-
[1]
LIU W, OH P, LIU X E, LEE M J, CHO W, CHAE S, KIM Y, CHO J. Nickel-rich layered lithium transition-metal oxide for high-energy lithium-ion batteries[J]. Angew. Chem. ‒Int. Edit., 2015, 54(15): 4440-4457 doi: 10.1002/anie.201409262
-
[2]
ELLIS B L, LEE K T, NAZAR L F. Positive electrode materials for Li-ion and Li-batteries [J]. Chem. Mater., 2010, 22(3): 691-714 doi: 10.1021/cm902696j
-
[3]
WANG D, LIU W H, ZHANG X H, HUANG Y, XU M B, XIAO W. Review of modified nickel-cobalt lithium aluminate cathode materials for lithium-ion batteries[J]. Int. J. Photoenergy, 2019, 2019: 1-13
-
[4]
LIN Y, LI Y, TANG M L, ZHAN L L, ZHAI Y X, CHEN W M, ZHOU M X, JI Y N, WANG P K. A review of high-capacity lithium-rich manganese-based cathode materials for a new generation of lithium batteries[J]. Inorg. Chim. Acta, 2024, 572: 122239 doi: 10.1016/j.ica.2024.122239
-
[5]
THACKERAY M M, JOHNSON C S, VAUGHEY J T, HA E I, HACKNEY S A. Advances in manganese-oxide 'composite' electrodes for lithium-ion batteries[J]. J. Mater. Chem., 2005, 15(23): 2257 doi: 10.1039/b417616m
-
[6]
RANA J, STAN M, KLOEPSCH R, LI J, SCHUMACHER G, WELTER E, ZIZAK I, BANHART J, WINTER M. Structural changes in Li2MnO3 cathode material for Li-ion batteries[J]. Adv. Energy Mater., 2014, 4(5): 1300998 doi: 10.1002/aenm.201300998
-
[7]
WANG Z, LU H Q, YIN Y P, SUN X Y, BAI X T, SHEN X L, ZHUANG W D, LU S G. FePO4-coated Li[Li0.2Ni0.13Co0.13Mn0.54]O2 with improved cycling performance as cathode material for Li-ion batteries[J]. Rare Met., 2017, 36(11): 899-904 doi: 10.1007/s12598-015-0647-6
-
[8]
YABUUCHI N, TAKEUCHI M, NAKAYAMA M, SHIIBA H, OGAWA M, NAKAYAMA K, OHTA T, ENDO D, OZAKI T, INAMASU T, SATO K, KOMABA S. High-capacity electrode materials for rechargeable lithium batteries: Li3NbO4-based system with cation-disordered rocksalt structure[J]. Proc. Natl. Acad. Sci. U. S. A., 2015, 112(25): 7650-7655 doi: 10.1073/pnas.1504901112
-
[9]
HE H C, LI H, BAI X T, CHANG Z H, GAO Z X, ZHANG X J, REN Z M. Comparative study of high-temperature cycling and thermal stability of LLOs and NCMs under medium-high voltage[J]. Energy Fuels, 2023, 37(9): 6854-6864 doi: 10.1021/acs.energyfuels.3c00845
-
[10]
THACKERAY M M, KANG S H, JOHNSON C S, VAUGHEY J T, HACKNEY S A. Comments on the structural complexity of lithium-rich Li1+xM1-xO2 electrodes (M=Mn, Ni, Co) for lithium batteries[J]. Electrochem. Commun., 2006, 8(9): 1531-1538 doi: 10.1016/j.elecom.2006.06.030
-
[11]
GAO K K, SUN C W, WANG Z L. Recent advances in high‑ performance lithium-rich manganese-based materials for solid-state lithium batteries[J]. Mater. Chem. Front., 2024, 8(19): 3082-3105 doi: 10.1039/D4QM00513A
-
[12]
GAUTHIER M, CARNEY T J, GRIMAUD A, GIORDANO L, POUR N, CHANG H H, FENNING D P, LUX S F, PASCHOS O, BAUER C, MAGLIA F, LUPART S, LAMP P, YANG S H. Electrode‑ electrolyte interface in Li-ion batteries: Current understanding and new insights[J]. J. Phys. Chem. Lett., 2015, 6(22): 4653-4672 doi: 10.1021/acs.jpclett.5b01727
-
[13]
BAO Y B, WANG J, QIAN Y X, DENG Y F, YANG X F, CHEN G H. An appropriate amount of new spinel phase induced by control synthesis for the improvement of electrochemical performance of Li-rich layered oxide cathode material[J]. Electrochim. Acta, 2020, 330: 135240 doi: 10.1016/j.electacta.2019.135240
-
[14]
WANG B, CUI J, LI Z J, WANG H, ZHANG D, WANG Q J, SUN H L, WU Y A. Review on comprehending and enhancing the initial Coulombic efficiency of Li-rich Mn-based cathode materials in lithium-ion batteries[J]. Mater. Chem. Front, 2023, 7(13): 2570-2594 doi: 10.1039/D3QM00064H
-
[15]
LI Q Y, NING D, ZHOU D, AN K, WONG D, ZHANG L J, CHEN Z H, SCHUCK G, SCHULZ C, XU Z J, SCHUMACHER G, LIU X F. The effect of oxygen vacancy and spinel phase integration on both anionic and cationic redox in Li-rich cathode materials[J]. J. Mater. Chem. A, 2020, 8(16): 7733-7745 doi: 10.1039/D0TA02517H
-
[16]
李钊, 王忠, 班丽卿, 王建涛, 卢世刚. 富锂锰基正极材料的表面改性研究进展[J]. 化学学报, 2019, 77(11): 1115-1128.LI Z, WANG Z, BAN L P, WANG J T, LU S G. Recent advances on surface modification of Li-and Mn-rich cathode materials[J]. Acta Chim. Sin., 2019, 77(11): 1115
-
[17]
ZHANG J C, ZHANG Q H, WONG D, ZHANG N A, REN G X, GU L, SCHULZ C, HE L H, YU Y, LIU X F. Addressing voltage decay in Li-rich cathodes by broadening the gap between metallic and anionic bands[J]. Nat. Commun., 2021, 12(1): 3071 doi: 10.1038/s41467-021-23365-9
-
[18]
