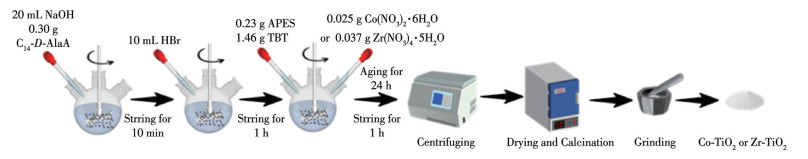
图 1.
不同催化剂的制备流程
Figure 1.
Preparation process of different catalysts

水环境中有机污染物(特别是抗生素类物质)的广泛残留对生态系统稳定性和人类健康构成严峻挑战。四环素(TC)作为典型抗生素污染物,因其化学稳定性高、生物累积性强,可通过污水排放、农田径流等途径持续存在于水环境中,这不仅会诱导耐药基因的传播,还会通过食物链放大效应威胁生态安全和公众健康[1-4]。传统水处理工艺(如生物降解、物理吸附)对这类难降解污染物的深度去除效率有限,难以满足日益严格的水质标准要求[5]。基于过硫酸钠/过氧单硫酸盐(PDS/PMS)活化产生强氧化性活性物种的高级氧化技术,凭借其高反应活性[如羟基自由基(·OH)氧化电位高达1.8~2.7 V]、宽pH适用范围(通常为2.0~10.0)及操作简便等优势,已成为降解顽固性有机污染物的有效手段[6-8]。
过渡金属活化过硫酸盐是当前高级氧化技术领域的研究焦点。尽管Mn基催化剂凭借其多价态特性(Mn3+/Mn4+)展现出优异的PDS活化性能,但Mn离子浸出导致的二次污染及潜在生物毒性严重限制了其实际应用前景[9-11]。相比之下,Ti基催化剂(特别是介孔TiO2)因其优异的环境相容性、高化学稳定性及丰富的表面羟基而备受关注。前期研究表明,介孔TiO2独特的非对称螺旋堆叠结构可产生显著缺陷结构,这能够促进界面电子转移过程,进而在无光照条件下通过氧空位(Ov)活化PDS生成单线态氧(1O2)及自由基[硫酸根自由基(SO4·-)/·OH][5, 9, 12]。然而,锐钛矿型TiO2在无光条件下普遍存在电子-空穴复合率高、对PDS的吸附能力较弱等固有缺陷,导致其活化效率难以满足实际需求[13]。近期研究指出,通过特定金属元素掺杂可有效调控TiO2的电子结构(如Ti3+/Ti4+氧化还原电势),优化其PDS活化路径,但不同掺杂元素对活性物种(自由基与非自由基)选择性生成的差异化调控机制尚未明晰,这严重制约了高性能催化剂的设计[9, 12]。
针对介孔TiO2催化剂在无光条件下活化PDS效率不足的关键问题,我们采用Co和Zr两种金属元素对其进行掺杂,制备介孔TiO2复合催化剂(Co-TiO2、Zr-TiO2)。系统分析了掺杂对催化剂微观形貌、比表面积、孔结构及Ov等的调控规律;以TC为目标污染物,定量评价了改性催化剂活化PDS的降解效能及反应动力学特征;通过猝灭实验和电子顺磁共振(EPR)原位检测,分析了Co/Zr掺杂体系主要活性物种(·OH和1O2)的差异及其反应路径,阐明了掺杂元素通过电负性差异(Co)或离子半径差异(Zr)调控反应机制的本质;并深入探讨了磷酸盐(PO43-)添加剂对优化氧化剂利用效率(ηRU)的作用机制。本研究可为设计高效、稳定的过硫酸盐活化催化剂提供理论依据,助力水环境中难降解污染物的深度去除。
钛酸四丁酯(TBT,分析纯)、N-十四烷基-D-丙氨酸(C14-D-AlaA,分析纯)、氢溴酸(HBr,分析纯)、三甲基[3-(三甲氧基硅烷基)丙基]氯化铵(APES,分析纯)、PDS(分析纯)、甲醇(MeOH,分析纯)、叔丁醇(TBA,分析纯)、L-组氨酸(L-his,分析纯)、重铬酸钾(K2Cr2O7,分析纯)、苯甲基亚砜(PMSO,分析纯)均购自上海麦克林生化科技有限公司。六水合硝酸钴(分析纯)、无水乙醇(分析纯)、浓硝酸(分析纯)均购自国药集团化学试剂有限公司。氢氧化钠(NaOH)购自上海凌峰化学试剂有限公司。五水合硝酸锆(分析纯)购自阿拉丁试剂有限公司。四环素(标准品)购自Sigma-Aldrich。
采用改进的水热法制备Co-TiO2和Zr-TiO2催化剂[14],如图 1所示。将0.30 g C14-D-AlaA加入三颈烧瓶中,再加入20 mL 0.05 mol·L-1的NaOH溶液。将烧瓶置于18 ℃的水浴锅中搅拌10 min至完全溶解,随后加入10 mL 0.1 mol·L-1的HBr溶液,继续搅拌反应1 h。在反应结束后的溶液中加入0.23 g APES和1.46 g TBT。制备纯TiO2时,不添加任何金属盐;制备掺杂催化剂时,在加入APES和TBT的同时加入0.025 g Co(NO3)2·6H2O或0.037 g Zr(NO3)4·5H2O,继续搅拌1 h。反应结束后,将混合溶液降温至5 ℃,然后陈化24 h。陈化后的固体产物经离心分离后进行真空干燥。将干燥后的固体在600 ℃高温下煅烧3 h并研磨,最终得到催化剂Co-TiO2和Zr-TiO2。
