应体系进行pH调节。实验结果显示,表观反应速率常数随pH变化呈现显著差异:随着pH由2提高至4,表观反应速率常数由0.016 6 min-1显著提升至0.025 8 min-1;而在中性至碱性条件下,pH由7升高至10时,表观反应速率常数则由0.015 3 min-1逐渐下降至0.007 9 min-1(图S2b)。尤其是在pH=10条件下,COD去除率明显降低,这可能与碱性环境下ROS生成能力减弱以及副反应加剧有关(图 6e)。综合上述结果可知,pH=4时体系具有最优的脱色效果和降解活性。
此外,还系统探究了电解质浓度对污染物去除性能的影响机制。如图 6f所示,在保持其他反应参数不变的条件下,考察了Na2SO4浓度在0.012 5~0.100 0 mol·L-1范围内的变化对降解动力学的影响。结果表明,电解质浓度对体系表观反应速率具有显著调控作用,适当提高电解质浓度有助于改善溶液导电性并促进电极反应的进行。当Na2SO4浓度为0.025 0 mol·L-1时,Ti/FeCoO/SnO2-Sb+PMS体系在150 min内即可实现MO的完全去除。从机理角度分析,在较低电解质浓度条件下,溶液的导电性不足限制了电化学过程中ROS(如·OH)的有效生成;而当电解质浓度超过0.025 0 mol·L-1后,尽管溶液电导率进一步提高,但MO的COD去除率并未出现显著提升,反而可能导致电解质资源浪费及运行成本增加。因此,综合考虑反应效率与经济性,0.025 0 mol·L-1 Na2SO4被确定为本体系中MO降解与脱色的最优电解质浓度。
进一步根据式S3计算体系的平均电流效率(average current efficiency,ACE),结果如图 7a所示。Ti/FeCoO/SnO2-Sb/PMS表现出最高的ACE值,表明该体系在MO矿化过程中具有更高的电能利用效率。值得注意的是,除MO外,该催化体系对MB、RhB及AR73等其他染料同样表现出高效的去除能力,这表明其具有良好的广谱适用性(图 7b、S3)。如图 7c所示,在Ti/FeCoO/SnO2-Sb/PMS体系催化反应后,MO溶液完全褪色,这主要归因于体系中生成的ROS对染料分子共轭结构的有效攻击与断裂,从而实现高效降解与矿化。
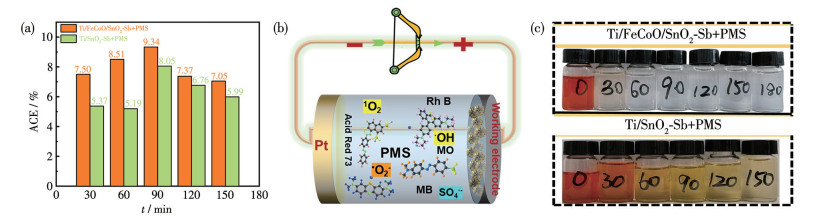
In c: the time from left to right was 0, 30, 60, 90, 120, 150, and 180 min, respectively.
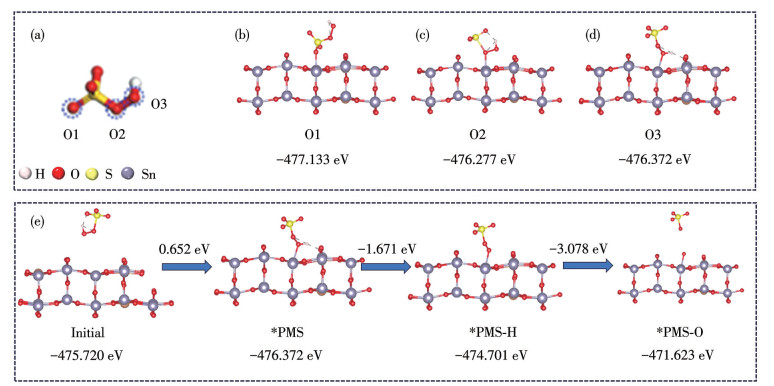
为进一步揭示PMS在SnO2-Sb表面的活化机理及其在电化学氧化过程中参与MO去除的关键反应步骤,采用DFT对相关结构进行了计算与分析。优化后的孤立PMS分子构型如图 8a所示,其中O1、O2和O3分别代表PMS分子中具有不同化学环境的氧原子。如图 8b~8d所示,分别构建了PMS分子通过末端氧O1、O2和O3位点吸附于SnO2-Sb(110)晶面表面Sn位点的吸附构型。当PMS通过O1位点与SnO2-Sb表面Sn原子形成配位吸附结构(PMS-O1/SnO2-Sb)时,其吸附能为-477.133 eV。而通过O2和O3位点形成的吸附构型(PMS-O2/SnO2-Sb和PMS-O3/SnO2-Sb)的吸附能分别为-476.277和-476.372 eV。相比之下,O1位点吸附构型具有更低的吸附能,表明SnO2-Sb表面Sn位点与PMS分子中O1原子的相互作用最为稳定,且该位点具有更强的供电子能力,有利于PMS分子的电子重排与活化。在此基础上,进一步计算PMS在SnO2-Sb表面发生电化学活化过程中的反应路径(图 8e)。首先,PMS分子通过其末端氧原子吸附于Sn位点,形成表面吸附中间体*PMS(PMS-SnO2-Sb),其相对吉布斯自由能为0.652 eV。随后,在外加电场作用下,*PMS发生质子耦合电子转移过程,形成表面质子化中间体 *PMS-H,其自由能降至-1.671 eV。最终,随着 *PMS-H结构进一步解离,生成了SO4·-及表面残留中间体*PMS-O,其自由能变化为-3.078 eV。上述结果表明,SnO2-Sb表面Sn位点能够稳定PMS的吸附并有效促进其逐步转化为高反应活性的自由基物种。所生成的ROS可进一步攻击MO分子,诱导其断链并转化为小分子产物,从而实现污染物的高效去除。
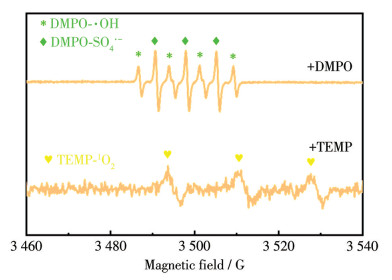
为进一步阐明Ti/FeCoO/SnO2-Sb+PMS体系中活性物种的生成及其在MO降解过程中的作用机理,采用电子顺磁共振(EPR)技术对反应体系进行了表征。以5,5-二甲基-1-吡咯啉-N-氧化物(5,5-dimethyl-1-pyrroline-N-oxide,DMPO)作为自旋捕获剂,在水溶液中获得了特征信号强度比为1∶2∶1∶2∶1∶2∶1的EPR谱图,该七重峰信号可归属于DMPO-·OH与DMPO-SO4·-自旋加合物,表明反应体系中同时生成了·OH和SO4·-。具体而言,位于中心位置的特征四重峰主要来源于DMPO-·OH,而相对外侧的信号则对应于DMPO-SO4·-,说明PMS在电化学作用下可被有效活化,产生多种强氧化性自由基物种。此外,以2,2,6,6-四甲基哌啶(2,2,6,6-tetramethylpiperidine,TEMP)作为单线态氧(1O2)的捕获剂,在Ti/FeCoO/SnO2-Sb+PMS体系中检测到由TEMP与1O2反应生成的2,2,6,6-四甲基哌啶-1-氧基(2,2,6,6-tetramethylpiperidin-1-oxyl,TEMPO)所对应的EPR信号(图 9)。该结果清晰表明体系中1O2的生成。综合EPR分析结果可知,在Ti/FeCoO/SnO2-Sb+PMS体系中,PMS可在电极表面被高效活化,同时产生·OH、SO4·-及1O2等多种ROS,这些活性物种协同作用,共同参与并主导了MO的降解过程。