YAN P F, ZHENG J M, CHEN T W, LUO L L, JIANG Y Y, WANG K, SUI M L, ZHANG J G, ZHANG S L, WANG C M. Coupling of electrochemically triggered thermal and mechanical effects to aggravate failure in a layered cathode[J]. Nat. Commun., 2018, 9(1): 2437 doi: 10.1038/s41467-018-04862-w
-
[19]
YAN P F, NIE A M, ZHENG J M, ZHOU Y G, LU D P, ZHANG X F, XU R, BELHAROUAK I, ZU X T, XIAO J, AMINE K, LIU J, GAO F, REZA S Y, ZHANG J G, WANG C M. Evolution of lattice structure and chemical composition of the surface reconstruction layer in Li1.2Ni0.2Mn0.6O2 cathode material for lithium ion batteries[J]. Nano Lett., 2015, 15(1): 514-522 doi: 10.1021/nl5038598
-
[20]
REN Z M, LI G H, BAI X T, HU W, LI X Y, HE H C, CHANG Z H, LIU Y, WANG Z Y, LIANG Z S, GUO L H, GAO Z X, WANG J T. A novel boron-doping approach for long-term cycling life of Li-rich layered cathode[J]. Electrochim. Acta, 2023, 461: 142630 doi: 10.1016/j.electacta.2023.142630
-
[21]
JIN Y L, XU Y L, REN F, REN P G. Mg-doped Li1.133Ni0.2 Co0.2Mn0.467O2 in Li site as high-performance cathode material for Li-ion batteries[J]. Solid State Ion., 2019, 336: 87-94 doi: 10.1016/j.ssi.2019.03.020
-
[22]
SHI Z J, WANG X, FENG W J, SONG C K, CHEN L J, LI M M. The effect on the properties of each element in the Li1.2Mn0.54Ni0.13Co0.13O2 material by the incorporation of Al[J]. Ionics, 2020, 26(12): 5951-5959 doi: 10.1007/s11581-020-03520-w
-
[23]
ZHANG P P, ZHAI X H, HUANG H, ZHOU J F, LI X B, HE Y P, GUO Z C. Suppression of structural phase transformation of Li-rich Mn-based layered cathode materials with Na ion substitution strategy[J]. Electrochim. Acta, 2020, 349: 136402 doi: 10.1016/j.electacta.2020.136402
-
[24]
HE W, YUAN D D, QIAN J F, AI X P, YANG H X, CAO Y L. Enhanced high-rate capability and cycling stability of Na-stabilized layered Li1.2[Co0.13Ni0.13Mn0.54]O2 cathode material[J]. J. Mater. Chem. A, 2013, 1(37): 11397 doi: 10.1039/c3ta12296d
-
[25]
LI T, XIA X Y, LIU J, LIU Z X, HU S, ZHANG L F, ZHENG Y W, WANG Z K, CHEN H L, PENG M J, QIAN T, YAN C L. Suppressing surface lattice oxygen evolution by fluorinated graphene-scaffolded lithium-rich manganese-based cathode for enhanced stability[J]. Energy Storage Mater., 2022, 49: 555-563 doi: 10.1016/j.ensm.2022.05.002
-
[26]
YU H N, XUE Z C, XUE Z Y, LUO Z Y, DING C X, HU G R, PENG Z D, CAO Y B, DU K. Inhibition of oxygen release and stabilization of the bulk structure of lithium-rich layered oxides by strong Mo—O covalent binding[J]. J. Mater. Chem. A, 2024, 12(1): 267-276 doi: 10.1039/D3TA05649J
-
[27]
AMZIL S, ZHANG Y, QIU T P, JIANG F, RAO H, FANG Z B, HUANG J J. {010} oriented Li1.2Mn0.54Co0.13Ni0.13O2@C nanoplate with high initial columbic efficiency[J]. J. Power Sources, 2021, 486: 229372 doi: 10.1016/j.jpowsour.2020.229372
-
[28]
李钊, 王忠, 李强, 班丽卿, 庄卫东, 卢世刚. 一种碳纳米管改性富锂锰基正极材料的策略[J]. 无机化学学报, 2019, 35(9): 1561-1569. doi: 10.11862/CJIC.2019.192LI Z, WANG Z, LI Q, BAN L Q, ZHUANG W D, LU S G. A strategy for carbon nanotubes modified lithium-manganese-rich cathode material[J]. Chinese J. Inorg. Chem., 2019, 35(9): 1561-1569 doi: 10.11862/CJIC.2019.192
-
[29]
LI Z, LI Q, ZHANG A B, WEN W, WANG L, WANG Z Y, WANG J T, LU S G, LI X, WANG Z. Tuning surface conductivity and stability for high-performance Li-and Mn-rich cathode materials[J]. New J. Chem., 2019, 43(47): 18943-18950 doi: 10.1039/C9NJ04531G
-
[30]
CHEN S, ZHENG Y, LU Y, SU Y F, BAO L Y, LI N, LI Y T, WANG J, CHEN R J, WU F. Enhanced electrochemical performance of layered lithium-rich cathode materials by constructing spinel-structure skin and ferric oxide islands[J]. ACS Appl. Mater. Interfaces, 2017, 9(10): 8669-8678 doi: 10.1021/acsami.6b14862
-
[31]
LI Z, LI Q, WU S J, ZHANG A B, ZHUO H X, ZHANG G N, WANG Z, WANG L, REN Z M, WANG J T. Enhanced electrochemical performance of Li-and Mn-rich cathode materials by particle blending and surface coating[J]. ChemistrySelect, 2020, 5(10): 3052-3061 doi: 10.1002/slct.201904290
-
[32]
LIU X H, WANG Z Y, ZHUANG W, BAN L Q, GAO M, LI W J, YIN Y P, WANG Z, LU S G. Li3PO4 modification on primary particle surface for high performance Li-rich layered oxide Li1.18Mn0.52Co0.15 Ni0.15O2 via a synchronous route[J]. New J. Chem., 2018, 42(6): 3999-4007 doi: 10.1039/C7NJ03337K
-
[33]
DENG Y P, FU F, WU Z G, YIN Z W, ZHANG T, LI J T, HUANG L, SUN S G. Layered/spinel heterostructured Li-rich materials synthesized by a one-step solvothermal strategy with enhanced electrochemical performance for Li-ion batteries[J]. J. Mater. Chem. A, 2016, 4(1): 257-263 doi: 10.1039/C5TA06945A
-
[34]
LI G H, REN Z M, LI A L, YU R, QUAN W, WANG C H, LIN T, YI D, LIU Y, ZHANG Q H, WANG J T, YU H J, SUN X L. Highly stable surface and structural origin for lithium-rich layered oxide cathode materials[J]. Nano Energy, 2022, 98: 107169 doi: 10.1016/j.nanoen.2022.107169
-
[35]
ZHAO X W, SHENG C C, CHANG Z, CHENG L, WU P, XU L, ZHOU Y M, HE P, TANG Y W, CAO X, ZHOU H S. Solid-state exfoliation growth mechanism of single-crystal Li-rich layered cathode materials[J]. Energy Storage Mater., 2025, 75: 104093 doi: 10.1016/j.ensm.2025.104093
-
[36]
GAO X G, WANG L, GUO J L, LI S H, ZHANG H Y, CHEN L, ZHANG Y, LAI Y Q, ZHANG Z A. Lattice engineering toward extraordinary structural stability of high-performance single-crystal Li-rich layered oxides cathodes[J]. Adv. Funct. Mater., 2024, 34(46): 2407692 doi: 10.1002/adfm.202407692
-
[37]
AI J, ZHAO X W, CAO X, XU L, WU P, ZHOU Y M, HE P, TANG Y W, ZHOU H S. Impact of lithium sources on growth process and structural stability of single-crystalline Li-rich layered cathodes[J]. Batteries Supercaps, 2025, 8(2): e202400425 doi: 10.1002/batt.202400425
-
[38]
WU T H, ZHANG X, WANG Y Z, ZHANG N, LI H F, GUAN Y, XIAO D D, LIU S Q, YU H J. Gradient "single-crystal" Li-rich cathode materials for high-stable lithium-ion batteries[J]. Adv. Funct. Mater., 2023, 33(4): 2210154 doi: 10.1002/adfm.202210154
-
[39]
LIU S Y, WANG Z L, HUANG Y K, NI Z J, BAI J R, KANG S F, WANG Y G, LI X. Fluorine doping and Al2O3 coating co-modified Li[Li0.20Ni0.133Co0.133Mn0.534]O2 as high performance cathode material for lithium-ion batteries[J]. J. Alloy. Compd., 2018, 731: 636-645 doi: 10.1016/j.jallcom.2017.09.341
-
[40]
CHEN C, GENG T F, DU C Y, ZUO P J, CHENG X Q, MA Y L, YIN G P. Oxygen vacancies in SnO2 surface coating to enhance the activation of layered Li-rich Li1.2Mn0.54Ni0.13Co0.13O2 cathode material for Li-ion batteries[J]. J. Power Sources, 2016, 331: 91-99 doi: 10.1016/j.jpowsour.2016.09.051
-
[41]
尹艳萍, 庄卫东, 王忠, 卢华权, 卢世刚. 不同锰源对富锂正极材料Li1.2Mn0.54Ni0.13Co0.13O2性能的影响[J]. 稀有金属, 2015, 39(10): 891-895.YIN Y P, ZHUANG W D, WANG Z, LU H Q, LU S G. Electrochemical characteristics of Li-rich cathode material Li1.2Mn0.54Ni0.13Co0.13O2 with different manganese raw materials[J]. Chinese Journal of Rare Metals, 2015, 39(10): 891-895
-
[42]
王忠, 卢华权, 尹艳萍, 庄卫东, 卢世刚. 锂离子电池富锂正极材料Li1.2Ni0.13Co0.13Mn0.54O2的合成及性能[J]. 稀有金属, 2016, 40(1): 38-42.WANG Z, LU H Q, YIN Y P, ZHUANG W D, LU S G. Synthesis and properties of Li-rich cathode material Li1.2Ni0.13Co0.13Mn0.54O2 in Li-ion batteries[J]. Chinese Journal of Rare Metals, 2016, 40(1): 38-42
-
[43]
XU G L, QIN Y, REN Y, CAI L, AN K, AMINE K, CHEN Z H. The migration mechanism of transition metal ions in LiNi0.5Mn1.5O4[J]. J. Mater. Chem. A, 2015, 3(24): 13031-13038 doi: 10.1039/C5TA02522B
-
[44]
WANG Z, YIN Y P, REN Y, WANG Z Y, GAO M, MA T Y, ZHUANG W D, LU S G, FAN A L, AMINE K, CHEN Z H. High performance lithium-manganese-rich cathode material with reduced impurities[J]. Nano Energy, 2017, 31: 247-257 doi: 10.1016/j.nanoen.2016.10.014
-
[45]
CHEN J H, FENG S Y, DENG J H, ZHOU Y F. Green synthesis of manganese-rich carbonate precursor with high sphericity for cobalt-free lithium-rich cathode materials through two-stage continuous coprecipitation method[J]. Ind. Eng. Chem. Res., 2025, 64(25): 12640-12653 doi: 10.1021/acs.iecr.5c00394
-
[46]
PURWAMARGAPRATALA Y, SYAHRIAL A Z, KARTINI E, SIMORANGKIR Y J B. Synthesis and characterization of precursors Ni0.5Mn0.4Co0.1(OH)2 by coprecipitation method[J]. J. Inst. Eng. India Ser. D, 2025, 106(1): 177-182 doi: 10.1007/s40033-023-00606-3
-
[47]
LI Y J, CHEN Y, LIU Y, CHU G W, SUN B C, CHEN J F. Growth mechanism and process intensification of Ni-Al-layered double hydroxide synthesized via coprecipitation[J]. AIChE J., 2025, 71(4): e18672 doi: 10.1002/aic.18672
-
[48]
尹艳萍, 卢华权, 王忠, 孙学义, 庄卫东, 卢世刚. 富锂层状正极材料Li1.2Mn0.54Ni0.13Co0.13O2的二次颗粒粒径对其倍率性能的影响[J]. 无机化学学报, 2015, 31(10): 1966-1970. doi: 10.11862/CJIC.2015.271YIN Y P, LU H Q, WANG Z, SUN X Y, ZHUANG W D, LU S G. Effect of different second particle size on rate capability of Li-rich layered cathode materials Li1.2Mn0.54Ni0.13Co0.13O2[J]. Chinese J. Inorg. Chem., 2015, 31(10): 1966-1970 doi: 10.11862/CJIC.2015.271
-
[49]
WARMAN J F, KARUNAWAN J, FLOWERI O, SURYADI P N, SANTOSA S P, ISKANDAR F. Revealing the dual role of ammonia in the hydroxide co-precipitation synthesis of cobalt-free nickel-rich LiNi0.9Mn0.05Al0.05O2 (NMA955) cathode materials for lithium-ion batteries[J]. Chem. ‒Asian J., 2025, 20(2): e202401080 doi: 10.1002/asia.202401080
-
[50]
PRETTENCIA L J, SOUNDARRAJAN E, MATHIARASU R R, KALAIVANI R, RAGHU S. Sol-gel route synthesis of high energy density Li[Li0.2Ni0.3Mn0.7]O2 cathode with controlled structure, morphology and enhanced electrochemical performance[J]. Energy Storage, 2023, 5(4): e427 doi: 10.1002/est2.427
-
[51]
ISLAM M, AHMED MD S, FAIZAN M, ALI B, BHUYAN M M, BARI G A K M R, NAM K W. Review on the polymeric and chelate gel precursor for Li-ion battery cathode material synthesis[J]. Gels, 2024, 10(9): 586 doi: 10.3390/gels10090586
-
[52]
PILLAI A M, SALINI P S, JOHN B, NAIR V S, JALAJA K, SUJATHA S A, DEVASSY M T. Cobalt-free Li-rich high-capacity cathode material for lithium-ion cells synthesized through sol-gel method and its electrochemical performance[J]. Ionics, 2022, 28(11): 5005-5014 doi: 10.1007/s11581-022-04725-x
-
[53]
HE X, WANG J, KLOEPSCH R, KRUEGER S, JIA H P, LIU H D, VORTMANN B, LI J. Enhanced electrochemical performance in lithium ion batteries of a hollow spherical lithium-rich cathode material synthesized by a molten salt method[J]. Nano Res., 2014, 7(1): 110-118 doi: 10.1007/s12274-013-0378-7
-
[54]
SHANG Y. Understanding the growth mechanism of lithium-rich layered oxides in molten salts route for high performance lithium-ion batteries[J]. Ceram. Int., 2025, 51(3): 2966-2973 doi: 10.1016/j.ceramint.2024.11.273
-
[55]
YAN L Y, GAO Y, CHEN M M, ZHAO C, YANG S C, CHEN C, LI J X, LI Y, WANG K H, WANG H, LI J J, ZHANG H T, MAO J. Preparation of single-crystal Li-rich Mn-based layered oxides with excellent electrochemical performance via simple stepwise high- temperature sintering[J]. ACS Appl. Mater. Interfaces, 2024, 16(44): 60166-60179 doi: 10.1021/acsami.4c11204
-
[56]
PULIDO R, NAVEAS N, GRABER T, RAÚL J. M P, FERNANDO A R, BRITO I, MORALES C, SORIANO L, PASCUAL L, MARINI C, JACOBO H M, MIGUEL M S. Hydrothermal control of the lithium-rich Li2MnO3 phase in lithium manganese oxide nanocomposites and their application as precursors for lithium adsorbents[J]. Dalton Trans., 2021, 50(31): 10765-10778 doi: 10.1039/D1DT01638E
-
[57]
FAN J M, LI G S, LUO D, FU C C, LI Q, ZHENG J, LI L P. Hydrothermal-assisted synthesis of Li-rich layered oxide microspheres with high capacity and superior rate-capability as a cathode for lithium-ion batteries[J]. Electrochim. Acta, 2015, 173: 7-16 doi: 10.1016/j.electacta.2015.05.028
-
[58]
陈志铭, 储爱民, 周子榆, 赵玉萍, 陈友明. 含碳雾滴燃烧制备微纳空心球型富锂锰基正极材料及性能研究[J]. 储能科学与技术, 2025, 14(4): 1362-1368.CHEN Z M, CHU A M, ZHOU Z Y, ZHAO Y P, CHEN Y M. Preparation and performance of Li-rich cathode material by carbon-containing droplet combustion[J]. Energy Storage Science and Technology, 2025, 14(4): 1362-1368
-
[59]
温涛, 李小成, 凡纪鹏, 邓一坤, 邹菁, 王海涛. 高镍锂离子电池三元正极材料的掺杂改性研究进展[J]. 石油化工高等学校学报, 2025, 38(3): 20-31.WEN T, LI X C, FAN J P, DENG Y K, ZOU J, WANG H T. Research progress on doping modification of ternary cathode materials for nickel-rich lithium ion batteries[J]. Journal of Petrochemical Universities, 2025, 38(3): 20-31
-
[60]
KUMAR D, KURIAN E, RAMESHA K. Synthesis of quasi-spherical LiNi0.86Mn0.1Co0.04O2 cathode particles via hydroxide coprecipitation: Influence of pH on precursor particle size in enhancing capacity and stability of Ni-rich cathode for lithium-ion batteries[J]. Energy Technol., 2025, 13(9): 2402418 doi: 10.1002/ente.202402418
-
[61]
KAN Y C, HU Y, CROY J, REN Y, SUN C J, HEALD S M, BAREÑO J, BLOOM I, CHEN Z H. Formation of Li2MnO3 investigated by in situ synchrotron probes[J]. J. Power Sources, 2014, 266: 341-346 doi: 10.1016/j.jpowsour.2014.05.032
-
[62]
KAN Y C, HU Y, REN Y, BAREÑO J, BLOOM I, SUN Y K, AMINE K, CHEN Z H. Differentiating allotropic LiCoO2/Li2Co2O4: A structural and electrochemical study[J]. J. Power Sources, 2014, 271: 97-103 doi: 10.1016/j.jpowsour.2014.07.178
-
[63]
CHEN Y X, LUO S L, LENG J, DENG S, YAN S, TIAN X, LI Y J, GUO J, LEI T X, ZHENG J C. Exploring the synthesis conditions and formation mechanisms of Li-rich layered oxides via solid-state method[J]. J. Alloy. Compd., 2021, 854: 157204 doi: 10.1016/j.jallcom.2020.157204
-
[64]
LI T T, SHI Z P, LI L, ZHANG Y B, LI Y, ZHAO J L, GU Q W, WEN W, QIU B, LIU Z P. Non-eutectic-salt reaction route towards morphological and structural rearrangement of Li-rich layered oxides for high-volumetric Li-ion batteries[J]. Chem. Eng. J., 2023, 474: 145728 doi: 10.1016/j.cej.2023.145728
-
[65]
YANG S Y, WANG X Y, YANG X K, LIU Z L, WEI Q L, SHU H B. High tap density spherical Li[Ni0.5Mn0.3Co0.2]O2 cathode material synthesized via continuous hydroxide coprecipitation method for advanced lithium-ion batteries[J]. Int. J. Electrochem., 2012, 2012: 323560
-
[66]
YING J R, JIANG C Y, WAN C R. Preparation and characterization of high-density spherical LiCoO2 cathode material for lithium ion batteries[J]. J. Power Sources, 2004, 129(2): 264-269 doi: 10.1016/j.jpowsour.2003.10.007
-
[67]
ZHANG H Y, GUO J L, GAO X G, LI S H, LAI Y Q, ZHANG Z A. Facile morphology modulation to enhance stability and fast kinetics of anionic redox for Li-rich layered oxides cathodes[J]. ACS Appl. Energy Mater., 2024, 7(3): 1234-1242 doi: 10.1021/acsaem.3c02785
-
[68]
WU C, BAN J Q, CHEN T, WANG J, HE Y D, WU Z G. Evolution path of precursor-induced high-temperature lithiation reaction during the synthesis of lithium-rich cathode materials[J]. ACS Omega, 2024, 9(13): 15191-15201 doi: 10.1021/acsomega.3c09567
-
[69]
VERDE M G, LIU H D, CARROLL K J, BAGGETTO L, VEITH G M, MENG Y S. Effect of morphology and manganese valence on the voltage fade and capacity retention of Li[Li2/12Ni3/12Mn7/12]O2[J]. ACS Appl. Mater. Interfaces, 2014, 6(21): 18868-18877 doi: 10.1021/am504701s
-
[70]
ZHENG J, LIU T C, HU Z X, WEI Y, SONG X H, REN Y, WANG W D, RAO M M, LIN Y, CHEN Z H, LU J, WANG C M, AMINE K, PAN F. Tuning of thermal stability in layered Li(NixMnyCoz)O2[J]. J. Am. Chem. Soc., 2016, 138(40): 13326-13334 doi: 10.1021/jacs.6b07771
-
[71]
REDEL K, KULKA A, WALCZAK K, PLEWA A, HANC E, MARZEC M, LU L, MOLENDA J. Origin of extra capacity in advanced Li-rich cathode materials for rechargeable Li-ion batteries[J]. Chem. Eng. J., 2021, 424: 130293 doi: 10.1016/j.cej.2021.130293
-
[72]
LI S H, LI H X, ZHANG H Y, ZHANG S, LAI Y, ZHANG Z A. Constructing stable surface structures enabling fast charging for Li-rich layered oxide cathodes[J]. Chem. Eng. J., 2022, 427: 132036 doi: 10.1016/j.cej.2021.132036
-
[73]
GRENIER A, KAMM G E, LI Y X, CHUNG H, MENG Y S, CHAPMAN K W. Nanostructure transformation as a signature of oxygen redox in Li-rich 3d and 4d cathodes[J]. J. Am. Chem. Soc., 2021, 143(15): 5763-5770 doi: 10.1021/jacs.1c00497
-
[74]
LEE J, DUPRE N, JEONG M, KANG S Y, AVDEEV M, GONG Y, GU L, YOON W S, KANG B. Fully exploited oxygen redox reaction by the inter-diffused cations in Co-free Li-rich materials for high performance Li-ion batteries[J]. Adv. Sci., 2020, 7(17): 2001658 doi: 10.1002/advs.202001658
-
[75]
HWANG J, MYEONG S, LEE E, JANG H, YOON M, CHA H, SUNG J, KIM M G, SEO D H, CHO J. Lattice-oxygen-stabilized Li-and Mn-rich cathodes with sub-micrometer particles by modifying the excess-Li distribution[J]. Adv. Mater., 2021, 33(18): 2100352 doi: 10.1002/adma.202100352
-
[76]
CHOI G, CHANG U, LEE J, PARK K, KWON H, LEE H, KIM Y I, SEO J H, PARK Y C, PARK I, KIM J, LEE S, CHOI J, YU B, SONG J H, SHIN H, BAEK S W, LEE S K, PARK H, JUNG K. Unraveling and regulating superstructure domain dispersion in lithium-rich layered oxide cathodes for high stability and reversibility[J]. Energy Environ. Sci., 2024, 17(13): 4634-4645 doi: 10.1039/D4EE00487F
-
[77]
HUA W B, CHEN M, SCHWARZ B, KNAPP M, BRUNS M, BARTHEL J, YANG X S, SIGEL F, AZMI R, SENYSHYN A, MISSIUL A, SIMONELLI L, ETTER M, WANG S N, MU X K, FIEDLER A, BINDER J R, GUO X D, CHOU S L, ZHONG B H, INDRIS S, EHRENBERG H. Lithium/oxygen incorporation and microstructural evolution during synthesis of Li-rich layered Li[Li0.2Ni0.2Mn0.6]O2 oxides[J]. Adv. Energy Mater., 2019, 9(8): 1803094 doi: 10.1002/aenm.201803094
-
[78]
PARK H, PARK H, SONG K, SONG S H, KANG S, KO K H, EUM D, JEON Y, KIM J, SEONG W M, KIM H, PARK J, KANG K. In situ multiscale probing of the synthesis of a Ni-rich layered oxide cathode reveals reaction heterogeneity driven by competing kinetic pathways[J]. Nat. Chem., 2022, 14(6): 614-622 doi: 10.1038/s41557-022-00915-2
-
[79]
WANG D W, ZHANG X, ZHONG G M, LI Y X, HONG C Y, DONG K J, CHEN C X, YANG Y. Temperature-dependence of calcination processes of Ni-rich layered oxides[J]. J. Power Sources, 2022, 529: 231258 doi: 10.1016/j.jpowsour.2022.231258
-
[80]
XIAO X, WANG L, LI J T, ZHANG B, HU Q, LIU J L, WU Y Q, GAO J H, CHEN Y B, SONG S L, ZHANG X Q, CHEN Z H, HE X M. Rational synthesis of high-performance Ni-rich layered oxide cathode enabled via probing solid-state lithiation evolution[J]. Nano Energy, 2023, 113: 108528 doi: 10.1016/j.nanoen.2023.108528
-
[81]
LIU J J, YUAN Y F, ZHENG J H, WANG L G, JI J, ZHANG Q, YANG L, BAI Z Y, LU J. Understanding the synthesis kinetics of single-crystal Co-free Ni-rich cathodes[J]. Angew. Chem. ‒Int. Edit., 2023, 62(20): e202302547 doi: 10.1002/anie.202302547
-
[82]
CAO K, SHEN T T, WANG K P, CHEN D M, WANG W L. Influence of different lithium sources on the morphology, structure and electrochemical performances of lithium-rich layered oxides[J]. Ceram. Int., 2017, 43(12): 8694-8702 doi: 10.1016/j.ceramint.2017.03.203
-
[83]
WANG P B, LUO M Z, ZHENG J C, HE Z J, TONG H, YU W J. Comparative investigation of 0.5Li2MnO3·0.5LiNi0.5Co0.2Mn0.3O2 cathode materials synthesized by using different lithium sources[J]. Front. Chem., 2018, 6: 159 doi: 10.3389/fchem.2018.00159
-
[84]
LI J, CAMARDESE J, GLAZIER S, DAHN J R. Structural and electrochemical study of the Li-Mn-Ni oxide system within the layered single phase region[J]. Chem. Mater., 2014, 26(24): 7059-7066 doi: 10.1021/cm503505b
-
[85]
CHEN M, CHEN D R, LIAO Y H, ZHONG X X, LI W S, ZHANG Y G. Layered lithium‑rich oxide nanoparticles doped with spinel phase: Acidic sucrose-assistant synthesis and excellent performance as cathode of lithium ion battery[J]. ACS Appl. Mater. Interfaces, 2016, 8(7): 4575-4584 doi: 10.1021/acsami.5b10219
-
[86]
PEI Y, CHEN Q, XIAO Y C, LIU L, XU C Y, ZHEN L, HENKELMAN G, CAO G Z. Understanding the phase transitions in spinel-layered-rock salt system: Criterion for the rational design of LLO/spinel nanocomposites[J]. Nano Energy, 2017, 40: 566-575 doi: 10.1016/j.nanoen.2017.08.054
-
[87]
HAO Z K, SUN H X, NI Y X, YANG G J, YANG Z, HAO Z M, WANG R H, YANG P K, LU Y, ZHAO Q, XIE W W, YAN Z H, ZHANG W, CHEN J. Suppressing bulk strain and surface O2 release in Li-rich cathodes by just tuning the Li content[J]. Adv. Mater., 2024, 36(1): 2307617 doi: 10.1002/adma.202307617
-
[88]
LIU P F, ZHANG H, HE W, XIONG T F, CHENG Y, XIE Q S, MA Y T, ZHENG H F, WANG L S, ZHU Z Z, PENG Y, MAI L Q, PENG D L. Lithium deficiencies engineering in Li‑rich layered oxide Li1.098Mn0.533Ni0.113Co0.138O2 for high‑stability cathode[J]. J. Am. Chem. Soc., 2019, 141(27): 10876-10882 doi: 10.1021/jacs.9b04974
-
[89]
HE D Y, TONG W X, ZHANG J, HUANG Z Y, CHEN Z W, YANG M L, WANG R, ZHAO W G, MA Z W, XIAO Y G. Structural insights into lithium-deficient type Li-rich layered oxide for high- performance cathode[J]. Chin. J. Struct. Chem., 2023, 42(5): 100060
-
[90]
CHEN C J, PANG W K, MORI T, PETERSON V K, SHARMA N, LEE P H, WU S H, WANG C C, SONG Y F, LIU R S. The origin of capacity fade in the Li2MnO3·LiMO2 (M=Li, Ni, Co, Mn) microsphere positive electrode: An operando neutron diffraction and transmission X-ray microscopy study[J]. J. Am. Chem. Soc., 2016, 138(28): 8824-8833 doi: 10.1021/jacs.6b03932
-
[91]
WEI Z N, SHI Z P, WEN X H, LI X, QIU B, GU Q W, SUN J, HAN Y Y, LUO H, GUO H C, XIA Y G, YIN C, CAI P J, LIU Z P. Eliminating oxygen releasing of Li-rich layered cathodes by tuning the distribution of superlattice domain[J]. Mater. Today Energy, 2022, 27: 101039 doi: 10.1016/j.mtener.2022.101039
-
[92]
ZHANG Y, CHEN Z F, SHI X Y, MENG C X, DAS P, ZHENG S H, PAN F, WU Z S. Regulation of 3d-transition metal interlayered disorder by appropriate lithium depletion for Li-rich layered oxide with remarkably enhanced initial Coulombic efficiency and stability[J]. Adv. Energy Mater., 2023, 13(5): 2203045 doi: 10.1002/aenm.202203045
-
[93]
WU Y, MA C, YANG J H, LI Z C, ALLARD L F, LIANG C D, CHI M F. Probing the initiation of voltage decay in Li-rich layered cathode materials at the atomic scale[J]. J. Mater. Chem. A, 2015, 3(10): 5385-5391 doi: 10.1039/C4TA06856D
-
[94]
SHEN C H, WANG Q, FU F, HUANG L, LIN Z, SHEN S Y, SU H, ZHENG X M, XU B B, LI J T, SUN S G. Facile synthesis of the Li-rich layered oxide Li1.23Ni0.09Co0.12Mn0.56O2 with superior lithium storage performance and new insights into structural transformation of the layered oxide material during charge-discharge cycle: In situ XRD characterization[J]. ACS Appl. Mater. Interfaces, 2014, 6(8): 5516-5524 doi: 10.1021/am405844b
-
[95]
ZHANG M K, LI J Y, PANG Q, HUA W B, DENG Y T, TANG M Q, PANG W K, WU Z G, ZHONG B H, XIAO Y, QIU L, GUO X D. Phase transition behavior during sintering process of Li-rich materials[J]. Adv. Energy Mater., 2025, 15(21): 2406031 doi: 10.1002/aenm.202406031
-
[96]
DEVARAJ A, GU M, COLBY R, YAN P, WANG C M, ZHENG J M, XIAO J, GENC A, ZHANG J G, BELHAROUAK I, WANG D, AMINE K, THEVUTHASAN S. Visualizing nanoscale 3D compositional fluctuation of lithium in advanced lithium-ion battery cathodes[J]. Nat. Commun., 2015, 6(1): 8014 doi: 10.1038/ncomms9014
-
[97]
YE D L, OZAWA K, WANG B, DENISA H J, ZOU J, SUN C H, WANG L Z. Capacity-controllable Li-rich cathode materials for lithium-ion batteries[J]. Nano Energy, 2014, 6: 92-102 doi: 10.1016/j.nanoen.2014.03.013
-
[98]
YE D L, WANG B, CHEN Y, HAN G, ZHANG Z, DENISA H J, ZOU J, WANG L Z. Understanding the stepwise capacity increase of high energy low-Co Li-rich cathode materials for lithium ion batteries[J]. J. Mater. Chem. A, 2014, 2(44): 18767-18774 doi: 10.1039/C4TA03692A
-
[99]
WEN X H, YIN C, QIU B, WAN L Y, ZHOU Y H, WEI Z N, SHI Z P, HUANG X, GU Q W, LIU Z P. Controls of oxygen-partial pressure to accelerate the electrochemical activation in Co-free Li-rich layered oxide cathodes[J]. J. Power Sources, 2022, 523: 231022 doi: 10.1016/j.jpowsour.2022.231022
-
[100]
LEE W, MUHAMMAD S, KIM T, KIM H, LEE E, JEONG M, SON S, RYOU J H, YOON W S. New insight into Ni-rich layered structure for next-generation Li rechargeable batteries[J]. Adv. Energy Mater., 2018, 8(4): 1701788 doi: 10.1002/aenm.201701788
-
[101]
DING Z P, XU M Q, LIU J T, HUANG Q, CHEN L B, WANG P, IVEY D G, WEI W F. Understanding the enhanced kinetics of gradient-chemical-doped lithium-rich cathode material[J]. ACS Appl. Mater. Interfaces, 2017, 9(24): 20519-20526 doi: 10.1021/acsami.7b02944
-
[102]
LIU J L, HOU M Y, YI J, GUO S S, WANG C X, XIA Y Y. Improving the electrochemical performance of layered lithium‑rich transition‑metal oxides by controlling the structural defects[J]. Energy Environ. Sci., 2014, 7(2): 705-714 doi: 10.1039/C3EE41664J
-
[103]
WAN X W, CHE W, ZHANG D Y, CHANG C K. Improved electrochemical performance of Mn-based Li-rich cathode Li1.4Mn0.61Ni0.18 Co0.18Al0.03O2.4 synthesized in oxygen atmosphere[J]. J. Alloy. Compd., 2021, 875: 159947 doi: 10.1016/j.jallcom.2021.159947
-
[104]
KNIGHT J C, MANTHIRAM A. Effect of nickel oxidation state on the structural and electrochemical characteristics of lithium-rich layered oxide cathodes[J]. J. Mater. Chem. A, 2015, 3(44): 22199-22207 doi: 10.1039/C5TA05703E
-
[105]
YANG J, XIA Y Y. Suppressing the phase transition of the layered Ni-rich oxide cathode during high-voltage cycling by introducing low-content Li2MnO3[J]. ACS Appl. Mater. Interfaces, 2016, 8(2): 1297-1308 doi: 10.1021/acsami.5b09938
-
[106]
ZHU J X, VO T, LI D S, LU R, KINSINGER N M, XIONG L, YAN Y S, KISAILUS D. Crystal growth of Li[Ni1/3Co1/3Mn1/3]O2 as a cathode material for high-performance lithium ion batteries[J]. Cryst. Growth Des., 2012, 12(3): 1118-1123 doi: 10.1021/cg200565n
-
[107]
MA Q X, PENG F W, LI R H, YIN S B, DAI C S. Effect of calcination temperature on microstructure and electrochemical performance of lithium-rich layered oxide cathode materials[J]. Mater. Sci. Eng. B‒Adv. Funct. Solid‒State Mater., 2016, 213: 123-130 doi: 10.1016/j.mseb.2016.04.010
-
[108]
NOMURA F, LIU Y B, TANABE T, TAMURA N, TSUDA T, HAGIWARA T, GUNJI T, OHSAKA T, MATSUMOTO F. Optimization of calcination temperature in preparation of a high capacity Li-rich solid-solution Li[Li0.2Ni0.18Co0.03Mn0.58]O2 material and its cathode performance in lithium ion battery[J]. Electrochim. Acta, 2018, 269: 321-330 doi: 10.1016/j.electacta.2018.03.027
-
[109]
HUANG Z Y, WANG X, FENG W J, LI W X, SHI Z J, LEI Z R. Effect of sintering temperature on the electrochemical performance of Li-rich Mn-based cathode material Li1.2Mn0.54Ni0.13Co0.13O2 by Co‑precipitation method[J]. J. Electroanal. Chem., 2021, 895: 115439 doi: 10.1016/j.jelechem.2021.115439
-
[110]
ZHENG Z H, BEI F L, HUI T, YU H Q, QIAN H, HUANG H H, CHE L D. Lithium salt preprocessing calcination strategy for a more stable layered structure of lithium-rich manganese-based cathodes[J]. J. Power Sources, 2024, 620: 235232 doi: 10.1016/j.jpowsour.2024.235232
-
[111]
YU F G, NONG J X, ZOU Z G, FENG M, ZHANG S C, LIANG F G, JIA S K, CHEN M. Enhanced electrochemical properties of Li1.2Mn0.54Co0.13Ni0.13O2 by modulation of surface oxygen vacancies[J]. J. Energy Storage, 2024, 98: 113149 doi: 10.1016/j.est.2024.113149
-
[112]
CHEN Y F, CHEN Z X, XIE K. Effect of annealing on the first- cycle performance and reversible capabilities of lithium-rich layered oxide cathodes[J]. J. Phys. Chem. C, 2014, 118(22): 11505-11511 doi: 10.1021/jp500138s
-
[113]
LUO D, LI G S, FU C C, ZHENG J, FAN J M, LI Q, LI L P. A new spinel-layered Li-rich microsphere as a high-rate cathode material for Li-ion batteries[J]. Adv. Energy Mater., 2014, 4(11): 1400062 doi: 10.1002/aenm.201400062
-
[114]
QIAN D N, XU B, CHI M F, MENG Y S. Uncovering the roles of oxygen vacancies in cation migration in lithium excess layered oxides[J]. Phys. Chem. Chem. Phys., 2014, 16(28): 14665-14668 doi: 10.1039/C4CP01799D
-
[115]
QIU B, ZHANG M H, WU L J, WANG J, XIA Y G, QIAN D N, LIU H D, HY S, CHEN Y, AN K, ZHU Y M, LIU Z P, MENG Y S. Gas-solid interfacial modification of oxygen activity in layered oxide cathodes for lithium-ion batteries[J]. Nat. Commun., 2016, 7(1): 12108 doi: 10.1038/ncomms12108
-
[116]
MA Q X, CHEN Z J, ZHONG S W, MENG J X, LAI F L, LI Z F, CHENG C, ZHANG L, LIU T F. Na-substitution induced oxygen vacancy achieving high transition metal capacity in commercial Li-rich cathode[J]. Nano Energy, 2021, 81: 105622 doi: 10.1016/j.nanoen.2020.105622
-
[117]
MOHAMAD A H, AKBAR S, MORRIS P. Role of oxygen vacancies in nanostructured metal-oxide gas sensors: A review[J]. Sens. Actuators B‒Chem., 2019, 301: 126845 doi: 10.1016/j.snb.2019.126845
-
[118]
TANG Z K, XUE Y F, TEOBALDI G, LIU L M. The oxygen vacancy in Li-ion battery cathode materials[J]. Nanoscale Horiz., 2020, 5(11): 1453-1466 doi: 10.1039/D0NH00340A
-
[119]
ANTONIO R P, SCHLEXER P, TOSONI S, PACCHIONI G. Increasing oxide reducibility: The role of metal/oxide interfaces in the formation of oxygen vacancies[J]. ACS Catal., 2017, 7(10): 6493-6513 doi: 10.1021/acscatal.7b01913
-
[120]
WANG K, QIU J M, HOU F C, YANG M, NIE K Q, WANG J O, HOU Y C, HUANG W Y, ZHAO W G, ZHANG P X, LIN J H, HU J T, PAN F, ZHANG M J. Unraveling the role of surficial oxygen vacancies in stabilizing Li-rich layered oxides[J]. Adv. Energy Mater., 2023, 13(32): 2301216 doi: 10.1002/aenm.202301216
-
[121]
MAJID F, LAI W H, SAFAEI J, WANG S J, HUANG Z F, MARLTON F, RUAN J F, SUN B, GAO H, OSTRIKOV K, NOTTEN P H L, WANG G X. Boosting the electrochemical performance of lithium-rich cathodes by oxygen vacancy engineering[J]. Batteries Supercaps, 2023, 6(7): e202300123 doi: 10.1002/batt.202300123
-
[122]
WEI A L, CHANG L J, LUO S H, CAO S Y, BI X L, YANG W, LIU J N, ZHANG F S. Preparation of LiNi0.5Mn1.5O4 cathode materials by non-constant temperature calcination and research on its performance[J]. Ionics, 2022, 28(2): 555-565 doi: 10.1007/s11581-021-04348-8
-
[123]
XU G F, WANG L Z, ZHAO J L, SHI B M, ZHANG X, WANG J T, LI N. Elevated stability of nickel-rich oxide cathode material with concentration gradient of transition metals via a novel size-controllable calcination method[J]. J. Alloy. Compd., 2022, 893: 162252 doi: 10.1016/j.jallcom.2021.162252
-
[124]
GAO T P, WONG K W, FUNG K Y, ZHANG W, NG K M. A rational three-step calcination strategy for synthesizing high-quality LiNi0.5Mn0.3Co0.2O2 cathode mater ials: The key role of suppressing Li2O formation[J]. Electrochim. Acta, 2018, 288: 153-164 doi: 10.1016/j.electacta.2018.09.012
-
[125]
ZHANG Y H, ZHANG D, WU L R, MA J, YI Q, WANG Z X, WANG X F, WU Z, ZHANG C, HU N F, HAW S C, CHEN J M, HU Z W, CUI G L. Stabilization of lattice oxygen in Li-rich Mn-based oxides via swing-like non-isothermal sintering[J]. Adv. Energy Mater., 2022, 12(43): 2202341 doi: 10.1002/aenm.202202341
-
[126]
ZHENG Z H, MA Z Y, TAO X L, HUI T, YU H Q, QIAN H, HUANG H H, CHE L D, BEI F L. Precisely designed 3-stage calcination strategy for lithium-rich manganese-based cathodes with improved cycling performance[J]. J. Power Sources, 2024, 623: 235497 doi: 10.1016/j.jpowsour.2024.235497

 下载:
下载:
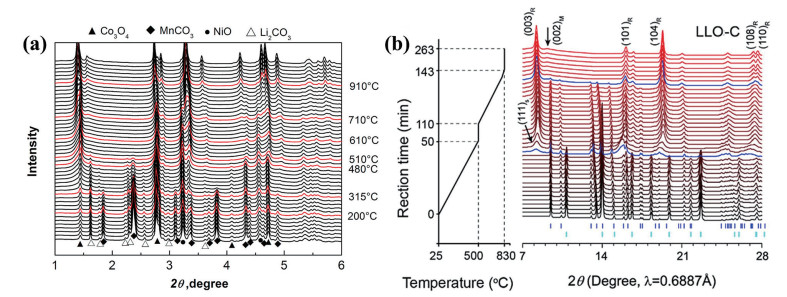

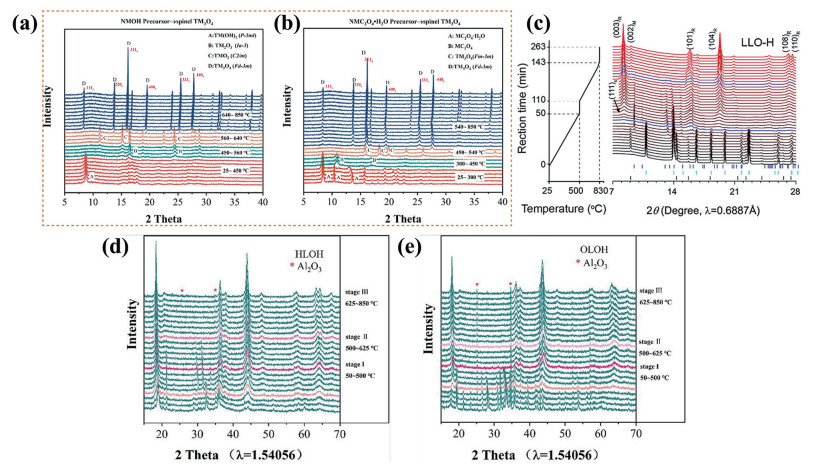
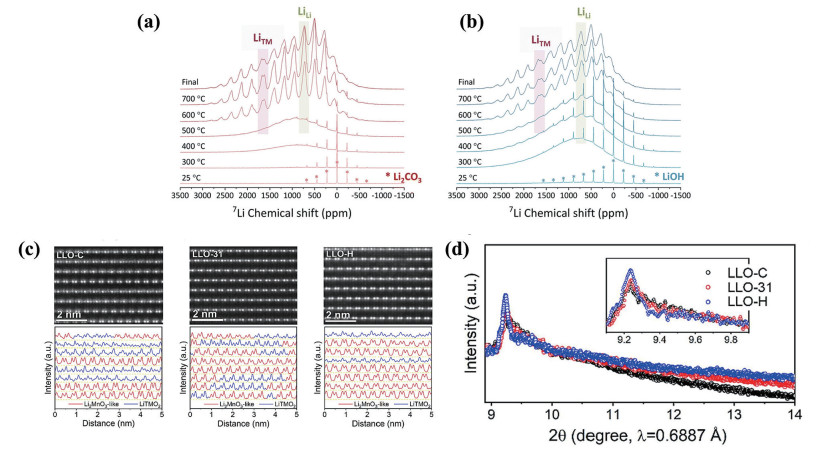
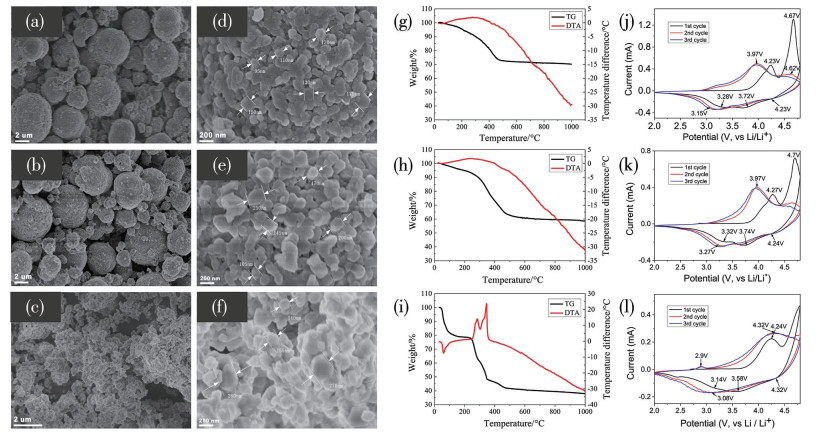

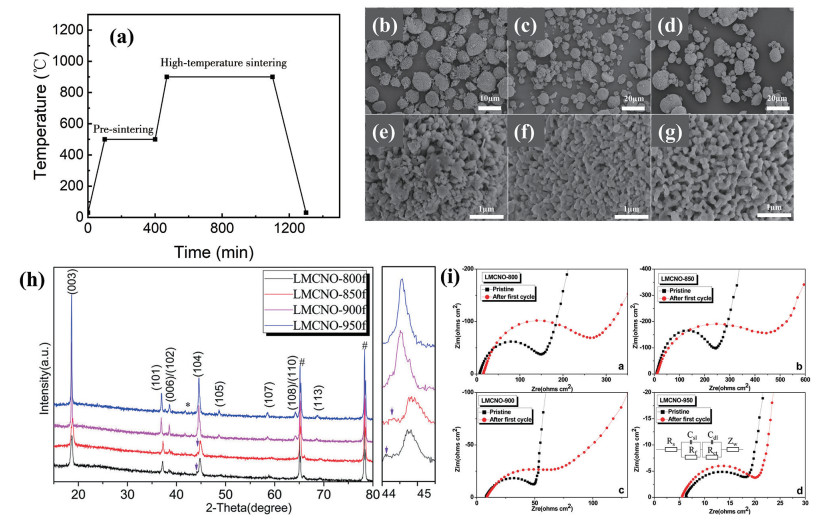
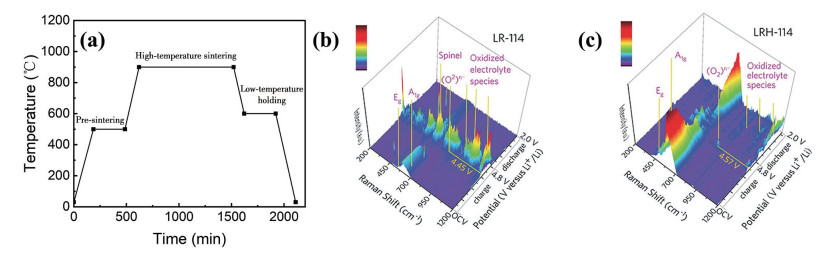
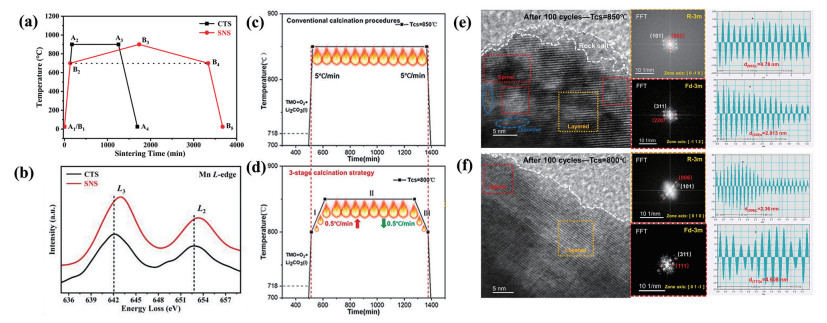

 下载:
下载:














 下载:
下载:
