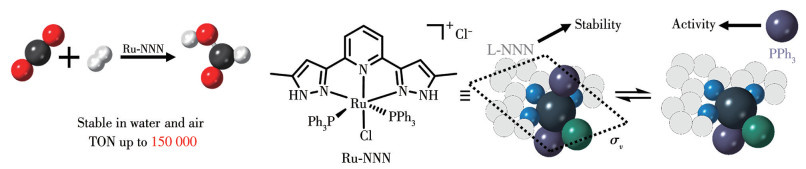
Scheme 1.
Constructing Ru-NNN complexes with both high catalytic activity and stability by using rigid ligands and weak ligands simultaneously
Efficient hydrogenation of CO2 realized by Ru-NNN complex
Huihua GONG , Tianhua CUI , Li JI , Liyuan ZHANG , Xueli ZHENG , Haiyan FU , Hua CHEN , Jiawei MAO , Ruixiang LI

To solve the ever-increasing energy and environmental issues, hydrogen energy serves as an important supplement due to its numerous advantages[1-3]. It is of great importance for hydrogen energy utilization to develop safe methods for hydrogen storage. Recently, formic acid has emerged as a promising liquid organic hydrogen carrier due to its low price, low toxicity, environmental friendliness, biodegradability, etc[4-7]. Catalytic hydrogenation of CO2 to formic acid is a green chemical reaction with high atom economy. However, CO2 hydrogenation to formic acid is strongly limited by thermodynamic constraints (ΔG298 K⊖=33 kJ·mol-1), and bases are widely used to neutralize formic acid and drive the reaction equilibrium toward the formate product. To realize efficient CO2 hydrogenation, it is essential to develop superior catalysts[8]. Many homogeneous catalysts have exhibited the potential for CO2 hydrogenation reactions[8-10].
To date, one of the best-performing catalysts for CO2 hydrogenation to formate is the iridium-pyridine-based PNP-pincer catalyst developed by Nozaki and colleagues[11], which achieved a remarkable turnover number (TON) of up to 3 500 000. Another notable catalyst is the ruthenium-pyridine-based PNP-pincer catalyst reported by Pidko and colleagues[12], exhibiting a high turnover frequency (TOF) of up to 1 100 000 h-1. However, even though PNP-pincer ligand-based homogeneous catalysts have demonstrated high efficiency for CO2 hydrogenation, these complexes are prone to oxidation due to the alkyl phosphines, which hinders their practical utilization. To solve this problem, pincer ligands without an alkylphosphine structure were introduced, including hydroxyl, amino, and heterocyclic rings containing N or O atoms, where C, N, or O atoms serve as the coordination sites[13-15]. However, these catalytic systems exhibit low catalytic efficiency. Therefore, we are committed to synthesizing non-phosphine pincer ligands and complexes that can efficiently and stably catalyze the hydrogenation of CO2 to formate under a wider range of conditions. It is generally believed that the catalytic hydrogenation activity is related to the vacant coordination sites provided by the active species during the reaction process. Typically, the easier it is for a complex to provide vacant sites, the higher its reactivity tends to be[16]. However, strongly rigid pincer ligands can form a stable complex structure during the reaction, making it difficult to create vacant coordination sites. On the other hand, one advantage of strongly rigid pincer ligands is their ability to stably chelate with the metal center throughout the reaction process, which effectively reduces the likelihood of metal center clustering, thereby enhancing catalyst stability. As a result, the complexes bearing rigid ligands often exhibit comparatively lower catalytic activity, while the complexes with non-rigid ligands face difficulties in balancing high activity with stability. To date, although various complexes integrating rigid and non-rigid ligands have been explored in catalyzing the hydrogenation of carbon dioxide[17-22], the complexes that have both good stability and high activity are rare[23]. Hence, it remains a tough challenge to synthesize complexes with high catalytic activity and excellent stability.
Herein, we synthesized Ru complexes bearing rigid pincer-type tridentate NNN ligands[24-25] and weakly coordinated triphenylphosphine (PPh3) (Scheme 1). PPh3 in the complexes is easily dissociated and forms the vacant coordination sites. The vacant coordination sites can, in return, enhance their catalytic activity. Furthermore, the presence of NNN ligands plays a crucial role in maintaining the stability of the Ru center, contributing to the overall robustness of the catalytic system. As a result, the synthesized Ru(Ⅱ)-NNN complex [Ru(L-NNN)Cl(PPh3)2]Cl (1, L-NNN=2,6-bis(5-methyl-1H-pyrazol-3-yl)pyridine) was not only quite stable in both water and air, but also showed high activity for CO2 hydrogenation towards formate, achieving a TON of up to 150 000.
NMR spectra were recorded on a Bruker AV Ⅱ-400 spectrometer at 400 MHz (1H NMR) and 162 MHz (31P NMR). Chemical shifts were reported downfield from internal Me4Si and external 85% H3PO4, respectively. All the solvents used for reactions were distilled under argon after drying over an appropriate drying agent. All other commercially available reagents were purchased from Aaladin, Adamas, Aldrich, and Alfa Aesar Chemical Company.
The ligand L-NNN was synthesized using pyridine-2, 6-dicarboxylic acid as the precursor[25]. In a round-bottom flask with a 250 mL capacity equipped with a reflux condenser apparatus, pyridine 2, 6-dicarboxylate (15 g) and anhydrous ethanol (120 mL) were combined and treated with the addition of acetyl chloride (25 mL), followed by a reaction at room temperature for 24 h. Upon completion of the reaction, the solid was removed by filtration, and the filtrate was neutralized with sodium carbonate solution, then extracted with ethyl acetate to yield a white solid (diethyl pyridine 2, 6-dicarboxylate). The diethyl pyridine 2, 6-dicarboxylate and ethanol sodium (8.8 g) were added to a toluene (60 mL) solution under an ice bath, followed by the gradual addition of a toluene solution of acetone (8.7 mL) to produce a yellowish turbid mixture. The reaction was then allowed to proceed at room temperature for 12 h. The mixture was filtered after the reaction to collect a light yellow solid, and the pH of the solid was neutralized to 1 with dilute hydrochloric acid before being extracted with ethyl acetate to obtain yellow solid 1, 1′-(pyridine-2, 6-diyl) bis(butane-1, 3-dione). Subsequently, in a 100 mL round-bottom flask, 1, 1′-(pyridine-2, 6-diyl)bis(butane-1, 3-dione) (2 mmol), glacial acetic acid (4 mL), and ethanol (30 mL) were stirred at room temperature for 30 min, followed by the addition of an ethanolic solution containing 4 mmol of hydrazine hydrate via a pressure-equalizing dropping funnel. The temperature was then raised to 65 ℃ to continue for 24 h. Upon completion, the solution was concentrated to around 10 mL, neutralized to pH=8 with sodium carbonate solution, and extracted with ethyl acetate to yield a faint yellow solid L-NNN. 1H NMR (400 MHz, DMSO-d6): δ 12.79 (s, 2H), 7.83 (t, J=7.7 Hz, 1H), 7.72 (d, J=7.8 Hz, 2H), 6.75 (s, 2H), 2.28 (s, 6H).
Ligand 2,6-bis(1, 5-dimethylpyrazol-3-yl)pyridine (L-NNN-Me) was prepared analogously to ligand L-NNN, except for using methyl hydrazine as the hydrazine reagent[24].
Complex 1 was synthesized according to the reported method[25]. A mixture of ligand L-NNN (1 mmol) and RuCl3·3H2O (1 mmol) was refluxed in EtOH (60 mL) for 5 h to give a reddish-brown precipitate. After the mixture had cooled to room temperature, it was filtered, washed with diethyl ether, and dried under vacuum. EtOH (30 mL) and PPh3 (2 mmol) were added to the filtered precipitate, and the mixture was refluxed for 6 h. After completion of the reaction, the solvent was removed under vacuum to get a reddish-brown solid. The resulting solid was dissolved in dichloromethane (DCM), followed by filtration to remove insoluble impurities, yielding a red solution. Upon addition of ether to the solution, orange solid Complex 1 was crystallized from the solution, with a yield of 50%. 1H NMR (400 MHz, DMSO-d6): δ 13.50 (s, 2H), 7.43-7.38 (m, 1H), 7.29 (t, J=7.2 Hz, 6H), 7.25-7.13 (m, 24H), 7.00 (d, J=7.8 Hz, 2H), 6.44 (s, 2H), 2.14 (s, 6H). 31P NMR (162 MHz, DMSO-d6): δ 33.6, 24.4, -6.9. Despite repeated attempts, the low solubility of the complex in all suitable NMR solvents prevented us from obtaining a high-quality 13C NMR spectrum.
A mixture of ligand L-NNN (1 mmol) and RuCl3·3H2O (1 mmol) was refluxed in EtOH (60 mL) for 5 h to get a reddish-brown precipitate. After the mixture was cooled to room temperature, it was filtered, washed with diethyl ether, and dried under vacuum. EtOH (30 mL) and PPh3 (1 mmol) were added to the filtered precipitate, and the mixture was refluxed for 6 h. After completion of the reaction, the solvent was removed under vacuum to obtain a reddish-brown solid. The resulting solid was dissolved in DCM, followed by filtration to remove insoluble impurities, yielding a red solution. Upon addition of ether to the solution, orange solid Complex 2 was crystallized from the solution, with a yield of 57%. 1H NMR (400 MHz, CD2Cl2): δ 15.09 (s, 4H), 7.33-7.17 (m, 32H), 6.89 (d, J=7.8 Hz, 4H), 6.03 (s, 4H), 2.45 (s, 12H). 31P NMR (162 MHz, CD2Cl2): δ 52.8. Due to the same reasons as those for Complex 1, high-quality 13C NMR spectra could not be obtained.
A mixture of ligand L-NNN-Me (1 mmol) and RuCl3·3H2O (1 mmol) was refluxed in EtOH (60 mL) for 5 h to get a reddish-brown precipitate. After cooling the precipitate to room temperature, it was filtered, washed with diethyl ether, and dried under vacuum. EtOH (30 mL) and PPh3 (2 mmol) were added to the filtered precipitate, and the mixture was refluxed for 6 h. After completion of the reaction, the solvent was removed under vacuum to obtain a reddish-brown solid. The resulting solid was dissolved in DCM, followed by filtration to remove insoluble impurities, yielding a red solution. Upon addition of ether to the solution, an orange solid complex, Ru-NNN-Me, was crystallized from the solution, with a yield of 61%. 1H NMR (400 MHz, CD2Cl2): δ 7.33 (t, J=7.8 Hz, 1H), 7.29-7.08 (m, 14H), 7.04-6.97 (m, 15H), 6.94-6.85 (m, 13H), 6.72 (s, 2H), 3.12 (s, 6H), 2.13 (s, 6H). 31P NMR (162 MHz, CD2Cl2): δ 43.8, 17.9, -5.6. For the same reason as for Complex 1, we were unable to obtain a high-quality 13C NMR spectrum.