接着对活性物种的生成机制以及MO的降解机制进行了讨论。在电场作用下,阳极表面金属活性位点(记为M)可与水分子发生相互作用,形成表面吸附态羟基自由基M(·OH),同时释放质子和电子(式1)。该过程表明电极表面能够直接促进·OH的生成,为后续氧化反应提供重要活性物种来源。与此同时,溶液中的PMS在电场和电极表面作用下被活化,形成高能态的*PMS(式2)。该活化过程为PMS进一步转化为多种强氧化性活性物种奠定了基础。随后,*PMS可通过电子转移过程进一步发生分解,生成SO4·-和·OH(式3)。在均相反应过程中,Co2+与Fe2+共同向PMS提供电子,引发其分解(式4和5)。随后,HSO5-将Fe3+/Co3+还原,生成过氧单硫酸根自由基(SO5·-)和Fe2+/Co2+(式6和7),从而实现了Fe2+/Fe3+与Co2+/Co3+之间的氧化还原循环。此外,PMS可以通过溶液中·OH的活化生成SO4·-(式8),而PMS的自分解导致1O2的生成(式9和10)。此外,电解质SO42-失去电子(e-)产生SO4·-(式11)。阴极的氧气还原反应导致溶液中形成·O2-(式12),同时,电活化使PMS也能够生成·O2-(式13)。基于上述反应机制分析,在电催化协同PMS活化过程中,体系内生成的·O2-与·OH发生反应,进而生成具有强氧化性的1O2(式14)。这一反应路径揭示了电催化降解与PMS活化之间的协同作用机制。体系内多种ROS的协同参与,包括·O2-、·OH、1O2等,共同构成了高效的氧化体系。这种多重活性物种参与的协同作用机制显著提升了MO的COD去除率,为有机污染物的高效去除提供了新的思路(式15)。
为明确Ti/FeCoO/SnO2-Sb+PMS体系中的活性物种构成,我们开展了猝灭实验。甲醇(MeOH)通常用作·OH与SO4·-的通用猝灭剂,而叔丁醇(TBA)则可特异性地猝灭·OH。根据式S4可得到MO的降解效率随时间变化的降解曲线。如图S4所示,当向反应体系加入TBA时,MO降解速率仅轻微降低,表明·OH的贡献较为有限。相比之下,MeOH表现出比TBA更显著的抑制作用,证实SO4·-在反应中发挥作用。此外,采用对苯醌(p-BQ)作为·O2-猝灭剂时,MO去除率受到轻微抑制,说明·O2-在降解过程中的作用较弱。而糠醛(FFA)对MO降解具有显著抑制效果,表明1O2可能参与主要反应过程,在30 min内MO的降解效率仅为51.3%。以上结果证明,Ti/ FeCoO/SnO2-Sb+PMS体系同时存在自由基与非自由基2种反应路径,其中SO4·-与1O2在降解过程中起主导作用。
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
|
|
(6) |
|
|
(7) |
|
|
(8) |
|
|
(9) |
|
|
(10) |
|
|
(11) |
|
|
(12) |
|
|
(13) |
|
|
(14) |
|
|
(15) |
通过安捷伦科技7100型液质联用仪(HPLC-MS)对MO及其催化产物在负和正离子化模式下进行了扫描和鉴定。基于MS谱图,使用安捷伦LC-MS质谱定性软件和个人化合物数据库与谱库(PCDL)(图S5),初步鉴定了含苯环化合物的种类。
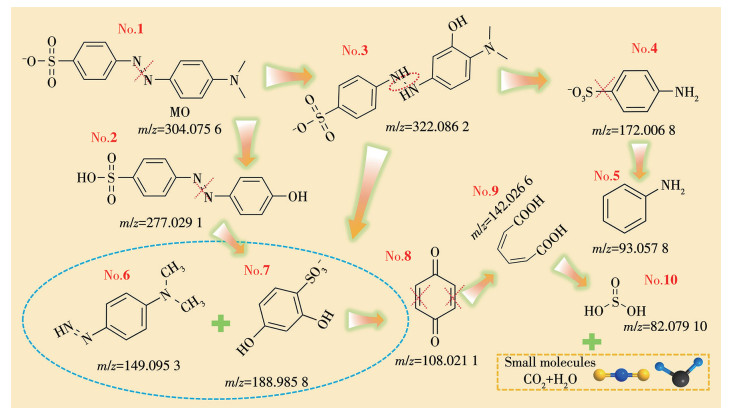
表S2显示了MO降解产物的不同质荷比、推定分子式及对应结构信息。MO分子是由偶氮键(—N=N—)与芳环构成的典型共轭体系,因此,其降解过程可理解为在·O2-、·OH、1O2和SO4·-活性物种参与下,共轭结构被逐步破坏并发生深度氧化的过程。基于HPLC-MS的检测结果,并结合关键芳香族中间体及羧酸类产物的出现规律,提出了如图 10所示的可能降解路径。反应初期,·OH优先攻击MO发色团中心(—N=N—),诱导偶氮键断裂,从而导致溶液快速脱色。化合物1被鉴定为MO,其前体[M-Na](m/z=304.075 6)仅在原始溶液及降解30 min的样品中检测到,说明MO在早期迅速发生结构转化[44]。随后,·OH进一步攻击电子云密度较高的二甲氨基取代芳环的对位,发生亲电加成并实现羟基取代,使芳环上的C—H键转化为C—OH键,生成单羟基化产物(m/z=322.086 2)。与此同时,连接磺酸基芳环与偶氮基团之间的C—N键发生断裂,导致含磺酸基片段脱离,并生成4-偶氮-N,N-二甲基苯胺(m/z=149.095 3)。对氨基苯磺酸(m/z=172.006 8)的检出表明偶氮键进一步彻底裂解,并伴随发生N-去甲基化过程(N—CH3键断裂)。在1O2等ROS的持续攻击下,芳环结构(如1,4-苯醌中间体)发生氧化开环,本质上表现为环内C=C与C—C键的断裂;生成的小分子羧酸(如草酸)继续经历C—C键裂解,最终实现矿化,转化为CO2和H2O。