采用型号为FEI Quanta FEG 650扫描电子显微镜(SEM,加速电压为10 kV)观察催化剂的形貌。使用FEI Tecnai G2 F20型透射电子显微镜(TEM,加速电压为200 kV)分析催化剂的微观结构。通过Micromeritics ASAP 2460型比表面积和孔径分析仪测定催化剂的比表面积和孔结构。采用Bruker D8 Advance X射线衍射仪(XRD,Cu Kα辐射源,λ=0.154 06 nm,工作电压为40 kV,电流为40 mA,扫描范围为10°~90°)分析催化剂的晶体结构。使用Thermo Scientific K-Alpha X射线光电子能谱仪(XPS,Al Kα辐射源,hν=1 486.6 eV)表征表面元素的化学状态。通过Bruker EMXplus-10/12电子顺磁共振波谱仪(微波频率为9.85 GHz,调制频率为100 kHz)检测Ov和活性物种的含量。
在避光条件下进行PDS活化降解TC实验[14]:取200 mL 20 mg·L-1的TC溶液于250 mL锥形瓶中,加入0.2 g催化剂,置于恒温磁力搅拌器(25 ℃)上避光预吸附60 min,以达到吸附-解吸平衡。随后加入0.571 4 g过二硫酸钠(Na2S2O8,10 mmol·L-1,即PDS)启动反应。反应过程中,于预定时间点(0、15、30、60、120、180、240、300、360 min)分别取样3 mL,立即加入过量无水乙醇猝灭反应,并高速离心分离(10 000 r·min-1,10 min)催化剂。取上清液,采用紫外可见分光光度计在波长352 nm处测定吸光度,根据预先建立的标准曲线计算TC的残余质量浓度。
采用下式确定TC降解过程中催化剂/PDS体系的ηRU[15],该指标定义为污染物(TC)的降解量(nTC,mol)与氧化剂(PDS)的消耗量(nPDS,mol)之间的化学计量比值,用以衡量氧化剂在降解污染物过程中的利用效率。
|
|
(1) |
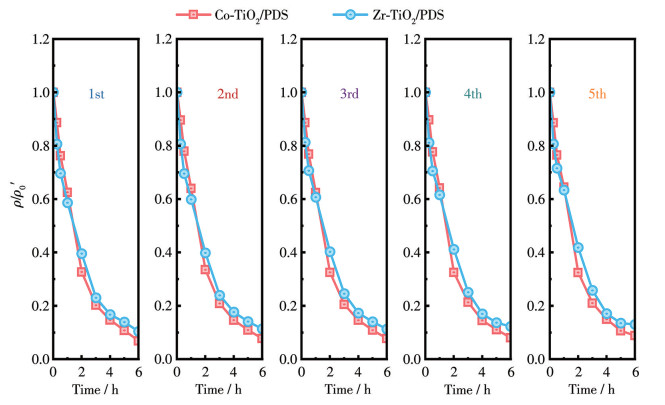
为评估催化剂的稳定性与可重复使用性,对Co-TiO2和Zr-TiO2进行了5次循环实验。每次PDS活化降解TC反应结束后,将催化剂离心分离,用去离子水反复洗涤至中性,于60 ℃烘干12 h,再投入下一轮降解实验,各轮反应条件保持一致。通过比较每次循环后TC的降解率,评价催化剂的重复使用性能。
为捕获不同活性物种,分别采用50 mmol·L-1的5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)溶液和20 mmol·L-1的2,2,6,6-四甲基哌啶-N-氧自由基(TEMP)溶液作为自旋捕获剂。在设定的反应时间点,从反应体系中取样并与相应自旋捕获剂迅速混合,随后将混合液转移至石英毛细管中,立即进行EPR测试。采用UV-2600(岛津)型紫外可见分光光度计测定TC的质量浓度。
为定量评估Co-TiO2/PDS与Zr-TiO2/PDS体系中主要活性物种(·OH与1O2)的稳态浓度及其表观反应动力学,进一步进行了探针实验。选用对活性物种具有特异性响应的探针化合物:硝基苯(NB)仅对·OH敏感,糠醇(FFA)对·OH、SO4·-和1O2均敏感。在催化降解体系中,于反应启动前分别加入NB或FFA,其初始质量浓度设定为0.5 mg·L-1(远低于TC的质量浓度,以避免对体系氧化机制产生显著干扰)。在反应过程中于预定时间点取样3 mL,立即加入过量无水乙醇猝灭反应,经高速离心后取上清液,采用高效液相色谱测定NB或FFA的残留质量浓度。色谱条件:C18反相色谱柱(4.6 mm×250 mm,5 μm),流动相为甲醇/水(体积比为60∶40),流速为1.0 mL·min-1,柱温为30 ℃,NB的检测波长为265 nm,FFA的检测波长为219 nm。通过拟合NB和FFA的降解曲线,得到其表观反应速率常数(kobs)。结合猝灭实验已明确的各体系主导活性物种,并依据相应的二级反应速率常数进行估算,得到·OH和1O2的表观反应速率常数及其相对稳态浓度。
为评估催化剂的金属离子溶出风险,采用电感耦合等离子体发射光谱仪(ICP-OES)对2种粉末催化剂活化PDS降解污染物后溶液中的Co、Zr质量浓度进行测定。
为进一步探究催化剂表面羟基对反应效率的影响,进行了磷酸盐调控实验。在PDS活化降解TC的体系中,反应启动后立即加入0.5 mmol磷酸二氢钠(NaH2PO4),以选择性地抑制催化剂的表面羟基。反应过程中保持体系pH为6.7~7.0(使用0.1 mol·L-1 HCl或NaOH溶液微调)。
为明确Co-TiO2/PDS与Zr-TiO2/PDS体系中起主导作用的活性物种,我们开展了猝灭实验,即通过投加特定猝灭剂以选择性消除目标活性物种。其中,MeOH用于猝灭·OH和SO4·-(浓度为PDS浓度的1 000倍),TBA用于猝灭·OH(浓度为PDS浓度的500倍),L-his用于猝灭1O2(浓度为PDS浓度的10倍);同时加入重铬酸钾与PMSO,两者浓度均为10 mmol·L-1,分别用于猝灭自由电子和高价金属物种。实验中PDS初始浓度为12 mmol·L-1,因所需猝灭剂浓度较高,均以适量体积直接加入体系。所有猝灭实验均在启动PDS反应前加入相应猝灭剂,其余条件均与1.4节降解实验保持一致。
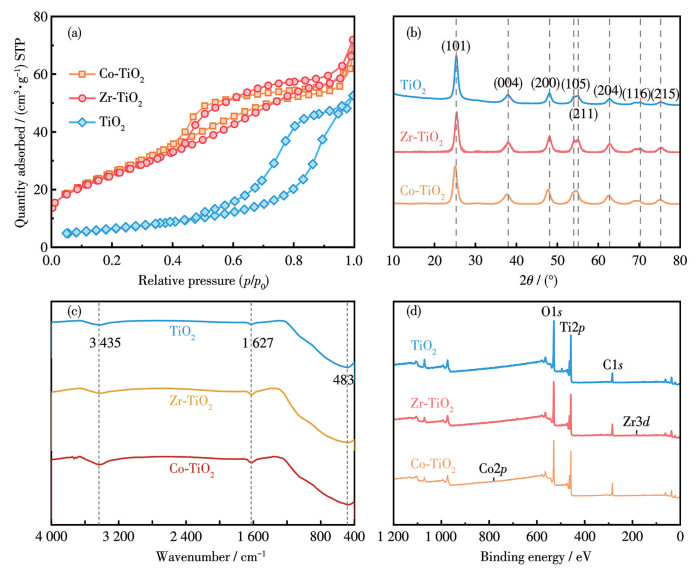
通过SEM对不同催化剂的微观形貌与结构进行表征(图 2)。SEM结果显示,掺杂后的催化剂呈现为大尺寸不规则的颗粒形貌,Co、Zr掺杂未显著改变TiO2基体的结构,TiO2颗粒表面未观察到明显的离散颗粒聚集,仅分布有极少的小颗粒。这表明掺杂的Co和Zr元素可能以氧化物纳米粒子的形式均匀分布在TiO2基体中[16]。

图 3a展示了3种催化剂的N2吸附-脱附等温线,三者均为具有H2型回滞环的Ⅳ型等温线。H2型回滞环通常与颗粒堆积孔或具有墨水瓶形孔的复杂孔道结构有关,表明催化剂具有明显的介孔结构。这种规整的孔道有利于反应物的传质与扩散,为催化反应提供了丰富的活性位点。表 1数据显示,Co-TiO2和Zr-TiO2的比表面积(SBET)分别为93和92 m2·g-1,显著高于未掺杂的TiO2(77 m2·g-1)。比表面积的增加为催化剂提供了更多反应活性位点,有利于污染物的吸附和传质过程[17-18]。
 下载:
导出CSV
下载:
导出CSV
| Sample | SBET / (m2·g-1) | Pore volume / (cm3·g-1) | Average pore diameter / nm |
| TiO2 | 77 | 0.243 5 | 6.4 |
| Co-TiO2 | 93 | 0.103 5 | 4.5 |
| Zr-TiO2 | 92 | 0.111 2 | 4.8 |
XRD图(图 3b)显示,Co-TiO2和Zr-TiO2中均未出现钴/锆氧化物的特征衍射峰。这可能归因于2种元素的掺杂量较低,且氧化物颗粒高度分散;也可能是掺杂元素进入了TiO2晶格,形成了取代掺杂。值得注意的是,Co-TiO2和Zr-TiO2的(101)晶面衍射峰位置相较于纯TiO2发生了偏移。由于Co3+(0.064 nm)和Zr4+(0.74 0 nm)的离子半径与Ti4+(0.068 nm)相近,这为两者进入TiO2晶格提供了可能,并且Zr4+离子半径大于Ti4+,取代掺杂导致晶格畸变和缺陷形成[19-22]。
Co-TiO2和Zr-TiO2的FTIR谱图如图 3c所示。3 435和1 627 cm-1处的吸收峰分别归属于样品表面吸附水分子的O—H伸缩振动和H—O—H弯曲振动。结合XRD结果可知,400~1 000 cm-1范围内的宽峰对应于锐钛矿型TiO2,其中Ti—O键的振动峰位于400~600 cm-1。纯TiO2的Ti—O峰位于483 cm-1,而Co-TiO2和Zr-TiO2中该峰分别位移至474和470 cm-1,表明Co和Zr取代了TiO2晶格中的部分Ti位点,证实了Co—O和Zr—O键的形成。此外,Ti—O峰向低波数移动,表明催化剂中Ov缺陷的增加,这可能是由于Co/Zr取代Ti后键能降低,晶格振动频率减小所致[23]。图 3d的XPS全谱图表明Co和Zr成功掺杂到TiO2中,这与XRD结果一致。
Ti2p、O1s、Co2p和Zr3d的高分辨XPS谱图如图 4所示。图 4a表明,Co-TiO2中Ti2p1/2和Ti2p3/2结合能低于纯TiO₂,而Zr-TiO2中Ti2p结合能相对于纯TiO2的偏移量小于0.2 eV,说明Co掺杂对TiO2电子结构的影响更为显著。此外,Co-TiO2和Zr-TiO2的Ti4+、Ti3+含量比(分别为1.059和1.013)略低于纯TiO2(1.27),这是由于掺杂离子对Ti4+发生了等价取代,并形成了Ti—O—Co和Ti—O—Zr键。Co与Ti之间显著的电负性差异导致TiO2晶格中的电子向Co迁移,造成局部电荷不平衡并产生Ov,这直接解释了Co-TiO2中Ti2p结合能降低的微观机制。而Zr与Ti之间的电负性差异较小,这使得Zr-TiO2中的电子转移效应较弱,其缺陷主要通过离子半径差异引起的晶格畸变形成[19-22]。
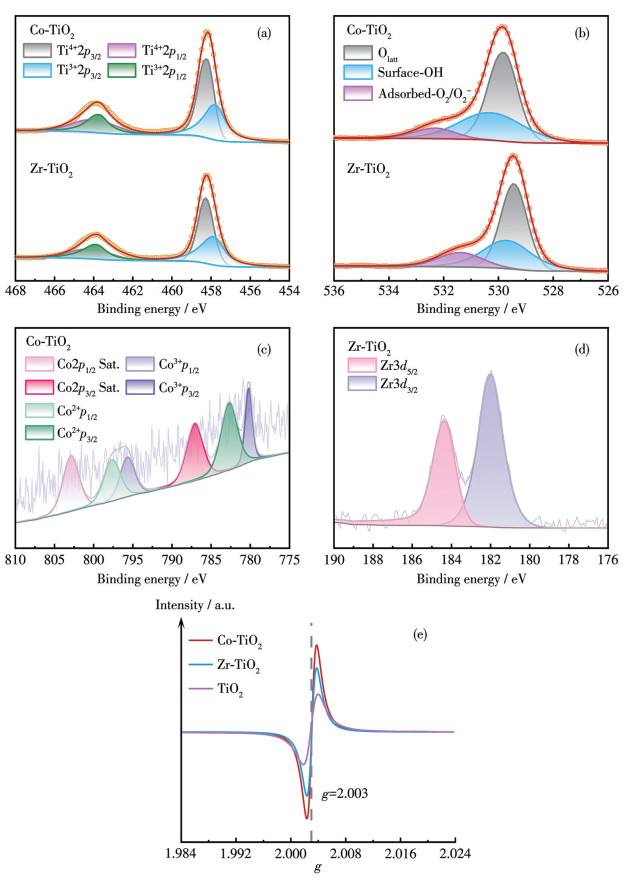