Catalytic CO2 hydrogenation was carried out in a Hastelloy Autoclave Reactor system equipped with a 25 mL cylinder. The catalyst was dissolved in a degassed aqueous solution (5 mL) of CsOH, along with the addition of 1 mL THF. The reactor was pressurized with 5 MPa of CO2/H2 (1:1, p/p) and heated at 90-170 ℃ for the appropriate time. DMF was added as an internal standard, while D2O was added as the solvent. Then, the formate was quantified by 1H NMR spectroscopy (Fig.S1, Supporting information).
To uncover the underlying mechanism, stoichiometric reactions were conducted with 30 μmol Ru-NNN Complex 1 and 4 mmol CsOH under 5 MPa of CO2/H2 (1:1, p/p) in the mixed solvent of THF-d8 and D2O (1 and 10 mL, respectively) at 140 ℃, in which the intermediates were monitored by NMR in situ.
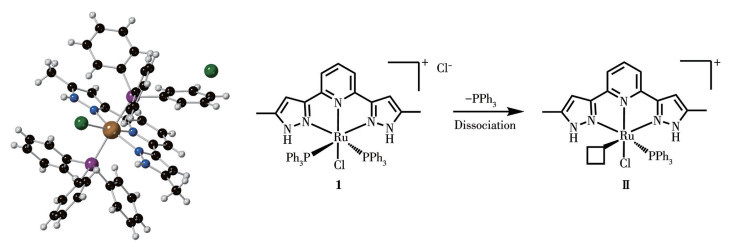
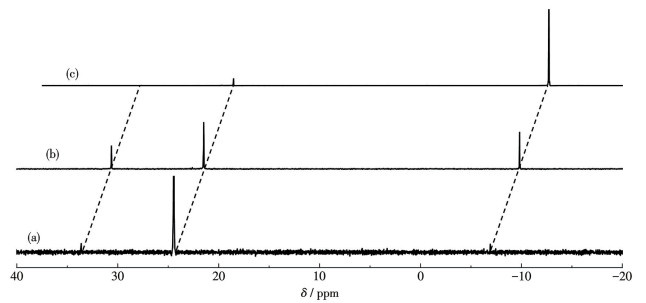
The Ru(Ⅱ)-NNN Complex 1 was synthesized by refluxing L-NNN in ethanol with an equal amount of RuCl3·3H2O and two equivalents of PPh3, following the method reported in the literature[25]. Ru(Ⅲ) can be reduced to Ru(Ⅱ) by ethanol under heating reflux conditions and form stable Ru(Ⅱ)-NNN complexes with ligands. Afterward, Complex 1 was crystallized by adding diethyl ether to the solution of dichloromethane[25]. The structure of Complex 1 has already been umambiguously confirmed by single-crystal X-ray diffraction in our previous work[25]. The crystal structure of Complex 1 is shown in Fig.1. The Ru(Ⅱ) center adopts a distorted octahedral coordination geometry with a meridionally coordinated L-NNN ligand and a chloride occupying the equatorial sites, while two trans-located PPh3 occupy the axial positions. The average Ru—P bond length is 0.238 4 nm, and the Ru—N bond length is 0.198 1 nm (pyridine). The complex exhibits a distinct mirror-symmetric structure with N—Ru—N as the σv symmetry plane, and the orientation of the phenyl groups in PPh3 also exhibits a symmetric structure. In the 31P NMR spectrum of Complex 1, a weakly characteristic peak of triphenylphosphine located at δ of -6.9 was observed, while the other weak single peak at δ of 33.6 might originate from the species (Ⅱ) after PPh3 dissociation (Fig.2a). Noteworthily, this phenomenon becomes more pronounced with increasing temperature (Fig.2b). After adding PPh3, the 31P NMR spectrum of Complex 1 (Fig.2c) only showed the characteristic peak of PPh3 (δ=-6.9) and the main peak of the Complex 1 (δ=23.8), indicating that the dissociation process was reversible. The rigidity of the aromatic heterocyclic structure in the NNN pincer ligands prevents bending or twisting, meaning their dissociation from the ruthenium ion requires simultaneous rupture of two or three coordination bonds. This kinetically high-energy barrier ensures strong chelation with the Ru center in a meridional fashion, greatly augmenting the complex′s stability. The influence of the trans-effect of phosphine and the cis-labilizing effect of the chloride facilitates the dissociation of PPh3 to provide a vacant site[25]. Probably, the PPh3, which is weakly coordinated compared to the rigid ligand L-NNN, can easily dissociate to provide vacant sites, contributing to the catalytic activity of Complex 1.
The catalytic performances of Ru Complex 1 were investigated in the mixed solvent of H2O/THF (5 mL/1 mL) for CO2 hydrogenation. Initially, we varied the type of base and screened multiple organic and inorganic bases, including 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), triethylamine, pentaethylene-hexamine, CsOH, NaOH, and KOH, based on the different types of bases with high reactivity reported in previous work[11-12,17-22]. We found that the complex exhibited the highest catalytic activity in the presence of CsOH, and there was a positive correlation between catalytic activity and the concentration of the base (Table 1 and S3). Cs+ ions exhibit stronger basicity in aqueous solutions compared to other alkali metal ions, which may more effectively facilitate the heterolytic cleavage of H2, thereby enhancing the reaction activity. Subsequently, we tested the catalytic reactivity at different reaction temperatures, increasing the temperature from 90 to 170 ℃ (Table 1, entries 4-12). The activity of Complex 1 varied significantly with temperature and peaked at 140 ℃, with a maximum TOF of 574 h-1 at 140 ℃ (Table 1, entry 10). The rigid L-NNN ligand endows the Complex 1 with high thermal stability. Afterward, we used 0.001 to 10 μmol of Complex 1 to react for 16 h at 140 ℃, and the TOF increased significantly with the decrease of complex concentration, reaching a maximum TOF of 9 347 h-1 (Table 1, entries 10, 16-19). Meanwhile, stability is another crucial parameter for ruthenium complexes, apart from their activity. We tested the catalytic activity of Complex 1 for CO2 hydrogenation at 140 ℃ at different time intervals ranging from 0.5 to 16 h. The TOF of Complex 1 did not change significantly (less than 5%) after 0.5, 1, 2, 4, 8, and 16 h (Table 1, entries 10, 13-15; Table S3, entries 3-4), indicating its excellent stability. Using 0.001 μmol of the catalyst, the TON of formate reached approximately 150 000 after 16 h, which was a high record among all Ru complexes (Table S2)[17-22]. Considering its excellent stability and high activity, Ru-NNN Complex 1 can be considered one of the best CO2 hydrogenation catalysts among all Ru complexes.
 下载:
导出CSV
下载:
导出CSV
| Entry | nCat. / μmol | Base | T / ℃ | t / h | TONb | TOFc / h-1 |
| 1 | 1 | NEt3 | 150 | 16 | 1 465 | 92 |
| 2 | 1 | DBU | 150 | 16 | 2 218 | 139 |
| 3 | 1 | KOH | 150 | 16 | 3 412 | 213 |
| 4 | 1 | CsOH | 150 | 16 | 6 213 | 388 |
| 5 | 1 | CsOH | 90 | 16 | 178 | 11 |
| 6 | 1 | CsOH | 100 | 16 | 512 | 32 |
| 7 | 1 | CsOH | 110 | 16 | 1 513 | 95 |
| 8 | 1 | CsOH | 120 | 16 | 2 746 | 172 |
| 9 | 1 | CsOH | 130 | 16 | 2 280 | 143 |
| 10 | 1 | CsOH | 140 | 16 | 9 176 | 574 |
| 11 | 1 | CsOH | 160 | 16 | 7 322 | 458 |
| 12 | 1 | CsOH | 170 | 16 | 4 894 | 306 |
| 13 | 1 | CsOH | 140 | 2 | 1 096 | 548 |
| 14 | 1 | CsOH | 140 | 4 | 2 245 | 561 |
| 15 | 1 | CsOH | 140 | 8 | 4 635 | 579 |
| 16 | 0.001 | CsOH | 140 | 16 | 149 558 | 9 347 |
| 17 | 0.01 | CsOH | 140 | 16 | 68 196 | 4 262 |
| 18 | 0.1 | CsOH | 140 | 16 | 32 116 | 2 007 |
| 19 | 10 | CsOH | 140 | 16 | 584 | 37 |
| 20d | 1 | CsOH | 140 | 16 | 10 304 | 644 |
| a General conditions: complex 1, base (10 mmol), pH2=pCO2=2.5 MPa, VTHF=1 mL, VH2O=5 mL; b TON=nformate/nRu; c TOF=TON/t; d 1 mmol LiOAc as additive. | ||||||
Furthermore, we increased the CO2 and H2 pressures, and the results showed that an increase in gas pressure could further enhance the reaction activity. When the CO2 and H2 pressure reached 3 MPa, the TOF increased to 704 h-1 (Table S3, entry 5) compared to the original conditions (Table 1, entry 10; Table S3, entry 12). These results suggest that the activity of Complex 1 has the potential for further improvement for CO2 hydrogenation.
After optimizing the reaction conditions, we aimed to further enhance the CO2 hydrogenation performance of Complex 1 with the help of additives. Various salt additives, including K3PO4, LiOAc, LiOTf, and KNO3, were introduced into the reaction system to improve its performance[26]. The results indicated that LiOAc exhibited the most significant promotional effect (Table 1, entry 20; Table S3, entries 6-8), achieving a maximum TOF of 644 h-1, representing a 12% improvement compared to the original value (Table 1, entry 10). The promotional effect of LiOAc in the reaction process could be attributed to two aspects: Li+ may function as a Lewis acid, while OAc- may act as an internal base. Bernskoetter′s work found that Li ions[27], as Lewis acids, can activate the M-OCOH intermediate, thereby enhancing the reaction activity. Hopmann′s work[28], through density functional theory (DFT) calculations, demonstrated that Li ions activate the process of CO2 insertion into the M—H bond, improving the reaction. Jessop′s research found that OAc- in RuCl(OAc)(PMe3)4 can serve as an internal base to promote the generation of the active species [RuH(PMe3)4]+[29]. In contrast, NO3- cannot have the same effect as OAc- due to its conjugate acid′s low pKa and the anion′s difficulty in donating electrons to the Ru center. Therefore, it can be concluded that the promotional effect of LiOAc on the reaction is a result of the combined action of both Lewis acid and internal base.
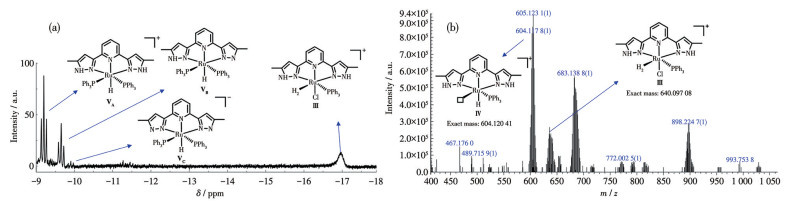
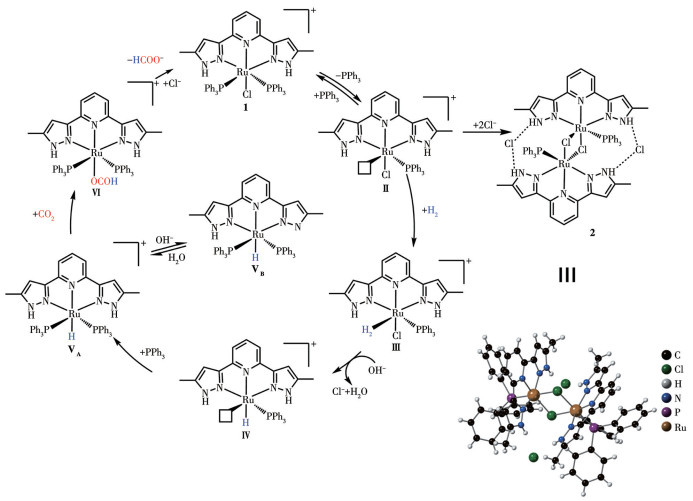
To investigate the reaction mechanism of the hydrogenation of CO2, we conducted activity tests for the catalytic reaction using stoichiometric Complex 1 in a mixed solvent of THF-d8 and D2O. During the catalytic reaction, the in-situ formation of several new species was revealed by the 1H NMR spectrum, exhibiting signals in the high-field region. This observation indicates the formation of metal-H species during the reaction (Fig.3a). The triplet peaks at around -9 {-9.17 (t, J=26.1 Hz, 1H), -9.65 (t, J=26.1 Hz, 1H), -9.90 (t, J=26.1 Hz, 1H)} and the broad peak at -17 indicate the formation of multiple distinct new species with significantly different coordination environments during the reaction. Based on literature reports[30], it is reasonable to infer that the triplet peaks at around -9 correspond to the Ru complex with symmetric two PPh3 groups formed by H substitution of Cl (intermediate ⅤA, δ=-9.17), as well as several structurally similar complexes formed by deprotonation of N—H on the pyrazole ring under alkaline conditions (ⅤB, δ=-9.65; ⅤC, δ=-9.90). The broad peak at -17 can be attributed to the Ru complex coordinated with hydrogen molecules (intermediate Ⅲ). Relevant evidence was also detected in the in situ HPLC-HRMS (Fig.3b). Peaks at m/z 640.097 8 and 604.117 8, corresponding to intermediate Ⅲ ([C31H30ClN5PRu]+, Exact mass: 640.097 08) and intermediate Ⅳ ([C31H29 N5PRu]+, Exact mass: 604.120 41) were identified in the HPLC-HRMS data. We speculate that the active species exists as a vacant coordination intermediate (Ⅱ) during the reaction, rather than as a Ru-Cl2 complex formed by the exchange of PPh3 and free chloride ions. This is because vacant coordination intermediates (Ⅱ) may possess higher reactivity than the Ru-Cl2 complex. To verify this speculation, we attempted to obtain Ru-Cl2 Complex 2. Interestingly, we observed that Complex 1 forms a reddish-brown solid when left to stand in solution for more than a week. So, we repeatedly recrystallized Complex 1 with DCM and diethyl ether. By taking advantage of the difference in solubility between PPh3 and the complex, we obtained Complex 2. Complex 2 was characterized using 1H NMR and 31P NMR spectra (Fig.S7 and S8) as well as single-crystal X-ray diffraction (Fig.S2). The single crystal structure reveals that Complex 2 is a dimer of two intermediate Ⅱ. The chemical shift of the 31P spectrum of Complex 2 (Fig.S8) also differs from that of the intermediate products generated in situ from Complex 1 (δ=33.6). When Complex 2 was added to the reaction along with two equivalents of PPh3, the catalytic activity for CO2 hydrogenation was significantly lower than that of Complex 1, with a TOF of only 63% of Complex 1 (Table S3, entry 9). On the other hand, adding excess PPh3 to the catalytic system of Complex 1 to inhibit its in-situ dissociation so that a decrease in reactivity was observed, with a TOF of only 75% of the original conditions (entry 10 in Table S3 vs entry 10 in Table 1). Therefore, it can be inferred that the high activity of Complex 1 is due to the formation of a vacant coordination site from the dissociation of PPh3 during the reaction. This dissociation results in the formation of intermediate Ⅱ, which serves as the active species. To eliminate the effect of the N—H functionality on the pyrazole ring during the reaction, we methylated the N—H on the pyrazole ring. Using a similar method to the synthesis of Complex 1, we synthesized Ru-NNN-Me. The catalytic activity of Ru-NNN-Me in the hydrogenation of CO2 to formate is comparable to that of Complex 1 and did not show significant deactivation (Table S3, entry 11). Therefore, we can conclude that the N—H functionality on the pyrazole does not have a positive effect on the catalytic reaction.
Data of the crystal structures show that the Ru—N bond lengths in complexes 1 and 2 are similar (Table S4), indicating that PPh3 has little influence on the charge of the central atoms in the complexes. It can be inferred that the abnormal changes in the Ru—P bond length in the Ru-NNN complexes 1 and 2 in this study are due to their special structures. This is supported by the crystal data of NNN complexes previously reported by the Yu′s group[31]. When the symmetry of the complex is broken, the bond length of the Ru—P bond is shortened[32] (Table S4). The slightly longer Ru—P bond length in Complex 1 (0.238 4 nm) compared to Complex 2 (0.228 3 nm) by about 4.4% may be attributed to the higher symmetry of Complex 1. The longer bond length in Complex 1 leads to the facile breaking of the Ru—P bond, contributing to the formation of the active intermediate Ⅱ with vacant coordination sites.
Based on the aforementioned characterization, we hypothesized potential intermediates and calculated their Gibbs free energies, along with those of the transition states, using Gaussian 09 and Shermo. The optimizations were performed at the PBE0/def2-SVP level. The Gibbs free energies of the intermediates were calculated at T=298.15 K and p=101.325 kPa using the Shermo program (Fig.4). DFT calculations reveal that the hydrogenation of carbon dioxide is thermodynamically feasible, with no excessively high energy barriers to prohibit the reaction. The free energy difference between the formation of the Ru-H2 (Ⅲ) intermediate from the Ru-PPh3 intermediate (Ⅱ) and the dimerization to form Complex 2 is minimal, favoring the latter. This suggests a slightly higher likelihood of forming Complex 2, which hinders the reaction′s progression. This aligns with the observed decrease in reactivity as Complex 2 forms.
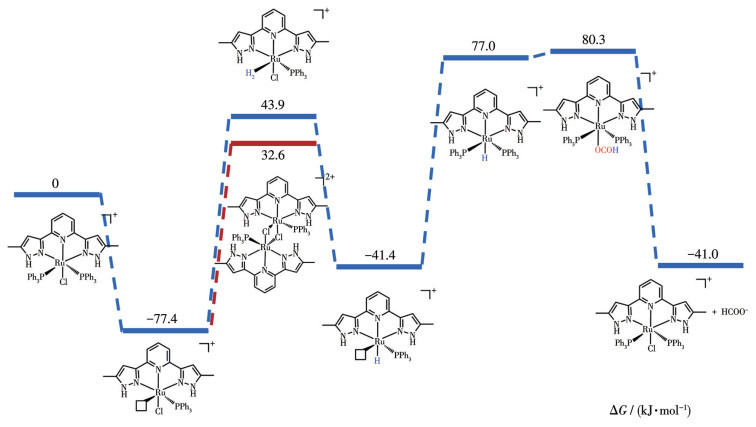
Based on the above results, we inferred the possible catalytic mechanism (Fig.5). First, Complex 1 partially dissociates one molecule of PPh3 and generates one molecule of active intermediate Ⅱ in solution. Intermediate Ⅱ may form the low-activity Complex 2 during the reaction. But due to the low concentration of intermediate Ⅱ, it is more likely to react with H2 to form the hydrogen molecular complex Ⅲ. Under alkaline conditions, CsOH assists intermediate Ⅲ in eliminating one molecule of HCl, yielding intermediate Ⅳ, H2O, and CsCl. Ⅳ is complexed with the free PPh3 in solution to form Ⅴ. The C=O bond in CO2 is activated by the Ru—H bond in intermediate Ⅴ through electrostatic interactions. Carbon dioxide is then inserted into the Ru—H bond of Ⅴ to form the Ru-OCOH intermediate Ⅵ. The HCOO- of Ⅵ is exchanged with Cl- in the solution to obtain the product formate and complete the catalyst regeneration.
In summary, we designed and synthesized a Ru-NNN Complex 1, which was applied to the hydrogenation of CO2 to formate, achieving relatively high catalytic activity (TON=150 000) compared to similar Ru complexes. By combining the crystal structure of Complex 2 with in-situ NMR and MS spectra, we inferred a potential reaction mechanism involving intermediate Ⅱ with empty coordination sites as the active intermediate. To further validate the plausibility of these intermediates, we conducted catalytic tests under various control conditions. By performing catalytic activity tests with an excess of PPh3 in a mixture with Complex 1, we confirmed that the high activity of Complex 1 was due to the vacant coordination sites generated by its in-situ dissociation. At the same time, the easy dissociation of PPh3 may be related to the symmetric structure of Complex 1, which requires further confirmation. This study showed that combining rigid and weak ligands in the catalysts simultaneously is a good strategy to solve the paradox of high activity and stability.
Supporting information is available at
JAIN I P. Hydrogen the fuel for 21st century[J]. Int. J. Hydrog. Energy, 2009, 34(17): 7368-7378 doi: 10.1016/j.ijhydene.2009.05.093
ABE J O, POPOOLA A P I, AJENIFUJA E, POPOOLA O M. Hydrogen energy, economy and storage: Review and recommendation[J]. Int. J. Hydrog. Energy, 2019, 44(29): 15072-15086 doi: 10.1016/j.ijhydene.2019.04.068
REN J W, MUSYOKA N M, LANGMI H W, MATHE M, LIAO S J. Current research trends and perspectives on materials-based hydrogen storage solutions: A critical review[J]. Int. J. Hydrog. Energy, 2017, 42(1): 289-311 doi: 10.1016/j.ijhydene.2016.11.195
TEICHMANN D, ARLT W, WASSERSCHEID P, FREYMANN R. A future energy supply based on liquid organic hydrogen carriers (LOHC)[J]. Energy Environ. Sci., 2011, 4(8): 2767-2773 doi: 10.1039/c1ee01454d
TEICHMANN D, STARK K, MÜLLER K, ZÖTTL G, WASSERSCHEID P, ARLT W. Energy storage in residential and commercial buildings via liquid organic hydrogen carriers (LOHC)[J]. Energy Environ. Sci., 2012, 5(10): 9044-9054 doi: 10.1039/c2ee22070a
NIERMANN M, BECKENDORFF A, KALTSCHMITT M, BONHOFF K. Liquid organic hydrogen carrier (LOHC)-Assessment based on chemical and economic properties[J]. Int. J. Hydrog. Energy, 2019, 44(13): 6631-6654 doi: 10.1016/j.ijhydene.2019.01.199
KIM C, LEE Y, KIM K, LEE U. Implementation of formic acid as a liquid organic hydrogen carrier (LOHC): Techno-economic analysis and life cycle assessment of formic acid produced via CO2 utilization[J]. Catalysts, 2022, 12(10): 1113 doi: 10.3390/catal12101113
JESSOP P G, JOÓ F, TAI C C. Recent advances in the homogeneous hydrogenation of carbon dioxide[J]. Coord. Chem. Rev., 2004, 248(21/22/23/24): 2425-2442
WEN J L, WANG F Y, ZHANG X M. Asymmetric hydrogenation catalyzed by first-row transition metal complexes[J]. Chem. Soc. Rev., 2021, 50(5): 3211-3237 doi: 10.1039/D0CS00082E
WANG D, ASTRUC D. The golden age of transfer hydrogenation[J]. Chem. Rev., 2015, 115(13): 6621-6686 doi: 10.1021/acs.chemrev.5b00203
TANAKA R, YAMASHITA M, NOZAKI K. Catalytic hydrogenation of carbon dioxide using Ir(Ⅲ)-pincer complexes[J]. J. Am. Chem. Soc., 2009, 131(40): 14168-14169 doi: 10.1021/ja903574e
FILONENKO G A, VAN PUTTEN R, SCHULPEN E N, HENSEN E J M, PIDKO E A. Highly efficient reversible hydrogenation of carbon dioxide to formates using a ruthenium PNP-pincer catalyst[J]. ChemCatChem, 2014, 6(6): 1526-1530 doi: 10.1002/cctc.201402119
STEEL P J. Ligand design in multimetallic architectures: Six lessons learned[J]. Accounts Chem. Res., 2005, 38(4): 243-250 doi: 10.1021/ar040166v
PERIS E, CRABTREE R H. Key factors in pincer ligand design[J]. Chem. Soc. Rev., 2018, 47(6): 1959-1968 doi: 10.1039/C7CS00693D
DURAND D J, FEY N. Computational ligand descriptors for catalyst design[J]. Chem. Rev., 2019, 119(11): 6561-6594 doi: 10.1021/acs.chemrev.8b00588
BRAUNSTEIN P, NAUD F. Hemilability of hybrid ligands and the coordination chemistry of oxazoline-based systems[J]. Angew. Chem. ‒Int. Edit., 2001, 40(4): 680-699 doi: 10.1002/1521-3773(20010216)40:4<680::AID-ANIE6800>3.0.CO;2-0
DAI Z J, LUO Q, CONG H J, ZHANG J, PENG T Y. New Ru(Ⅱ) N′NN′-type pincer complexes: Synthesis, characterization and the catalytic hydrogenation of CO2 or bicarbonates to formate salts[J]. New J. Chem., 2017, 41(8): 3055-3060 doi: 10.1039/C6NJ03855G
THAI T T, THERRIEN B, SÜSS-FINK G. Arene ruthenium oxinato complexes: Synthesis, molecular structure and catalytic activity for the hydrogenation of carbon dioxide in aqueous solution[J]. J. Organomet. Chem., 2009, 694(25): 3973-3981 doi: 10.1016/j.jorganchem.2009.09.008
SANZ S, AZUA A, PERIS E. ‘(η6-Arene)Ru(Bis-NHC)’ complexes for the reduction of CO2 to formate with hydrogen and by transfer hydrogenation with iPrOH[J]. Dalton Trans., 2010, 39(27): 6339-6343 doi: 10.1039/c003220d
SCOTT M, BLAS MOLINOS B, WESTHUES C, FRANCIÒ G, LEITNER W. Aqueous biphasic systems for the synthesis of formates by catalytic CO2 hydrogenation: Integrated reaction and catalyst separation for CO2-scrubbing solutions[J]. ChemSusChem, 2017, 10(6): 1085-1093 doi: 10.1002/cssc.201601814
KOTHANDARAMAN J, CZAUN M, GOEPPERT A, HAIGES R, JONES J P, MAY R B, PRAKASH G K S, OLAH G A. Amine-free reversible hydrogen storage in formate salts catalyzed by ruthenium pincer complex without pH control or solvent change[J]. ChemSusChem, 2015, 8(8): 1442-1451 doi: 10.1002/cssc.201403458
HUFF C A, SANFORD M S. Catalytic CO2 hydrogenation to formate by a ruthenium pincer complex[J]. ACS Catal., 2013, 3(10): 2412-2416 doi: 10.1021/cs400609u
HE X C, LI Y Q, FU H Y, ZHENG X L, CHEN H, LI R X, YU X J. Synthesis of unsymmetrical N-heterocyclic carbene-nitrogen-phosphine chelated ruthenium(Ⅱ) complexes and their reactivity in acceptorless dehydrogenative coupling of alcohols to esters[J]. Organometallics, 2019, 38(8): 1750-1760 doi: 10.1021/acs.organomet.9b00071
ZHU Z, ZHANG J, FU H Y, YUAN M L, ZHENG X L, CHEN H, LI R X. Construction of pincer-type symmetrical ruthenium(Ⅱ) complexes bearing pyridyl-2, 6-pyrazolyl arms: Catalytic behavior in transfer hydrogenation of ketones[J]. RSC Adv., 2014, 4(95): 52734-52739 doi: 10.1039/C4RA07524B
CHEN Y Y, CUI T H, CHEN H, ZHENG X L, FU H Y, LI R X. Pyrazole-pyridine-pyrazole (NNN) ruthenium(Ⅱ) complex catalyzed acceptorless dehydrogenation of alcohols to aldehydes[J]. Dalton Trans., 2023, 52(35): 12368-12377 doi: 10.1039/D3DT01430D
BERNSKOETTER W H, HAZARI N. Reversible hydrogenation of carbon dioxide to formic acid and methanol: Lewis acid enhancement of base metal catalysts[J]. Accounts Chem. Res., 2017, 50(4): 1049-1058 doi: 10.1021/acs.accounts.7b00039
ZHANG Y Y, MACINTOSH A D, WONG J L, BIELINSKI E A, WILLIARD P G, MERCADO B Q, HAZARI N, BERNSKOETTER W H. Iron catalyzed CO2 hydrogenation to formate enhanced by Lewis acid co-catalysts[J]. Chem. Sci., 2015, 6(7): 4291-4299 doi: 10.1039/C5SC01467K
PAVLOVIC L, HOPMANN K H. Understanding the influence of Lewis acids on CO2 hydrogenation: The critical effect is on formate rotation[J]. Organometallics, 2023, 42(20): 3025-3035 doi: 10.1021/acs.organomet.3c00342
GETTY A D, TAI C C, LINEHAN J C, JESSOP P G, OLMSTEAD M M, RHEINGOLD A L. Hydrogenation of carbon dioxide catalyzed by ruthenium trimethylphosphine complexes: A mechanistic investigation using high-pressure NMR spectroscopy[J]. Organometallics, 2009, 28(18): 5466-5477 doi: 10.1021/om900128s
GALAN B R, BIGELOW J O, DOUGHERTY W G, KASSEL W S, HULLEY E B, HELM M L, RAKOWSKI DUBOIS M, APPEL A M, LINEHAN J C. Operando mechanistic studies of CO2 hydrogenation by ruthenium complexes using high-pressure NMR spectroscopy[J]. ACS Catal., 2023, 13(23): 15611-15619 doi: 10.1021/acscatal.3c03908
YE W J, ZHAO M, DU W M, JIANG Q B, WU K K, WU P, YU Z K. Highly active ruthenium(Ⅱ) complex catalysts bearing an unsymmetrical NNN ligand in the (asymmetric) transfer hydrogenation of ketones[J]. Chem. ‒Eur. J., 2011, 17(17): 4737-4741 doi: 10.1002/chem.201002039
MISHRA D, NASKAR S, CHATTOPADHYAY S K, MAJI M, SENGUPTA P, DINDA R, GHOSH S, MAK T C W. Synthesis, crystal structure determination, spectroscopic and electrochemical studies of trans-[Ru(PPh3)2(bbpH2)Cl]Cl·CHCl3·H2O (bbpH2=2,6-bis(benzimidazolyl) pyridine)-An infinite double columnar supramolecule in the solid state[J]. Transit. Met. Chem., 2005, 30(3): 352-356 doi: 10.1007/s11243-004-5860-3

Scheme 1 Constructing Ru-NNN complexes with both high catalytic activity and stability by using rigid ligands and weak ligands simultaneously

Figure 1 Crystal structure of Complex 1 and active intermediate (Ⅱ) (Crystal structure of Complex 1 was obtained from the reference [25].)

Figure 2 (a) 31P NMR spectrum of Complex 1; (b) 31P NMR spectrum of Complex 1 at 50 ℃; (c) 31P NMR spectrum of the mixture of Complex 1 and PPh3 (162 MHz, CDCl3)

Figure 4 Gibbs free energy profiles for CO2 hydrogenation catalyzed by the Ru-NNN complex

Figure 5 Possible catalytic mechanism for CO2 hydrogenation catalyzed by the Ru-NNN complex
Table 1. Catalytic activity of complex 1 for hydrogenation of CO2 to formatea
| Entry | nCat. / μmol | Base | T / ℃ | t / h | TONb | TOFc / h-1 |
| 1 | 1 | NEt3 | 150 | 16 | 1 465 | 92 |
| 2 | 1 | DBU | 150 | 16 | 2 218 | 139 |
| 3 | 1 | KOH | 150 | 16 | 3 412 | 213 |
| 4 | 1 | CsOH | 150 | 16 | 6 213 | 388 |
| 5 | 1 | CsOH | 90 | 16 | 178 | 11 |
| 6 | 1 | CsOH | 100 | 16 | 512 | 32 |
| 7 | 1 | CsOH | 110 | 16 | 1 513 | 95 |
| 8 | 1 | CsOH | 120 | 16 | 2 746 | 172 |
| 9 | 1 | CsOH | 130 | 16 | 2 280 | 143 |
| 10 | 1 | CsOH | 140 | 16 | 9 176 | 574 |
| 11 | 1 | CsOH | 160 | 16 | 7 322 | 458 |
| 12 | 1 | CsOH | 170 | 16 | 4 894 | 306 |
| 13 | 1 | CsOH | 140 | 2 | 1 096 | 548 |
| 14 | 1 | CsOH | 140 | 4 | 2 245 | 561 |
| 15 | 1 | CsOH | 140 | 8 | 4 635 | 579 |
| 16 | 0.001 | CsOH | 140 | 16 | 149 558 | 9 347 |
| 17 | 0.01 | CsOH | 140 | 16 | 68 196 | 4 262 |
| 18 | 0.1 | CsOH | 140 | 16 | 32 116 | 2 007 |
| 19 | 10 | CsOH | 140 | 16 | 584 | 37 |
| 20d | 1 | CsOH | 140 | 16 | 10 304 | 644 |
| a General conditions: complex 1, base (10 mmol), pH2=pCO2=2.5 MPa, VTHF=1 mL, VH2O=5 mL; b TON=nformate/nRu; c TOF=TON/t; d 1 mmol LiOAc as additive. | ||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们