我们成功构建了新型Ti/FeCoO/SnO2-Sb复合电极,其核心在于通过引入FeCoO中间层,实现了电极微观结构与催化性能的协同优化。该设计不仅使电极表面形成了更为致密、均匀的纳米级结构,有效抑制了颗粒粗化,更显著提升了电极表面的电子传输效率,从而在电催化活化PMS降解MO的过程中,实现了30 min内完全脱色的高效性能。相较于传统的Ti/SnO2-Sb+PMS电极,Ti/FeCoO/SnO2-Sb+PMS复合电极体系的性能优势与机理独特性体现在3个方面:(1) 复合体系能同步产生·O2-、·OH、SO4·-和1O2多种活性物种,并实现SO4·-的高效生成,这是单一SnO2-Sb+PMS体系所不具备的关键优势。(2) 机理研究证实,直接电子转移过程在降解中占据主导地位,这为污染物的快速、高效去除提供了更为直接的路径。(3) FeCoO层与SnO2-Sb催化层的结合,建立了“微观结构优化—电子传输增强—PMS活化效率提升”的清晰构效关系。本研究中提出的“中间层调控”策略不仅为高性能阳极设计提供了新思路,更为通过电催化与高级氧化技术联用,实现难降解有机废水的高效、低成本处理奠定了理论依据并展示了可行的技术范式。
刘永鑫, 李星辰, 刘鸿嘉, 李丹妮, 张涛, 陈曦. Fe3O4向MIL-100(Fe)的转化对活化过硫酸盐降解抗生素性能的增强作用[J]. 无机化学学报, 2025, 41(12): 2503-2513 doi: 10.11862/CJIC.20250169LIU Y X, LI X C, LIU H J, LI D N, ZHANG T, CHEN X. Enhancement effect of Fe3O4 conversion to MIL-100(Fe) on activation of persulfate for degradation of antibiotic[J]. Chinese J. Inorg. Chem., 2025, 41(12): 2503-2513 doi: 10.11862/CJIC.20250169
HONG Y X, HUANG Z, XU J L, XUE Y W, FENG H, ZHANG T, LU Z Y, XIAO J F, ZHANG Q, HONG J M. Surging efficient PMS activation through a COF-MOF dual immobilized catalytic platform: Synergy of enhanced electron transfer and adsorption-PMS activation[J]. Sep. Purif. Technol., 2024, 345: 127376 doi: 10.1016/j.seppur.2024.127376
LIU S Q, ZHANG Z C, HUANG F, LIU Y Z, FENG L, JIANG J, ZHANG L Q, QI F, LIU C. Carbonized polyaniline activated peroxymonosulfate (PMS) for phenol degradation: Role of PMS adsorption and singlet oxygen generation[J]. Appl. Catal. B‒Environ., 2021, 286: 119921 doi: 10.1016/j.apcatb.2021.119921
YU D, HE J H, XIE T P, XU Q, CHEN H Y, XIANG B. Switching the adsorption sites of PMS on SrCoO2.52 to enhance catalytic performance[J]. J. Mater. Chem. A, 2024, 12(2): 1274-1283 doi: 10.1039/D3TA06102G
ASIF M B, JI B X, MAQBOOL T, ZHANG Z H. Algogenic organic matter fouling alleviation in membrane distillation by peroxymonosulfate (PMS): Role of PMS concentration and activation temperature[J]. Desalination, 2021, 516: 115225 doi: 10.1016/j.desal.2021.115225
LI A Q, YANG Y, BAI X N, BAO H B, HE M, ZENG X Z, WANG Y J, LI F, QIN S J, YANG W J, LI X M. Trimetallic MOF-derived Fe-Mn-Sn oxide heterostructure enabling exceptional catalytic degradation of organic pollutants[J]. J. Colloid Interface Sci., 2025, 679: 232-244 doi: 10.1016/j.jcis.2024.10.098
LIU W L, SU X X, WU Y F, YI G Y, GUO X K, SHI S B, ZHANG C X, ZHANG Y L. A comprehensive review of PbO2 electrodes in electrocatalytic degradation of organic pollutants[J]. Environ. Res., 2025, 279: 121885 doi: 10.1016/j.envres.2025.121885
CHEN Z X, LU Y, LIU X Y, LI J Q, LIU Q X. Novel magnetic catalysts for organic pollutant degradation via contact electro-catalysis[J]. Nano Energy, 2023, 108: 108198 doi: 10.1016/j.nanoen.2023.108198