Co-TiO2和Zr-TiO2的O1s谱图如图 4b所示。根据先前研究[22],这些峰可分别拟合为晶格氧(Olatt)、表面羟基氧和吸附氧物种(O2、O2-)。可以发现,Co和Zr的掺杂使表面羟基氧的相对原子含量从纯TiO2的23.11%分别提升至35.10%(Co-TiO2)和31.82%(Zr-TiO2)。这种变化可归因于Co和Zr掺杂对TiO2晶格的影响。为直接定量表征催化剂的Ov浓度,进行了EPR测试(图 4e)。EPR谱图显示,所有样品在g=2.003处均出现明显的Ov顺磁信号。对比信号强度发现,2种金属掺杂后催化剂的Ov浓度均高于未掺杂TiO2,而且Co-TiO2的Ov浓度高于Zr-TiO2。这直接证实了金属掺杂有效提升了TiO2的Ov浓度,且Co掺杂诱导产生Ov的能力强于Zr掺杂。Ov在PDS活化过程中能够起到关键作用,其能够在PDS活化中作为活性位点,通过夺取PDS的电子进而发挥路易斯酸位点的作用,从而促进PDS的吸附和活化,加速反应中活性氧物种(ROS)的生成[24-25]。此外,Ov的存在能够加速超氧自由基(·O2-)的歧化反应,从而生成氧化能力更强、氧化剂利用率更高的1O2。
Co-TiO2的Co2p高分辨谱图和Zr-TiO2的Zr3d高分辨谱图如图 4c、4d所示。Co2p高分辨谱图中位于797.36和782.58 eV处的2个特征峰及位于787.18和802.98 eV处的2个卫星峰证明了Co2+的存在,795.78和780.08 eV处的2个特征峰则证明了Co3+的存在[26]。通过计算可知Co2+、Co3+的含量比为3.25,这不仅促进了催化剂中Ov的形成,也能够通过氧化还原反应促进PDS的活化[27]。Zr3d谱图中位于184.38和181.98 eV处的2个特征峰分别归于Zr3d5/2和Zr3d3/2[21],这表明Zr-TiO2中的Zr主要以Zr4+的形式存在。
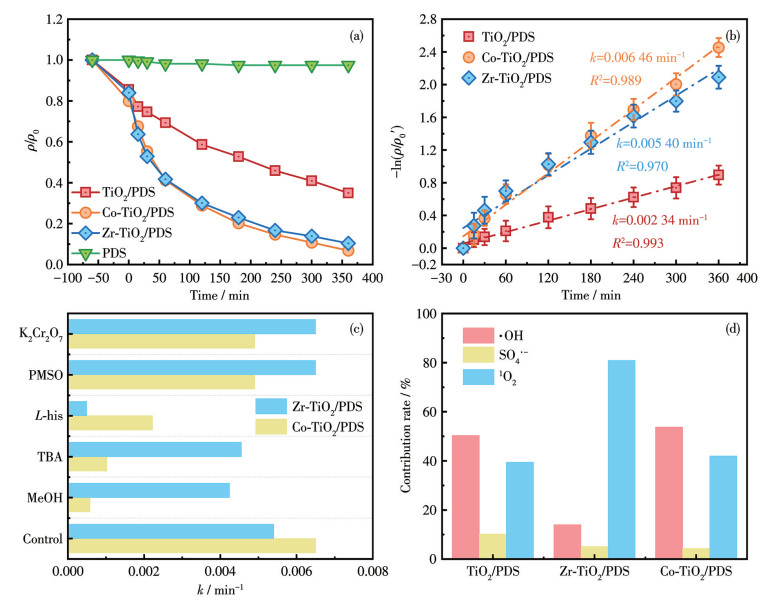
以TC为目标污染物,对比了Co-TiO2/PDS和Zr-TiO2/PDS体系的催化氧化性能,结果如图 5所示。由图可知,没有催化剂时,纯PDS体系几乎不具备降解性能。与TiO2/PDS体系(65.0%)相比,6 h内Co-TiO2/PDS和Zr-TiO2/PDS体系的降解率分别提高至93.1%和89.6%(图 5a)。通过采用准一级动力学模型对降解曲线进行拟合,获得了各体系的反应速率常数(k,图 5b)。所有动力学拟合均基于3次平行实验的数据,图中误差条代表标准偏差。所有样品的拟合优度(R2)均大于0.97,表明准一级动力学模型能很好地描述TC的降解过程。Co-TiO2/PDS和Zr-TiO2/PDS体系的反应速率常数分别为0.006 46和0.005 40 min-1,分别是TiO2/PDS体系(0.002 34 min-1)的2.8倍和2.4倍。由此可见,Co、Zr的掺杂显著增强了TiO2对PDS的活化能力,这归因于以下原因:Co-TiO2和Zr-TiO2具有较大的比表面积和较小的孔径,这有利于污染物的吸附分解和活性物种的生成;此外,Co、Zr的掺杂明显提高了催化剂的Ov含量,大量Ov的形成促进了电子转移,从而显著提升了催化反应活性。
为评估催化剂对TC的矿化性能,测定了降解实验前后反应体系的总有机碳(TOC)含量。为消除吸附影响,TOC测定前对反应后悬浮液超声处理0.5 h,使吸附的有机物充分解吸,从而消除吸附作用对TOC测量结果的影响。结果发现,Co-TiO2/PDS和Zr-TiO2/PDS体系降解TC的TOC去除率分别为89.5%和81.3%,与分光光度法得到的去除率接近,说明2种催化剂都能彻底矿化TC。
根据猝灭实验结果(图 5c)可知,K2Cr2O7与PMSO对降解过程的影响作用较弱,表明在Co-TiO2/PDS与Zr-TiO2/PDS两种体系中,降解过程并非主要由直接电子转移或高价金属物种所主导。在Co-TiO2/PDS体系中,MeOH的加入使TC的准一级反应速率常数从0.006 46 min-1下降至0.000 58 min-1;而TBA与L-his的影响作用相对较弱,对应反应速率常数为0.001 02和0.002 22 min-1。这说明在该体系中,·OH是主要的活性物种,同时有少量SO4·-的参与。
在Zr-TiO2/PDS体系中,L-his对降解的影响作用最为显著,使TC反应速率常数由0.005 40 min-1降至0.00 049 min-1;而MeOH与TBA的影响效果较弱,对应反应速率常数分别为0.004 41和0.004 58 min-1,表明该体系以1O2为主导活性物种。进一步通过贡献率分析(图 5d)可得,在Co-TiO2/PDS体系中,·OH、SO4·-和1O2对TC降解的贡献率分别为53.7%、4.3%和42.0%;而在Zr-TiO2/PDS体系中,1O2的贡献率高达80.9%,·OH与SO4·-的贡献率合计仅为19.1%。
为定量验证Co-TiO2/PDS和Zr-TiO2/PDS体系中主要活性物种的差异,采用探针实验测定了·OH和1O2在不同催化体系中的表观反应速率常数(kobs),并据此估算其稳态浓度(分别为c·OH, ss和
为评估Co、Zr掺杂TiO2催化剂在废水处理过程中的环境安全性,考察了降解反应后金属离子的溶出行为。采用ICP-OES检测了2种掺杂催化剂活化PDS降解TC后金属离子的溶出浓度。结果表明,2种催化剂在降解TC后,金属离子溶出量极低。Co离子溶出浓度为4.2 μg·L-1,远低于我国《生活饮用水卫生标准》(GB 5749—2022)中规定的Co限值(1.0 mg·L-1);Zr离子溶出质量浓度为3.1 μg·L-1,处于极低水平,且该标准未对Zr浓度作出规定,进一步表明其环境风险可忽略。与其他文献报道的Ti基催化剂溶出水平相比,我们制备的催化剂展现出优异的环境安全性。为评估催化剂的稳定性,对Co-TiO2和Zr-TiO2进行了5次循环使用实验。如图 6所示,在5次循环降解实验中,Co-TiO2/PDS体系对TC的降解率由首次的93.1%下降至第5次的91.2%,Zr-TiO2/PDS体系的降解率则由首次的89.6%下降至第5次的87.0%。表明2种催化剂均具有良好的可重复使用性。
利用猝灭实验和EPR检测进一步探究了掺杂引起的活性物种变化。在EPR测试中,通过对比标准谱图对特征信号进行归属:以DMPO为捕获剂时,强度比为1∶2∶2∶1的四重峰为DMPO-·OH加合物的特征信号;以TEMP为捕获剂时,强度比为1∶1∶1的三重峰为TEMP-1O2加合物的特征信号(图 7)。在Co-TiO2/PDS体系的EPR谱图中,DMPO-·OH四重峰信号相较于TiO2/PDS体系明显增强,表明Co掺杂促进了·OH的生成。而在Zr-TiO2/PDS体系中检测到明显的TEMP-1O2的三重峰信号,表明体系中存在1O2生成。在Zr-TiO2/PDS体系中同样可检测到DMPO-·OH的信号峰,说明体系中仍存在·OH的生成,但其信号峰强度要明显弱于Co-TiO2/PDS体系中DMPO-·OH的信号峰。TiO2/PDS、Co-TiO2/PDS和Zr-TiO2/PDS体系的TC降解主要依赖于自由基和1O2的协同作用。Co-TiO2/PDS和Zr-TiO2/PDS体系中的优势活性物种不一致,Co-TiO2/PDS和TiO2/PDS体系均以·OH为主要活性物种,1O2为次要活性物种,而Zr-TiO2则与二者相反。EPR测试结果进一步证实,与TiO2/PDS体系相比,Co-TiO2/PDS和Zr-TiO2/PDS体系中活性物种的种类并未发生根本变化,主要差异体现在各物种的浓度上。Co-TiO2/PDS体系中产生了更多的·OH和SO4·-,这归因于Co对PDS的高效活化能力,即使在相对较低的含量下也能显著增强自由基的生成。然而Zr作为氧化还原惰性金属,并不能显著增强PDS的活化性能,因此Zr-TiO2/PDS体系与TiO2/PDS体系在自由基生成能力上差别不大。EPR结果表明,在3个体系中均可检测到TEMP-1O2特征信号,但其信号强度由大到小的顺序为TiO2/PDS、Co-TiO2/PDS、Zr-TiO2/PDS。这说明1O2在各体系中均有生成,但瞬时生成量并不完全取决于Ov浓度。结合EPR结果可知,金属掺杂显著提高了催化剂中的Ov浓度,且Co-TiO2的Ov浓度高于Zr-TiO2,二者均高于未掺杂TiO2,表明Ov有利于活性氧物种的活化过程。然而,1O2是否成为主导活性物种还受到掺杂金属电子结构的调控。Zr4+为氧化还原惰性金属,更倾向于通过Ov位点介导的非自由基路径活化PDS,从而使1O2在污染物降解中占主导地位;而Co则易发生Co3+/Co2+氧化还原循环,更有利于生成SO4·-和·OH等自由基物种,导致Co-TiO2/PDS体系以自由基路径为主。因此,尽管Zr-TiO2体系中1O2的EPR信号强度并非最高,其在反应中的相对贡献率却最大,体现了不同掺杂金属对活性物种反应路径的选择性调控作用。
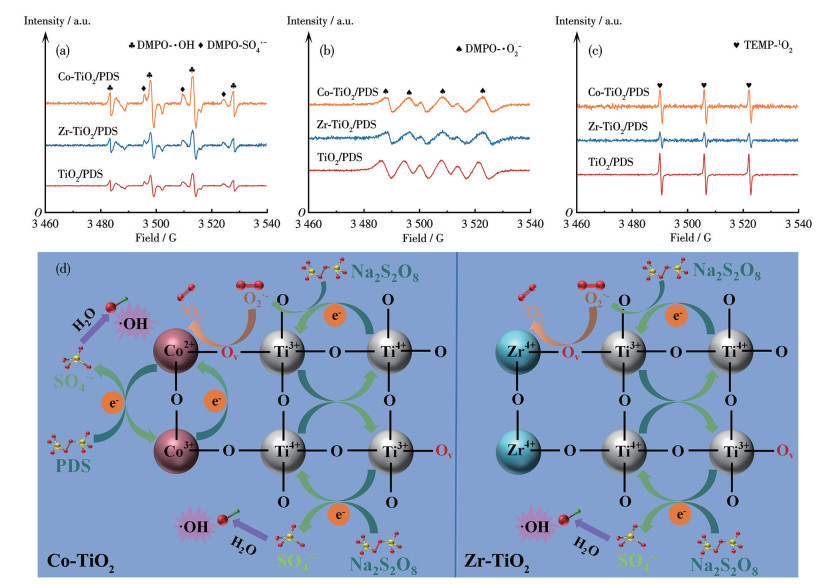
(a) DMPO-·OH and DMPO-SO4·-; (b) DMPO-·O2-; (c) TEMP-1O2.
基于以上分析,Co-TiO2/PDS和Zr-TiO2/PDS体系表现出差异化的活性物种生成路径(图 7d)。在Co-TiO2/PDS体系中,·OH的生成主要依赖于Co3+/Co2+氧化还原反应的循环(图 4c)。催化剂表面的Co2+直接将电子转移至吸附的PDS,引发其O—O键断裂,产生SO4·-,后者可进一步转化为·OH。该路径与猝灭实验和EPR结果相符。在Zr-TiO2/PDS体系中,1O2的生成则主要通过非自由基路径。Zr4+掺杂诱导产生的丰富Ov作为路易斯酸位点,优先吸附并活化PDS。吸附的PDS经由双电子转移过程,形成不稳定的过氧中间体,最终分解为1O2。该路径与1O2特异性猝灭实验和EPR结果相符。
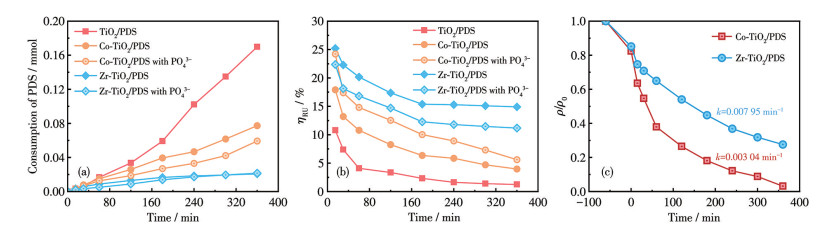
图 8展示了PDS活化行为与体系ηRU的关联性。可以发现,与TiO2/PDS体系相比,Co-TiO2/PDS和Zr-TiO2/PDS在实现更高TC降解速率的同时表现出更低的PDS消耗量,致使Co-TiO2/PDS和Zr-TiO2/PDS体系的ηRU显著高于TiO2/PDS体系(图 8b)。值得注意的是,Co-TiO2的Ov浓度高于Zr-TiO2,但其ηRU反而低于后者。这是因为Co-TiO2/PDS体系中·OH(半衰期约为10-9 s)和1O2(半衰期约为10-6 s)浓度显著更高,但因二者传质距离极短,高浓度下易发生自猝灭效应,导致活性物种在攻击TC前失活,进而降低ηRU[28]。为验证该结论,实验中添加0.5 mmol磷酸盐(磷酸二氢钠)以选择性地抑制催化剂的表面羟基[29](溶液pH维持在6.7~7.0)。结果显示,在Co-TiO2/PDS体系中,TC降解率由89.6%降至72.4%,反应速率常数由0.005 40 min-1降至0.003 04 min-1;而Zr-TiO2/PDS体系呈现反向效应:TC降解率提升至96.9%,反应速率常数增至0.007 95 min-1(图 8c)。尽管磷酸盐的添加均降低了两体系的PDS消耗量(图 8a),但ηRU变化趋势截然相反:Co-TiO2/PDS体系的ηRU显著提升,Zr-TiO2/PDS体系则受到抑制(图 8b)。这表明磷酸盐通过调控表面羟基,有效缓解了Co-TiO2/PDS体系的自猝灭效应,从而优化了反应剂利用效率。
Co-TiO2/PDS与Zr-TiO2/PDS体系的主要活性物种不同,导致二者对TC分子的初始攻击位点和路径在理论上存在差异[14, 30]。1O2作为亲电试剂更倾向于攻击TC分子中富含电子的N原子[亲电Fukui指数(f -)最高],而·OH作为非选择性自由基可攻击包括N原子在内的多个位点。然而,由于2个催化体系中均存在多种自由基(·OH、SO4·-、·O2-)与非自由基(1O2)的协同作用,且TC分子中的N原子均是各类活性物种最易攻击的位点[自由基Fukui指数(f 0)和f -均最高],这使得在实际降解过程中,不同体系产生的中间产物谱图较为复杂,难以清晰区分单一物种主导下的路径差异。更重要的是,TOC分析结果表明,2种体系均能实现对TC的高效矿化(6 h矿化率均大于75%),最终产物均为CO2和H2O等无机小分子。考虑到本文的研究重点在于阐明掺杂调控的活性物种的生成机制,且关于·OH与1O2对TC降解路径差异的详细讨论在我们前期的相关工作中[30]已有系统阐述,因此未重复展示详细的中间产物鉴定数据,而是聚焦于主导活性物种的鉴别与反应机理的解析。
通过Co和Zr掺杂改性介孔TiO2催化剂,系统探究了掺杂对其活化过硫酸盐的影响机制。结果表明,Co、Zr掺杂显著提升了催化剂的比表面积和氧空位浓度,其中Zr掺杂主要通过晶格畸变实现,而Co掺杂则促进了电子迁移。在TC降解实验中,Co-TiO2/PDS和Zr-TiO2/PDS体系分别达到93.1%和89.6%的降解率,其反应速率常数分别是未掺杂TiO2/PDS体系的2.8倍和2.4倍。活性物种分析表明,Co-TiO2/PDS体系以·OH为主要降解路径,而Zr-TiO2/PDS体系则以1O2为主要活性物种,揭示了不同掺杂元素对反应路径的显著调控差异。本研究阐明了过渡金属掺杂对钛基介孔催化剂活化PDS性能的调控机制,特别是明确了Co与Zr掺杂在提升活性、调控反应路径及微观作用机制方面的差异,为设计高效、环境友好的过硫酸盐活化催化剂提供了重要的理论依据,有助于推动基于过硫酸盐的高级氧化技术在水处理领域的实际应用。
PÉREZ-MOYA M, GRAELLS M, CASTELLS G, AMIGO J, ORTEGA E, BUHIGAS G, PEREZ L M, MANSILLA H D. Characterization of the degradation performance of the sulfamethazine antibiotic by photo-Fenton process[J]. Water Res., 2010, 44(9): 2533-2540
LI R, DONG H R, TIAN R, CHEN J, XIE Q Q. Activation of sulfite by different Fe-O-based nanomaterials for oxidative removal of sulfamethazine in aqueous solution[J]. Sep. Purif. Technol., 2020, 250: 117230 doi: 10.1016/j.seppur.2020.117230
TURCIOS A E, PAPENBROCK J. Biofiltration of the antibacterial drug sulfamethazine by the species chenopodium quinoa and its further biodegradation through anaerobic digestion[J]. J. Environ. Sci., 2019, 75: 54-63 doi: 10.1016/j.jes.2018.02.022
张欢, 王记江, 范广, 唐龙, 岳二林, 白超, 王潇, 张玉琦. 一种用于检测四环素和对硝基苯酚的高稳定性镉(Ⅱ)金属有机骨架[J]. 无机化学学报, 2024, 40(3): 646-654.ZHANG H, WANG J J, FAN G, TANG L, YUE E L, BAI C, WANG X, ZHANG Y Q. A highly stable cadmium(Ⅱ) metal-organic framework for detecting tetracycline and p-nitrophenol[J]. Chinese J. Inorg. Chem., 2024, 40(3): 646-654
WANG J L, WANG S Z. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants[J]. Chem. Eng. J., 2018, 334: 1502-1517 doi: 10.1016/j.cej.2017.11.059
XU W Q, NI C C, DENG N, HUANG X. Underestimated role of hydroxyl radicals for bromate formation in persulfate-based advanced oxidation processes[J]. Environ. Res., 2024, 252: 118870 doi: 10.1016/j.envres.2024.118870
XIANG S X, DONG H R, LI Y J, XIAO J Y, DONG Q X, HOU X Z, CHU D D. A comparative study of activation of peroxymonosulfate and peroxydisulfate by greigite (Fe3S4) for the degradation of sulfamethazine in water[J]. Sep. Purif. Technol., 2022, 290: 120873 doi: 10.1016/j.seppur.2022.120873
朱羽科, 蒋孝佳, 陈嘉奕, 付小航, 吴礼光, 王挺. 过硫酸盐激活协同可见光催化降解四环素的机理和降解路径[J]. 无机化学学报, 2023, 39(10): 1857-1868. doi: 10.11862/CJIC.2023.164ZHU Y K, JIANG X J, CHEN J Y, FU X H, WU L G, WANG T. Mechanism and degradation pathway of tetracycline degradation by persulfate activation coupled with visible light photocatalysis[J]. Chinese J. Inorg. Chem., 2023, 39(10): 1857-1868 doi: 10.11862/CJIC.2023.164
WANG H T, LONG Z Y, CHEN R Y, ZHANG H Y, SHI H F, CHEN Y G. Boosting PMS activation over BiVO4 piezo-photocatalyst to rapidly degrade tetracycline: Intermediates and mechanism[J]. Sep. Purif. Technol., 2024, 331: 125598 doi: 10.1016/j.seppur.2023.125598
CHEN P, CHENG Z L, ZHANG X Z, YAN C Q, WEI J, QIU F C, LIU Q Y. Fe-Mn bimetallic catalyst to activate peroxymonosulfate (PMS) for efficient degradation of tetracycline: Mechanism insights and application for pharmaceutical wastewater[J]. J. Clean. Prod., 2024, 445: 141365 doi: 10.1016/j.jclepro.2024.141365
王洋, 郑晓芹, 刘洋, 张凯, 寇佳慧, 孙林兵. 基于限阈空间制备Mn单原子催化剂及其电催化析氧性能[J]. 无机化学学报, 2024, 40(11): 2175-2185. doi: 10.11862/CJIC.20240165WANG Y, ZHENG X Q, LIU Y, ZHANG K, KOU J H, SUN L B. Preparation of Mn single-atom catalyst based on confined space and its electrocatalytic oxygen evolution performance[J]. Chinese J. Inorg. Chem., 2024, 40(11): 2175-2185 doi: 10.11862/CJIC.20240165
HU P D, LONG M C. Cobalt-catalyzed sulfate radical-based advanced oxidation: A review on heterogeneous catalysts and applications[J]. Appl. Catal. B‒Environ., 2016, 181: 103-117 doi: 10.1016/j.apcatb.2015.07.024
YAN Z Y, ZHAO X Y, ZHONG C K, GAO W Y, FENG Y J, LIU J. Decomposition of tetracycline with peroxymonosulfate activated by an ordered hierarchical pores Fe-N/C catalyst rich in FeNx sites: Insights into singlet oxygen mechanism[J]. Sep. Purif. Technol., 2025, 354: 129489 doi: 10.1016/j.seppur.2024.129489
陈京乐, 黄睿, 吴礼光, 傅智慧, 朱宏远, 王挺, 陈华丽, 李春娟. Ti基和Mn基介孔催化剂活化过硫酸钠降解有机污染物的反应机制[J]. 环境科学学报, 2024, 44(4): 22-35.CHEN J L, HUANG R, WU L G, FU Z H, ZHU H Y, WANG T, CHEN H L, LI C J. Reaction mechanism of Ti-based and Mn-based mesoporous catalysts for activating sodium persulfate to degrade organic pollutants[J]. Acta Scientiae Circumstantiae, 2024, 44(4): 22-35
YANG X Y, CAI J S, WANG X N, LI Y F, WU Z X, WU W D, CHEN X D, SUN J Y, SUN S P, WANG Z H. A bimetallic Fe-Mn oxide-activated oxone for in situ chemical oxidation (ISCO) of trichloroethylene in groundwater: Efficiency, sustained activity, and mechanism investigation[J]. Environ. Sci. Technol., 2020, 54(6): 3714-3724 doi: 10.1021/acs.est.0c00151
QIN X F, LI H B, YU Y Y, YANG Y, WANG K X, MA T, NIE X Q, BAI Y L, ZHANG R Y. Ornidazole degradation based on peroxymonosulfate activation induced by oxygen vacancies (OV)-enriched Cu-Co-TiO2: Coexistence of free-radical and non-radical pathways[J]. Process. Saf. Environ., 2024, 192: 1008-1025 doi: 10.1016/j.psep.2024.10.099
GUO S, YANG Z X, ZHANG H L, YANG W, LI J, ZHOU K. Enhanced photocatalytic degradation of organic contaminants over CaFe2O4 under visible LED light irradiation mediated by peroxymonosulfate[J]. J. Mater. Sci. Technol., 2021, 62: 34-43
GUO T, JIANG L S, WANG K, LI Y, HUANG H X, WU X Y, ZHANG G K. Efficient persulfate activation by hematite nanocrystals for degradation of organic pollutants under visible light irradiation: Facet-dependent catalytic performance and degradation mechanism[J]. Appl. Catal. B‒Environ., 2021, 286: 119883 doi: 10.1016/j.apcatb.2021.119883
LI X Y, LIU J H, LV R L, CHU Y Y, LV L, LU J H, ZHANG W M. Novel carbon-coated zirconium oxide nanocomposites enable ultrahigh oxidant utilization efficiency for selective degradation of organic contaminants[J]. Chem. Eng. J., 2023, 463: 142369 doi: 10.1016/j.cej.2023.142369
LI X Y, LV R L, ZHANG W M, LI M Y, LU J H, REN Y, YIN Y, LIU J H. Amorphous zirconium oxide activates peroxymonosulfate for selective degradation of organic compounds: Performance, mechanisms and structure-activity relationship[J]. Water Res., 2023, 228: 119363 doi: 10.1016/j.watres.2022.119363
ZHENG W S, SUN Y, GU Y P. Catalysis and adsorption of Zr-doped Fe3O4 nanoparticles provide a new strategy for diazinon removal and phosphorus recovery from aqueous solution[J]. J. Environ. Chem. Eng., 2022, 10(2): 107153 doi: 10.1016/j.jece.2022.107153
ZHANG H, AN Q, SU Y, QUAN X, CHEN S. Co3O4 with upshifted d-band center and enlarged specific surface area by single-atom Zr doping for enhanced PMS activation[J]. J. Hazard. Mater., 2023, 448: 130987 doi: 10.1016/j.jhazmat.2023.130987
DAS K, SHAILESH S N, KUMAR M, DE S K. Morphology dependent luminescence properties of Co doped TiO2 nanostructures[J]. J. Phys. Chem. C, 2009, 113(33): 14783-14792 doi: 10.1021/jp9048956
WANG S D, ZHU J Y, LI T, GE F, ZHANG Z H, ZHU R L, XIE H J, XU Y. Oxygen vacancy-mediated CuCoFe/tartrate-LDH catalyst directly activates oxygen to produce superoxide radicals: Transformation of active species and implication for nitrobenzene degradation[J]. Environ. Sci. Technol., 2022, 56(12): 924-7934
WU L Y, SUN Z Q, ZHEN Y F, ZHU S S, YANG C, LU J, TIAN Y, ZHONG D, MA J. Oxygen vacancy-induced nonradical degradation of organics: Critical trigger of oxygen (O2) in the Fe-Co LDH/peroxymonosulfate system[J]. Environ. Sci. Technol., 2021, 55(22): 15400-15411 doi: 10.1021/acs.est.1c04600
ZHENG W S, SUN Y, GU Y P. Assembly of UiO-66 onto Co-doped Fe3O4 nanoparticles to activate peroxymonosulfate for efficient degradation of fenitrothion and simultaneous in-situ adsorption of released phosphate[J]. J. Hazard. Mater., 2022, 436: 129058 doi: 10.1016/j.jhazmat.2022.129058
YAO Y Y, WANG C H, YANG Y P, ZHANG S, YAN X, XIAO C M, ZHOU Y J, ZHU Z G, QI J W, SUN X Y, LI J S. Mn-Co dual sites relay activation of peroxymonosulfate for accelerated decontamination[J]. Appl. Catal. B‒Environ., 2023, 330: 122656 doi: 10.1016/j.apcatb.2023.122656
ZHOU X Q, ZHANG L Y, WANG H, LU W W, ZHANG R C, LI H X, LI J, WEI X F. The regulation of generation rate facilitates the win-win of ROS yield and utilization efficiency in heterogeneous persulfate catalytic oxidation system[J]. Sep. Purif. Technol., 2023, 323: 124389 doi: 10.1016/j.seppur.2023.124389
ALI J, WENLI L, SHAHZAD A, IFTHIKAR J, AREGAY G G, SHAHIB L L, ELKHLIFI Z, CHEN Z L, CHEN Z Q. Regulating the redox centers of Fe through the enrichment of Mo moiety for persulfate activation: A new strategy to achieve maximum persulfate utilization efficiency[J]. Water Res., 2020, 181: 115862 doi: 10.1016/j.watres.2020.115862
林福伟, 蒋孝佳, 陈京乐, 吴礼光, 陈华丽, 王挺. 催化活性组分对氧化型缓释材料修复地下水中四环素的影响机制[J]. 环境科学学报, 2025, 45(7): 317-327.LIN F W, JIANG X J, CHEN J L, WU L G, CHEN H L, WANG T. Influence mechanism of catalytic active components on the remediation of tetracycline in groundwater by oxidation-type sustained-release materials[J]. Acta Scientiae Circumstantiae, 2025, 45(7): 317-327

图 2 (a) TiO2、(b) Co-TiO2和(c) Zr-TiO2的SEM照片
Figure 2 SEM images of (a) TiO2, (b) Co-TiO2, and (c) Zr-TiO2

图 3 样品的(a) N2吸附-脱附等温线、(b) XRD图、(c) FTIR谱图和(d) XPS全谱图
Figure 3 (a) N2 adsorption-desorption isotherms, (b) XRD patterns, (c) FTIR spectra, and (d) XPS survey spectra of samples

图 4 Co-TiO2和Zr-TiO2的(a) Ti2p、(b) O1s、(c) Co2p、(d) Zr3d高分辨率XPS谱图; (e) Co-TiO2和Zr-TiO2的EPR谱图
Figure 4 (a) Ti2p, (b) O1s, (c) Co2p, and (d) Zr3d high-resolution XPS spectra of Co-TiO2 and Zr-TiO2; (e) EPR spectra of Co-TiO2 and Zr-TiO2

图 5 不同催化剂活化PDS体系的降解性能: TC的降解曲线(a)及相应的准一级动力学拟合曲线(b); 猝灭实验结果(c); 活性物种贡献率(d)
Figure 5 Degradation performances of different catalyst/PDS systems: degradation curves of TC (a) and corresponding pseudo-first-order kinetic fitting curves (b); quenching experiment results (c); reactive species contribution rates (d)

图 6 Co-TiO2/PDS和Zr-TiO2/PDS体系的稳定性实验
Figure 6 Stability experiment of Co-TiO2/PDS and Zr-TiO2/PDS systems

图 7 (a~c) 不同催化剂体系中EPR谱图对比; (d) 活性物种生成机制示意图
Figure 7 (a-c) Comparison of EPR spectra in different catalytic systems; (d) Schematic diagram of reactive species generation mechanism
(a) DMPO-·OH and DMPO-SO4·-; (b) DMPO-·O2-; (c) TEMP-1O2.

图 8 PO43-添加对不同体系的影响: (a) PDS消耗量; (b) ηRU; (c) TC降解曲线
Figure 8 Effects of PO43- addition on different systems: (a) PDS consumption; (b) ηRU; (c) TC degradation curves
表 1 不同催化剂的比表面积、孔体积和孔径
Table 1. Specific surface areas, pore volumes, and pore sizes of different catalysts
| Sample | SBET / (m2·g-1) | Pore volume / (cm3·g-1) | Average pore diameter / nm |
| TiO2 | 77 | 0.243 5 | 6.4 |
| Co-TiO2 | 93 | 0.103 5 | 4.5 |
| Zr-TiO2 | 92 | 0.111 2 | 4.8 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