YIN F, LIU J H, ZHANG Y, LIU M N, WANG L Y, YU Z C, YANG W H, ZHANG J, LONG Y Z. Contact-electro-catalysis for organic pollutants degradation based on 2D fluorinated graphite[J]. Adv. Funct. Mater., 2024, 34(41): 2406417 doi: 10.1002/adfm.202406417
LI Y, HUANG C X, REN D Z, ZHANG M J, HUO Z B. Sn-based precursors dependence for electrocatalytic degradation of organic pollutants on Ti/SnO2-Sb electrode[J]. J. Environ. Chem. Eng., 2024, 12(3): 112868 doi: 10.1016/j.jece.2024.112868
BRAXTON E, FOX D J, BREEZE B G, TULLY J J, LEVEY K J, NEWTON M E, MACPHERSON J V. Electron paramagnetic resonance for the detection of electrochemically generated hydroxyl radicals: Issues associated with electrochemical oxidation of the spin trap[J]. ACS Meas. Sci. Au, 2023, 3(1): 21-31 doi: 10.1021/acsmeasuresciau.2c00049
XUE Y D, WANG Y T, PAN Z H, SAYAMA K. Electrochemical and photoelectrochemical water oxidation for hydrogen peroxide production[J]. Angew. Chem.‒Int. Edit., 2021, 60(19): 10469-10480 doi: 10.1002/anie.202011215
XU T, FU L Y, LU H Y, ZHANG M Y, WANG W L, HU B N, ZHOU Y H, YU G. Electrochemical oxidation degradation of rhodamine B dye on boron-doped diamond electrode: Input mode of power attenuation[J]. J. Clean. Prod., 2023, 401: 136794 doi: 10.1016/j.jclepro.2023.136794
HE K K, WU M H, SHEN W X, FANG C, YANG X, WANG Y, ZHANG Y W, CHEN L, WANG Q, WAN B, ZHANG Z F. Binder-free high-pressure, high-temperature surface-porous boron-doped polycrystalline diamond for electrochemical degradation of organic pollutants[J]. Diamond Relat. Mater., 2025, 159: 112744 doi: 10.1016/j.diamond.2025.112744
LI C, ZHU X, YANG S S, TIAN S L, LI Y J, LI B, PAN Z L, LI H B. Novel strategy for the efficient degradation of organic contaminants using porous graphite electrodes: Synergistic mechanism of anodic and cathodic reactions[J]. Chem. Eng. J., 2022, 429: 132340 doi: 10.1016/j.cej.2021.132340
MUKIMIN A, NINGSIH K, MUHAMMAD F, GHOZALI A A, ROCHMAN F F. A novel electrochemical advanced oxidation technology using a low-cost graphite plate anode for batik-printing wastewater treatment[J]. J. Environ. Chem. Eng., 2025, 13(5): 117759 doi: 10.1016/j.jece.2025.117759
XIA K X, DENG D L, ZHOU X, TANG B B, LIU S L, QIN T H, ZOU Y, ZHANG J Z. Efficient electrocatalytic oxidation of tetracycline in water using a SnO2-Sb/FTO electrode[J]. J. Water Process. Eng., 2025, 70: 107090 doi: 10.1016/j.jwpe.2025.107090
HU Z Y, GUO C, WANG P, GUO R, LIU X W, TIAN Y. Electrochemical degradation of methylene blue by Pb modified porous SnO2 anode[J]. Chemosphere, 2022, 305: 135447 doi: 10.1016/j.chemosphere.2022.135447
LV Y J, AN P, ZHANG A, REN P X, MA T, WANG Y J, LIANG D W, LUO D. A triboelectric-electromagnetic hybrid nanogenerator enhancing electrochemical oxidation for organic pollutant degradation[J]. J. Mater. Chem. A, 2025, 13(35): 29092-29100 doi: 10.1039/D5TA00194C
ZHU Y F, CHEN M T, FENG Y R, AI Q, LIU Y M, YAN Y R, LI Q L, LOU J. Hierarchical porous PbO2 electrode for electro-degradation of various contaminants[J]. Small Struct., 2025, 6(4): 2400389 doi: 10.1002/sstr.202400389
TIEN H N, MWAZIGHE F M. Preparation of Ti/SnO2-Sb/La-βPbO2 electrode and its application in the degradation of some pollutants including prednisolone and 8-hydroxyquinoline[J]. Chemosphere, 2023, 333: 138933 doi: 10.1016/j.chemosphere.2023.138933
LI X H, SHAO S L, YANG Y, MEI Y, QING W H, GUO H, PENG L E, WANG P, TANG C Y. Engineering interface with a one-dimensional RuO2/TiO2 heteronanostructure in an electrocatalytic membrane electrode: Toward highly efficient micropollutant decomposition[J]. ACS Appl. Mater. Interfaces, 2020, 12(19): 21596-21604 doi: 10.1021/acsami.0c02552
LIU S Q, LIU R Q, ZHANG Y H, HAN W Q, LI J S, SUN X Y, SHEN J Y, WANG L J. Development of a 3D ordered macroporous RuO2 electrode for efficient pyrazole removal from water[J]. Chemosphere, 2019, 237: 124471 doi: 10.1016/j.chemosphere.2019.124471
RU M X, LANG K, XIAO X D, WANG H, MENG H Y, JIANG B J. Efficient electrocatalytic degradation of tetracycline using NiCo2O4/MnO2 composite electrode: Synergistic enhancement mechanisms and environmental toxicity assessment[J]. J. Environ. Chem. Eng., 2025, 13(2): 115947 doi: 10.1016/j.jece.2025.115947
HONG X D, LI Y, WANG X, LONG J P, LIANG B. Carbon nanosheet/MnO2/BiOCl ternary composite for degradation of organic pollutants[J]. J. Alloy. Compd., 2022, 891: 162090 doi: 10.1016/j.jallcom.2021.162090
CHEN J X, JIANG Y Y, FENG Y X, YANG S, SHANG X R. Efficient electrochemical oxidation of ofloxacin by IrO2-RuO2-TiO2/Ti anode: Parameters optimization, kinetics and degradation pathways[J]. Environ. Pollut., 2025, 374: 126216 doi: 10.1016/j.envpol.2025.126216
ASFAHA Y G, ZEWGE F, YOHANNES T, KEBEDE S. Application of hybrid electrocoagulation and electrooxidation process for treatment of wastewater from the cotton textile industry[J]. Chemosphere, 2022, 302: 134706 doi: 10.1016/j.chemosphere.2022.134706
WEI J, LIU Y, WU X. A cyclone reactor of electrochemical advanced oxidation processes using PbO2 anode and H2O2 electrosynthesis cathode[J]. Water Res., 2023, 245: 120629 doi: 10.1016/j.watres.2023.120629
XIE J, MA J, ZHANG C, KONG X, WANG Z, WAITE T D. Effect of the presence of carbon in Ti4O7 electrodes on anodic oxidation of contaminants[J]. Environ. Sci. Technol., 2020, 54(8): 5227-5236 doi: 10.1021/acs.est.9b07398
HOU K K, ZHU G W, FENG Y J, LIU Y M, QUAN X. Enhanced electrochemical oxidation of perfluorooctanoic acid on Ti/SnO2-Sb electrode by surface morphology regulation[J]. Chin. Chem. Lett., 2024, 35(3): 108704
GUO T, CHEN X, YIN L F. Recent advancements in modified SnO2-Sb electrodes for electrochemical treatment of wastewater[J]. J. Mater. Chem. A, 2024, 12(8): 4397-4420
TANG C B, ZHANG W Y, GUO X X, WANG T, YU L H, XUE J Q, YIN X Y, ZHENG N. Fabrication of long-acting Ti/TiN/SnO2-Sb anodes for electro-oxidative degradation of hydroxychloroquine[J]. Sep. Purif. Technol., 2025, 373: 133547
ZHONG J X, JIANG H, WANG Z L, YU Z G, WANG L Z, MUELLER J F, GUO J H. Efficient photocatalytic destruction of recalcitrant micropollutants using graphitic carbon nitride under simulated sunlight irradiation[J]. Environ. Sci. Ecotechnol., 2021, 5: 100079
MURAUSKAS T, KUBILIUS V, RAUDONIS R, SKAPAS M, PLAUSINAITIENE V. Structure modification, evolution, and compositional changes of highly conductive La: BaSnO3 thin films annealed in vacuum and air atmosphere[J]. Nanomaterials, 2022, 12: 2408
MAO Y Z, CHEN R N, YOU H H, LIU Y, LUAN S R, HOU L, GAO F M. Advanced performance of S and N Co-doped Sb@CNFs with a 3D conductive network as superior lithium-ion battery anodes[J]. J. Alloy. Compd., 2022, 904: 164000
GARBASSI F. XPS and AES study of antimony oxides[J]. Surf. Interface Anal., 1980, 2: 165-169
LIM J, YANG Y, HOFFMANN M R. Activation of peroxymonosulfate by oxygen vacancies-enriched cobalt-doped black TiO2 nanotubes for the removal of organic pollutants[J]. Environ. Sci. Technol., 2019, 53(12): 6972-6980
PAN S J, LI H, WANG T Y, FU Y, WANG S N, XIE Z S, WEI L, LI H, LI N. Er-doping enhances the oxygen evolution performance of cobalt oxide in acidic medium[J]. ACS Catal., 2024, 14(18): 13814-13824
WU L Y, SUN Z Q, ZHEN Y F, ZHU S S, YANG C, LU J, TIAN Y, ZHONG D, MA J. Oxygen vacancy-induced nonradical degradation of organics: Critical trigger of oxygen (O2) in the Fe-Co/peroxymonosulfate system[J]. Environ. Sci. Technol., 2021, 55(22): 15400-15411
LI S S, ZHOU M H, WU H Z, SONG G, JING J, MENG N, WANG W. High-efficiency degradation of carbamazepine by the synergistic electro-activation and bimetals (FeCo@NC) catalytic-activation of peroxymonosulfate[J]. Appl. Catal. B‒Environ., 2023, 338: 123064
YAO B, YU Y G, WANG Z, YANG J, ZHOU Y Y, DIONYSIOU D D. Electro-induced activation of persulfate over N-doped porous carbon decorated with Fe/Ni bimetals for organic pollutants enhanced degradation: Synergism of electro-activation and catalytic activation[J]. Chem. Eng. J., 2023, 476: 146769
YANG H M, LIANG J T, ZHANG, L, LIANG Z H. Electrochemical oxidation degradation of methyl orange wastewater by Nb/PbO2 electrode[J]. Int. J. Electrochem. Sci., 2016, 11(2): 1121-1134
WANG G R, LIU Y, YE J W, LIN Z F, YANG X J. Electrochemical oxidation of methyl orange by a Magnéli phase Ti4O7 anode[J]. Chemosphere, 2020, 241: 125084
NGUYEN C H, FU C C, JUANG R S. Degradation of methylene blue and methyl orange by palladium-doped TiO2 photocatalysis for water reuse: Efficiency and degradation pathways[J]. J. Clean. Prod., 2018, 202: 413-427

图 1 Ti/FeCoO/SnO2-Sb复合电极的制备路线示意图
Figure 1 Schematic diagram of the route of Ti/FeCoO/SnO2-Sb composite electrode

图 2 Ti/Fe-Co(PLED)电极的(a) SEM图及(b) 对应区域的放大图; Ti/FeCoO电极的(c) SEM图及(d) 对应区域的放大图; Ti/FeCoO电极的(e) TEM图与(f) HRTEM图; (g) Ti/FeCoO电极中元素的STEM-EDS映射图; Ti/FeCoO/SnO2-Sb电极的(h) SEM图及(i) 对应区域的放大图; (j) Ti/FeCoO/SnO2-Sb电极的HRTEM图; (k) Ti/FeCoO/SnO2-Sb电极中元素的STEM-EDS映射图
Figure 2 (a) SEM images of Ti/Fe-Co(PLED) electrode and (b) the corresponding magnified view; (c) SEM images of Ti/FeCoO electrode and (d) the corresponding magnified view; (e) TEM image and (f) HRTEM image of Ti/FeCoO electrode; (g) STEM-EDS elemental mappings for the Ti/FeCoO electrode; (h) SEM images of Ti/FeCoO/SnO2-Sb electrode and (i) the corresponding magnified view; (j) HRTEM image of the Ti/FeCoO/SnO2-Sb electrode; (k) STEM-EDS elemental mappings for the Ti/FeCoO/SnO2-Sb electrode

图 3 (a) Ti/Fe-Co(PLED)、Ti/FeCoO和Ti/FeCoO/SnO2-Sb电极的XRD图; (b) Ti/Fe-Co(PLED)和Ti/FeCoO电极的XRD放大图
Figure 3 (a) XRD patterns of Ti/Fe-Co(PLED), Ti/FeCoO, and Ti/FeCoO/SnO2-Sb electrodes; (b) Enlarged XRD patterns of Ti/Fe-Co(PLED) and Ti/FeCoO electrodes

图 4 Ti/FeCoO/SnO2-Sb电极的Sn3d (a)、O1s和Sb3d (b)XPS谱图; Ti/FeCoO电极的XPS全谱图(c)及Fe2p (d)、Co2p (e)和O1s (f)XPS谱图
Figure 4 Sn3d (a), O1s and Sb3d (b) spectra of Ti/FeCoO/SnO2-Sb electrode; XPS survey spectrum (c), and Fe2p (d), Co2p (e), and O1s (f) XPS spectra of Ti/FeCoO electrode

图 5 Ti/SnO2-Sb和Ti/FeCoO/SnO2-Sb电极的(a) LSV和(b) CV曲线; (c) Ti/SnO2-Sb和(d) Ti/FeCoO/SnO2-Sb电极在非法拉第区域内、不同扫描速率下的CV曲线; Ti/SnO2-Sb和Ti/FeCoO/SnO2-Sb电极的(e) EIS和(f) j-t曲线
Figure 5 (a) LSV and (b) CV curves for Ti/SnO2-Sb and Ti/FeCoO/SnO2-Sb electrode; CV curves of (c) Ti/SnO2-Sb and (d) Ti/FeCoO/SnO2-Sb electrodes in the non-Faradaic region at different scan rates; (e) EIS and (f) j-t curves of Ti/SnO2-Sb and Ti/FeCoO/SnO2-Sb electrodes

图 6 Ti/FeCoO/SnO2-Sb+PMS和Ti/SnO2-Sb+PMS体系的(a) COD去除率和(d) COD去除的拟一级动力学拟合曲线; (b) 电流密度、(c) 初始MO质量浓度、(e) 初始溶液pH和(f) PMS浓度对COD去除率的影响
Figure 6 (a) COD removal efficiencies and (d) pseudo-first-order kinetic fitting curves for COD removal of Ti/FeCoO/SnO2-Sb/PMS and Ti/SnO2-Sb/PMS systems; Effect of (b) current density, (c) initial MO mass concentration (e) initial solution pH, and (f) PMS concentration on COD removal efficiency
Inset: the molecular structure of MO; Reaction conditions in a and d: pollutant concentration: 20 mg·L-1, j=50 mA·cm-2, and pH=4, cPMS=0.100 0 mmol·L-1.

图 7 (a) 不同体系的ACE; (b) Ti/FeCoO/SnO2-Sb阳极活化PMS降解印染废水过程中活性物种的生成机理示意图; (c) 在Ti/FeCoO/SnO2-Sb+PMS (上)和Ti/SnO2-Sb+PMS (下)体系中MO溶液随反应时间变化的照片
Figure 7 (a) ACE of different systems; (b) Schematic illustration of active species generation during PMS activation by the Ti/FeCoO/SnO2-Sb anode for textile wastewater degradation; (c) Photographs of MO solutions treated in the Ti/FeCoO/SnO2-Sb+PMS (upper) and Ti/SnO2-Sb+PMS (lower) systems at different reaction times
In c: the time from left to right was 0, 30, 60, 90, 120, 150, and 180 min, respectively.

图 8 DFT计算:(a) PMS的理论模型; (b~d) PMS吸附在SnO2-Sb电极上的优化构型; (e) PMS在SnO2-Sb电极表面的示意图
Figure 8 DFT calculations: (a) theoretical model of PMS; (b-d) optimized configurations of PMS adsorbed on the SnO2-Sb electrode; (e) schematic diagram of the PMS onto the surface of the SnO2-Sb electrode

图 9 在Ti/FeCoO/SnO2-Sb+PMS系统中获得的DMPO-·OH、DMPO-SO4·-和TEMP-1O2 EPR谱图
Figure 9 EPR spectra of DMPO-·OH, DMPO-SO4·-, and TEMP-1O2 obtained in the Ti/FeCoO/SnO2-Sb+PMS system

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:
