图 1.
AADC PET显像剂的结构
Figure 1.
Structure diagrams of AADC PET radiotracers

多巴胺(dopamine,DA)是哺乳动物大脑内含量丰富的一种重要的单胺类神经递质,调控着中枢神经系统(CNS)中多种生理功能[1]。多巴胺的合成、存储、释放、传导、再吸收等过程的异常都将导致多巴胺系统功能紊乱,进而导致帕金森病(Parkinson disease,PD)和其他运动障碍、精神分裂症(schizophrenia,SZ)、躁狂、抑郁、药物滥用和饮食失调等多种神经精神疾病[2]。多巴胺系统包含不同靶点,如芳香族氨基酸脱羧酶(aromatic amino acid decarboxylase,AADC)、多巴胺转运体(dopamine transporter,DAT)、囊泡单胺转运蛋白2(vesicular monoamine transporter,VMAT2) 以及多巴胺受体(dopamine receptors,D1R、D2R、D3R、D4R、D5R),这些靶点从不同的生理角度反映着多巴胺系统的功能情况。通过总结这些靶点在疾病过程中的变化规律可以为疾病早期诊断、分期以及治疗提供依据。正电子发射计算机断层扫描(positron emission tomography,PET)具有高灵敏度与分辨率,能定量、无创、可视化地跟踪体内分子水平的生理生化过程,通过PET成像评估这些突触前和突触后多巴胺系统靶点的变化情况为评估多巴胺能神经末梢功能改变提供有效手段,对于疾病早期诊断、治疗、预后等意义重大[3]。多巴胺系统PET成像的关键是探针分子化学结构的设计。这些分子的化学结构设计通常参考体内天然存在的化学物质或者已在临床使用的药物的化学结构类型,以及来自计算机辅助药物设计(computeraided drug design,CADD)手段筛选出的分子[4-5],通过对这些化学分子进行修饰和放射性标记得到PET显像剂并用于成像研究。然而这些化合物受代谢稳定性、体内亲和性、非特异性摄取等诸多因素的影响,其中仅有很小的一部分显像剂可以进入临床研究阶段[6]。多年来,针对多巴胺系统各靶点特异性示踪剂的开发与应用一直都是研究热点,本文将从化学分子的筛选、修饰、优化过程以及应用等方面对多巴胺系统各靶点显像剂进行综述。
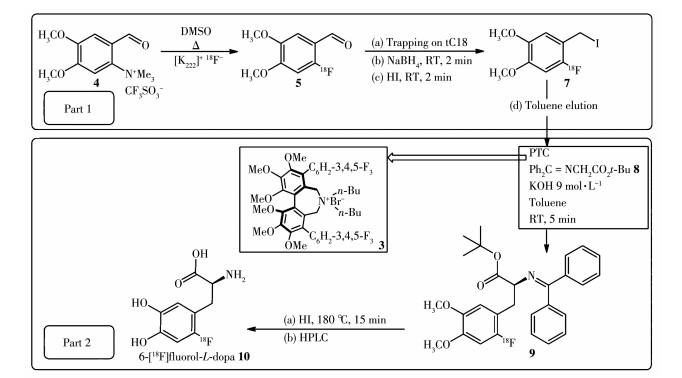
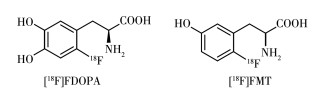
DA是一种天然存在的儿茶酚胺,不能直接通过被动运输传送过血脑屏障(blood brain barrier,BBB)。多巴胺在脑内由L-酪氨酸(L-tyrosine,Tyr)通过一系列酶促途径在多巴胺能神经元末端合成,其中AADC负责将酪氨酸羟化酶催化酪氨酸产生的多巴(L-3,4-dihydroxyphenylalanine,L-DOPA)转化成多巴胺,并储存在突触囊泡中,AADC是多巴胺系统突触前的优良生物标志物。儿茶酚类的多巴类似物在早期被选为靶向AADC的PET示踪剂探针分子,即在L-DOPA的苯环6位上引入放射性核素18F得到[18F]FDOPA,如图 1所示。然而在实际操作中,[18F] FDOPA的合成过程相当复杂,放化产率也较低。随着[18F]FDOPA在神经精神疾病以及神经内分泌瘤中越来越多的应用,其放射性标记工艺也不断在优化。目前关于放射性标记工艺优化主要基于亲核18F氟化方法,该方法相比于亲电18F氟化方法具有无需载体添加、比活度大、产量大的优点。目前,应用较多的方法是用[18F]F-亲核芳香取代硝基前体苯环上吸电子离去基团硝基之后,再通过多步反应合成最终产物。很多研究者通过改变前体来提高标记产率,如图 2所示为Libert等以三甲氨基藜芦醛三氟甲磺酸盐(4)为前体进行[18F]F-亲核芳香取代后再进行醛还原和卤化反应,然后进行不对称烷基化反应,最后用氢碘酸水解得到[18F]FDOPA,获得的放化产率较高(大于36%)[7]。为简化标记步骤也有人尝试用镍配位前体、二芳基碘盐前体和三甲基锡前体,以及运用金属催化方法等,但这些方法也存在前体制作困难或者反应过程不稳定的缺陷,还需进行改进[8]。
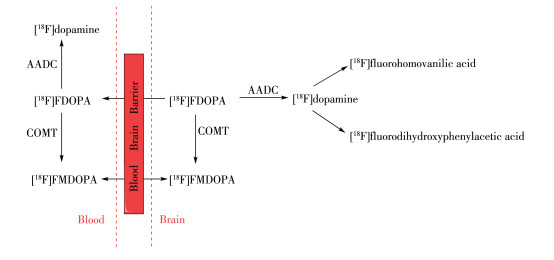
进入中枢神经系统的[18F]FDOPA在AADC的作用下转化成[18F]dopamine被摄取到多巴胺神经元神经末端的突触囊泡中储积起来。未被多巴胺神经元突触囊泡储存的[18F]dopamine在酶的作用下快速代谢成[18F]fluorohomovanilic acid而排出脑外。因此在代谢一段时间后,放射性集中在脑内纹状体等富含多巴胺神经元的区域,纹状体的放射性摄取与内源性多巴胺浓度正相关,反映了AADC的活性以及黑质多巴胺能神经元的完整性[9]。该药物已被FDA (food and drug administration)批准,用于临床PD的早期诊断与病程进展监测。然而儿茶酚甲氧基转移酶(catechol-omethyl transferase,COMT)作用产生的代谢物6-[18F]fluoro-L-3-methyoxy-4-hydroxyphenylalanine([18F]FMDOPA)会穿过血脑屏障进入脑内导致脑内背景信号升高,使药代动力学分析复杂化[10],如图 3所示为[18F]FDOPA体内代谢过程。目前,临床常用基于参考区的Patlak图形近似模型得到流入常数Ki的参数图,以其代表[18F]FDOPA在各区域摄取速率从而进行定量评估[11]。在PD患者中,[18F] FDOPA的Ki值在临床表现最明显的肢体对侧纹状体中显著降低,且壳核比尾状核受影响更大[12]。对PD患者的纵向[18F]FDOPA PET研究显示,PD进展过程中,尾状核多巴胺末端退化率为每年3.5%,壳核为每年8.9%[13]。针对[18F]FDOPA脑内代谢物干扰的问题提出的改进办法则是选择不受COMT作用的分子进行标记,为此研究者们把目标锁定在了如图 1所示的非儿茶酚类的化学分子6-[18F]fluoro-L-meta-tyrosine([18F]FMT),它被转运蛋白运送通过血脑屏障,并被AADC脱羧成6-[18F]fluorotyramine,再被神经元内的单胺氧化酶(monoamine oxidase,MAO) 氧化为6-[18F]fluoro-3-hydroxyphenylacetic acid([18F] FHPAA)并最终停留在脑组织里。与[18F]FDOPA相比,[18F]FMT对多巴胺能末梢的丧失或具有更高灵敏性[14]。
DAT位于多巴胺神经元末端的突触前膜,负责将突触间隙的多巴胺收回,并传递回细胞质,这使得多巴胺可以重新被包裹进囊泡或者直接分解代谢以维持突触的正常生理功能[15]。多巴胺能末端损伤导致释放到突触间隙的多巴胺减少,为维持突触多巴胺水平的平衡,DAT会代偿性减少,因此在多巴胺失活和循环过程中,DAT为多巴胺能系统的密度和结构完整性提供了一个很好的神经化学标志物,在一定程度上反映了突触前的多巴胺能功能,DAT减少的测量可能是反映多巴胺神经元损失程度的一个有用的指标[16]。
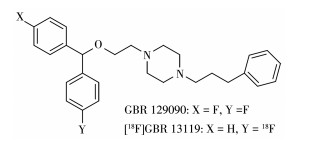
DAT显像剂的开发源于DAT抑制剂,包括抗精神病药物如GBR12909(1-(2 - [bis(4 - fluorophenyl) methoxy]ethyl)-4-(3-phenylpropyl)piperazine)、诺米芬辛(nomifensine,2-methyl-4-phenyl-1,2,3,4-tetrahy-droisoquinolin-8-amine),以及神经兴奋剂,如可卡因、安非他命(苯基乙丙胺)。早期研究将GBR12909中的一个对氟苯环替换为苯环得到的GBR13119进行18F标记(图 4),该显像剂在猴脑纹状体和小脑快速摄取,且体内稳定性良好,但由于其高亲脂性以及高非特异性摄取使其未能得到进一步应用[17]。在可卡因分子中的N-甲基或甲酯上进行11C标记,虽然该显像剂可在体内与DAT结合但其过快的药代动力学特性和N-脱烷基化限制了其用于DAT定量研究。后续DAT显像剂开发除保留可卡因对DAT的亲和力外,还通过对化合物修饰,达到改善化合物的代谢特性的目的。Clarke等研究证明将可卡因中托品烷3β位的苯甲酸基用苯基取代得到的3β-苯基托品烷类配体具有比可卡因更高的DAT结合亲和力,并且避免了原先苯甲酸酯水解的代谢途径,具有更好的代谢稳定性[18]。研究发现3β-苯基托品烷类配体的3β-芳环上的取代基和取代模式、2β取代基以及N-取代基的性质对配体识别位点的相互作用都很重要[19]。目前,图 5所示的应用较多的3β-苯基托品烷类DAT PET显像剂都是在3β-芳环上引入尺寸较小且具有亲脂性卤素基团或甲基,并在N-取代基引入甲基、氟丙基、丙基、烯丙基基团,这些化合物对DAT有着良好的结合力[20]。N-取代基为甲基时对DAT有最优亲和力,N-取代基为氟丙基、丙基、烯丙基时对DAT的亲和力虽然稍逊色于甲基但仍有纳摩尔级亲和力,且降低了对其他单胺类转运体如五羟色胺转运体(serotonin transporter,SERT)、去甲肾上腺素转运体(norepinephrine transporter,NET)的亲和力[19-21]。
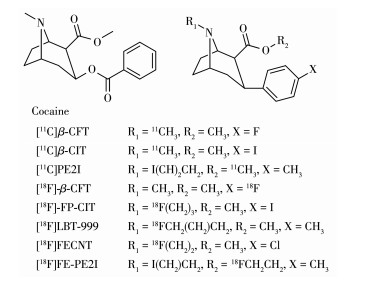
3β-苯基托品烷类化合物具有多个适合11C、18F放射性标记的位点,并且其中很多成功标记的显像剂都具有良好亲和力、药代动力学性质。11C标记位点有2种,N-11C-甲基化标记以及O-11C-甲基化标记,如[11C]CFT、[11C]β-CIT、[11C]PE2I等。但O-11C-甲基化标记很多时候比N-11C-甲基化标记更有优势,因为前体很容易在温和的条件下通过酯水解合成,羧酸前体可以通过简单的水萃取分离[22]。相比于11C,18F产生的正电子能量较低,在组织中湮灭距离短(约2.4 mm),因而能得到高分辨率图像,这使得18F标记的化合物一直备受关注。18F标记的DAT显像剂有些在3β苯环对位通过芳香族亲核取代反应或亲电氟化进行放射性标记,如[18F]-β-CFT。有些在N-烷烃链采用两步法进行亲核取代进行标记,如[18F]-FP-CIT、[18F]LBT-999、[18F]FECNT,用18F标记的[18F]氟乙基磺酸盐、[18F]氟丙基碘盐等与前体进行的两步法标记更好地解决了直接用18F离子标记前体的一步法产率过低的问题。类似的还有对2β酰基氧进行氟烷基化,常用2-[18F]fluoroethyl bromide等进行标记,如[18F]FE-PE2I[23-25]。
目前临床诊断PD更多基于对病史、症状体征、左旋多巴药物治疗反应的综合评估,缺乏诊断的金标准,然而PD发病机制极为复杂,寻找有效的生物标志物是PD诊断的关键。虽然外周血外泌体以及炎症因子等生物标志物的研究取得了一定进展,但不能反映PD的本质变化,对于PD早期诊断缺乏灵敏度与准确性。PD的主要病理改变为黑质多巴胺能神经元的大量变性丢失,然而这些病理改变却远远早于临床症状的出现,当出现明显临床症状时早已错过了最佳治疗时间,因此早期诊断对于延缓PD进程以及改善患者生活质量都十分重要,DAT PET显像恰好可以评估纹状体突触前多巴胺能神经元的密度,以此反映黑质神经元丢失的严重程度,从而及时发现病理变化,帮助PD的早期诊断[26]。早期的3β-苯基托品烷类DAT显像剂[11C]CFT为PD的早期诊断、进展情况以及鉴别提供了重要帮助[23, 27]。相比于正常对照组(normal control,NC)与特发性震颤患者(essential tremor,ET),PD患者与多系统萎缩患者(multiple system atrophy,MSA)的壳核与尾状核的[11C]CFT摄取严重降低。联合[18F]FDG可以实现对PD与MSA的精确鉴别,MSA患者小脑与脑桥的[18F]FDG摄取明显下降而PD患者无明显变化,实现对临床难以区分的疾病的精确鉴别。在PD早期诊断与进展程度的研究上,[11C]CFT也有着重要价值,相比于NC,轻度PD患者的尾状核/小脑摄取比明显降低(NC vs轻度PD:1.92±0.33 vs 2.82±0.43,P < 0.01),但是壳核/小脑摄取值未见明显下降,并且中度-重度PD患者的尾状核/小脑(轻度PD vs中度-重度PD:1.92±0.33 vs 1.53±0.10,P < 0.05)以及壳核/小脑(轻度PD vs中度- 重度PD:1.87±0.36 vs 1.43± 0.11,P < 0.05)的摄取比均明显低于轻度PD患者[28]。Nurmi等对PD患者进行2年的随访发现,PD患者壳核与尾状核的DAT密度年递减率分别为13.1% 与12.5%,然而健康同龄人递减率为2.1% 与2.8%[29]。基于[11C]CFT的结构,一些11C、18F标记的DAT配体也陆续被开发,在特异性、亲和力、代谢性质等方面都有不断的改进。[18F]-FP-CIT在特异性以及动力学速度上较[11C]CFT都有所提高,Park等对9例临床不确定帕金森病患者(未能诊断是PD还是药物诱导的帕金森综合征)进行[18F]-FP-CIT扫描,根据PET结果其中有6例表现为DAT可用性下降,并被确诊为PD,并且与一段时间之后的随访结果一致,说明DAT PET成像在区分不确定帕金森综合征上具有优势[30]。第二代显像剂[18F]FE-PE2I更快速的药物动力学性质使其在靶区达到平衡的时间较短以及其优良的代谢特性使其能成为临床上有力的研究工具[31-32], 在PD诊断方面与商用化使用的单光子发射计算机断层扫描(SPECT)显像剂[123I]FP-CIT效果相当,[18F]FE-PE2I的不可置换结合电位(non-displaceable binding potential,BPND)与[123I]FP-CIT的标准摄取值比率(SUVR)在DAT分布的主要脑区类内相关系数(intra-class correlation coefficient,ICC)非常高,即尾状体(ICC=0.923)、壳核(ICC=0.922)和纹状体(ICC=0.946)[33]。[18F]FE-PE2I所需的PET扫描时间更短(注射后30 min进行10 min静态成像),从而可以提高患者舒适度[34]。而且PET相比于SPECT具有更高的灵敏度和空间分辨率,所得图像的边界更清晰,使用[18F]FE-PE2I PET探针进行成像有助于优化[123I]FP-CIT的SPECT图像在边界位置(小于10%) 模糊的问题从而更准确勾画感兴趣区(region of interest,ROI)[35]。另一个第二代显像剂[18F]LBT-999对DAT具有更高特异性和亲和力,人体研究显示能区分PD患者与健康人的壳核、尾状核的摄取差异,但还存在轻微脱氟的问题[36]。目前3-苯基托品烷类显像剂普遍存在与其它单胺类转运体(SERT、NET)选择性不足的问题,不过DAT在基底神经节的浓度远超这些转运体,因此纹状体区域的定量不受太大影响,但无法解释纹状体外区域的DAT的变化情况。表 1展示了几类典型DAT PET探针在动力学、代谢稳定性、非特异摄取以及选择性等方面的比较[17]。
 下载:
导出CSV
下载:
导出CSV
| Radiotracer | Advantage | Disadvantage |
| [18F]GBR13119 | Rapid kinetics, the metabolite not penetrating the blood-brain barrier, low affinity for the norepinephrine transporter | High lipophilicity, high non-specific uptake |
| [11C]cocaine | Be used to study the pharmacological properties related to cocaine | Unstable metabolism, rapid brain clearance, poor selectivity, BBB-penetrating metabolites |
| [11C]CFT | Good in-vivo stability, selective for other monoamine transporters | Slow kinetics, difficultly in reaching dynamic equilibrium |
| [11C]β-CIT | Good metabolic stability | High affinity for other monoamine transporters, the slow kinetics make it difficult to reach dynamic equilibrium |
| [11C]PE2I | High specificity, selective for other monoaminetransporters (above 30), stable brain metabolism | Slow kinetics, BBB-penetrating metabolites, long time to peak concentration |
| [18F]-β-CFT | Good in-vivo stability, selective for other monoamine transporters | Low kinetics, slow radio-uptake ratio, long time to peak concentration (225 min for striatum) |
| [18F]-FP-CIT | High specificity, fast kinetics, no lipophilic metabolites | Poor selectivity for other monoamine transporters, slight in-vivo defluorination |
| [18F]LBT-999 | High selectivity for other monoamine transporters, high specificity, high signal to noise ratio | Slight in-vivo defluorination, high affinity between radioactive metabolite and DAT which causing difficulties in quantification |
| [18F]FECNT | High selectivity for other monoamine transporters, high specificity, high signal to noise ratio | BBB-penetrating metabolites |
| [18F]FE-PE2I | High specificity, fast kinetics, high selectivity for serotonin transporters, fewer brain metabolites | — |
DA对觉醒、注意、运动、知觉、动机和情绪的神经生理控制起着重要作用。它的作用是由5种主要的多巴胺受体亚型介导的[37]。多巴胺受体通过与G蛋白偶联来与一些调节神经元功能的膜或细胞质效应分子(通常是酶、转运体或离子通道)相互作用。按照初级结构和药理性质的相似性归类,可以把多巴胺受体分为D1类受体(D1R、D5R)和D2类受体(D2R、D3R、D4R)。多巴胺受体与SZ、PD等神经精神疾病以及可卡因成瘾机制关系密切。靶向多巴胺受体的PET显像剂可以在活体水平研究多巴胺受体密度、分布和功能变化,从而为疾病诊断及分子机制研究提供帮助[38]。
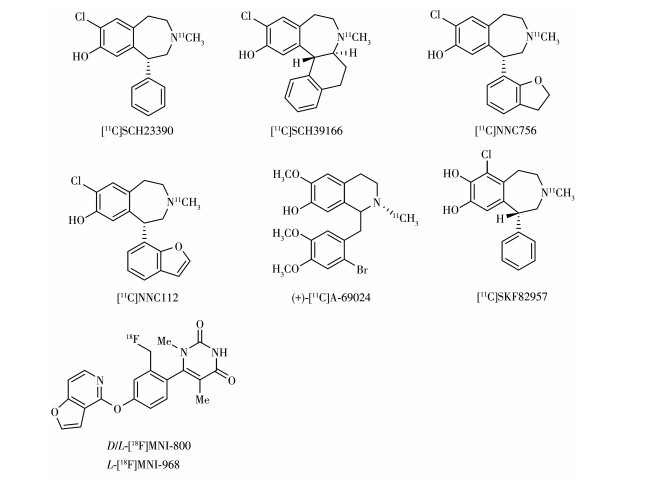
D1R是大脑中含量最丰富多巴胺受体亚型,其分布广泛,在基底节密度较高,在皮质区域也有少量分布但密度仅有纹状体的1/5~1/3[38]。目前已开发了多种D1R PET示踪剂,其中[11C]SCH23390、[11C] NNC112(图 6)已广泛应用于临床研究。
D1R显像剂多为如图 7所示的苯基苯并吖庚因结构,其独特药理特性主要归功于β-儿茶酚乙氨基的模块,这种结构模仿了儿茶酚胺化合物DA,被认为对D1R的亲和力有很大贡献[1]。早期的D1R显像剂为用[11C]碘甲烷对苯基苯并氮杂化合物(R)-(+)-8-chloro-2,3,4,5-tetrahydro-5-phenyl-1H-3-benzazepin-7-ol进行N-烷基化标记得到[11C]SCH23390,雄性食蟹猴PET扫描显示其能够快速入脑,并且在纹状体有明显浓聚,但存在与五羟色胺受体2(serotonin 5- HT2 receptors,5-HT2R)亲和力高以及脑清除过快的缺陷[39]。同系列但构象更加限制的化合物[11C] SCH39166的出现改善了[11C]SCH23390代谢过快的问题,并且降低了对5-HT2R的亲和力(仅有[11C] SCH23390的1/35),食蟹猴注射[11C]SCH39166后18 min用非放射性配体SCH23390进行抑制,纹状体摄取明显减少,说明[11C]SCH39166对D1R有明显特异性摄取且结合可逆。但[11C]SCH39166和[11C] SCH23390的纹状体/小脑低摄取比导致信噪比低。在[11C]SCH23390苯并氮杂结构的骨架上,设计了C-1位苯并二氢呋喃取代的化合物[11C]NNC756,猴子注射示踪剂后60 min时纹状体/小脑摄取比可达8,注射后130 min时高达11,在皮质的摄取明显低于[11C]SCH23390[40],但其示踪剂动力学较慢,在人体内60 min时脑内分布仍达不到平衡,并且在脑新皮层区还与5-HT2R结合。之后,在C-1位以苯并呋喃替代NNC756的C-1位的苯并二氢呋喃的示踪剂[11C] NNC112效果更优于[11C]NNC756,它对D1R有高亲和力,纹状体/小脑摄取比高。5-HT2R配体酮色林抑制实验显示[11C]NNC112皮层的摄取不来自与5-HT2R的结合,并且非特异摄取少,不仅适合在纹状体成像也适合受体密度低的新皮层等区域成像。[11C]NNC112在SZ等精神疾病的研究中发挥重要作用[41]。从未进行过药物治疗的SZ患者皮层区域的[11C]NNC112摄取明显高于健康受试者,但进行过药物治疗且停药一段时间的SZ患者[11C]NNC112摄取相比于健康受试者没有明显增加,然而经长期抗精神病药物(舒必利)治疗的重度残留期SZ患者[11C] NNC112摄取显著降低,这提示SZ患者D1R数量的增加与疾病本身有关,并可能通过慢性抗精神病药物治疗使D1R数量正常化,然而长期的药物治疗导致D1R密度下降[42-43]。一些非苯基苯并氮杂结构(1-benzyltetrahydroiso quinolines)的化合物如(+)-[11C]A-69024也对D1R有高特异性和选择性,(+)-[11C]A-69024在脑内被快速摄取,并且在脑内清除速率适中(清除半衰期16.1 min),纹状体/小脑的摄取比在30 min时达到最高值7.4[44-45]。
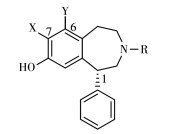
D1R同时具有高亲和态与低亲和态,拮抗剂与D1R结合时不区分高低亲和态,但与激动剂结合则展现这2种亲和态并且优先与受体高亲和态结合,但高亲和态被认为是多巴胺受体的功能状态,因此研究疾病与正常状态下高亲和态受体密度的区别更具生理意义。D1R激动剂PET示踪剂是评估D1R高亲和态的有用成像工具,可以提供更多重要的体内功能信息。在C-7位用卤素取代OH可以调控配体对D1R在激动与拮抗之间更偏向拮抗作用,早期开发的显像剂多为此结构,因此多为D1R拮抗剂。对于[11C]SKF 82957,其在C-7位保留OH基团,具有激动剂活性,在C-6位引入Cl提高了其选择性,该显像剂对D1R有高亲和性和选择性,其激动剂属性使得对高亲和态D1R的测量更为敏感。慢性酒精处理的大鼠相比于控制组,嗅结节/小脑以及纹状体/小脑的[11C]SKF 82957摄取比分别增加67%与58%,然而拮抗剂[11C]SCH23390的摄取比仅增加21% 与17%,因此,[11C]SKF 82957的体内测量说明了慢性乙醇增加了嗅结节与纹状体中高亲和态D1R[46]。但其产生的亲脂性代谢物会穿过血脑屏障,对信噪比造成一定影响[47-48]。近年来,也有 18F标记的显像剂出现,Barret等对以苄基氟为主体的化合物进行 18F标记得到外消旋体[18F]MNI-800与其对映异构体[18F]MNI-968,这些新构型化合物也具有高特异性与选择性,代谢稳定性良好,并有希望用于人体研究[49]。图 6所示为文中介绍的D1R显像剂的结构式。
D2类受体在纹状体、边缘系统、黑质、丘脑、皮层等都有分布,其通过与抑制型G蛋白(Gi/o)偶联从而抑制胞内环腺苷酸的合成[50-51]。D2类受体家族在神经疾病的研究中一直备受关注,如图 8所示的探针[11C]raclopride和[18F]fallypride已广泛用于临床研究PD、SZ等脑神经疾病。
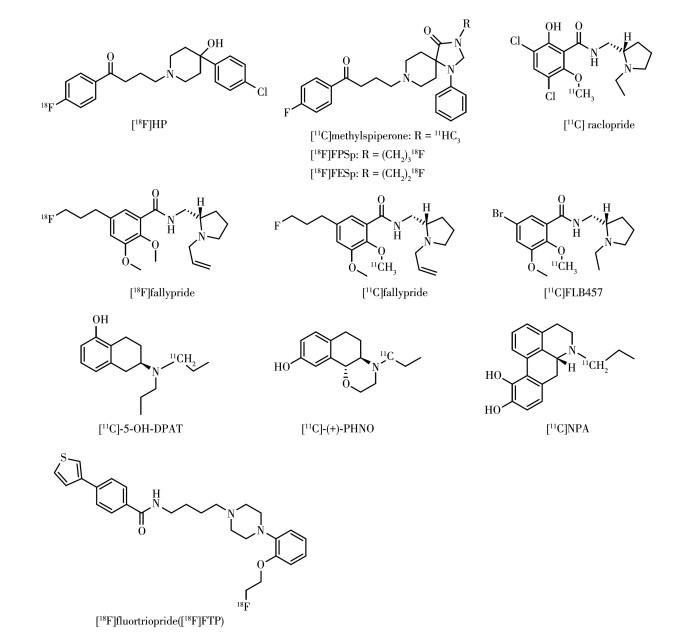
D2R显像剂在所有多巴胺受体显像剂类文献中报道得最多。早期的显像剂基于对氟丁苯酮的抗精神病药物氟哌啶醇(haloperidol,HP)与螺哌隆(spiperone,SP),这2种药物中都含有F原子,18F替换19F对药物理化性质无太大影响,丁苯酮链的长度对DA受体结合亲和力优良并且有良好的D2R/D4R亚型选择性[52]。对氟苯的结构能够伸入到配体与受体结合的口袋深处,在受体底部疏水裂缝与氨基酸残基通过CH-π相互作用、疏水相互作用等结合。体内成像结果显示[18F]HP清除过快且脑内非特异性过高,不适合体内成像。HP中哌啶上的叔羟基虽然有助于增加D2R亲和性但不是D2R亲和力的主要贡献因素。不同于HP,SP在叔羟基相应位置连接刚性结构的三氮杂螺结构,G蛋白偶联受体(Gprotein-coupled receptor,GPCR)结构解析说明该刚性结构对于SP在D2R扩展结合口袋的结合十分重要[53],通过N-[11C]甲基化对SP标记得到的[11C]meth-ylspiperone,它入脑量多。灵长类动物PET显示其可快速聚集在尾状核和壳核,人体研究显示注射后70~130 min,壳核/小脑摄取比可达4,该探针在1983年用于人脑PET成像,这也是第一个用于人体研究的受体类PET显像剂。但由于 11C半衰期短,很难测量配体受体间的一些动力学参数如特异性结合的最大值[54]。使用半衰期更长的核素 18F可以解决这个问题。18F标记的SP类似物为N-[18F]氟丙基螺哌隆([18F]FPSp)和N-[18F]氟乙基螺哌隆([18F]FESp)[55]。但丁苯酮类探针与5-HT2R等单胺类受体结合也高,随着基于苯酰胺结构的放射性配体的出现,对D2R与单胺类受体的选择性有了提高,药物的体内药代动力学性质也得到改善。苯酰胺类探针的设计源自典型抗精神病药物舒必利,其对D2R亲和力高,对其他受体亲和力较小[56]。苯酰胺类探针结构为苯甲酰胺通过1个亚甲基连接1个四氢吡咯环,在苯环上引入烷烃链、甲氧基、卤素原子、羟基等基团以及改变吡咯烷氮上烷基的长度对化合物进行修饰,对舒必利修饰首先得到了雷氯必利(raclopride)和依匹必利(epidepride)。[11C]raclopride是一种选择性但非特异性D2R显像剂,其对D3R也有一定亲和力。注射[11C]raclopride后纹状体/小脑的摄取比值随时间平稳上升,20 min后达4,脑内其他区域非特异性摄取很少。基于多种优良性能,[11C]raclopride已广泛用于人类D2R/D3R PET研究,此外,[11C]raclopride的结合具有可逆性并且其对内源多巴胺的敏感性较强,可用于研究内源多巴胺水平变化的影响。在条件刺激下(观看可卡因吸食视频),瘾君子的壳核与尾状核的[11C]raclopride的特异性结合均显著减少,减少程度与受试者描述的可卡因渴望程度(与多巴胺分泌增加有关)正相关[57]。对epidepride进一步修饰,将其苯环5位的碘用氟丙基取代得到FPMB,即(S)-N-[(1-ethyl-2-pyrrolidinyl)methyl]-5-(3[18F]fluoropropyl)-2,3-dimethoxybenzamid,该丙基链的长度对于保持D2R亲和力是最佳的,然而[18F]FPMB的亲脂性太低导致入脑量过少,因而通过将[18F]FPMB烷基链延长从而增加亲脂性,并且还要保留对D2R的亲和性,然而将5-氟丙基替换为5-氟丁基却导致D2R亲和力的下降,于是将目标转向吡咯烷的N,因为N-烷基链可能可以忍受微小的变化。有研究显示将舒必利上的乙基换为烯丙基使得D2R亲和力增加了15倍[58],于是将FPMB吡咯烷N上的乙基替换为烯丙基得到的[18F]fallypride,这不仅提高了亲脂性,大大增加了入脑量,并且亲和力也提高了8倍(FPMB:Ki=0.26 nmol·L-1,fallypride:Ki=0.03 nmol· L-1)[59],能对D2R数量更少的外纹状体区域成像,它解释了氯氮平的治疗效果可能更多源自与外纹状体区域多巴胺受体的结合[60-61]。[18F]fallypride也广泛用于PD的研究,PD患者较健康受试者的扫描结果显示腹侧中脑、杏仁核、丘脑、海马体、苍白球、尾状核的受体结合力分别下降39%、33%、30%、22%、16%、11%,而壳核与腹侧纹状体变化微小。Fisher等用[18F]fallypride研究跑步对尚未接受药物治疗的早期PD患者D2R的影响,研究发现,PD患者接受高强度跑步训练后脑内[18F]fallypride的受体结合力(binding potential,BP)显著增加(训练前:5.58±0.43,训练后:11.0±1.09),而未接受训练的对照组PD患者BP值未有明显改变,健康受试者训练前后BP值也未有明显改变,接受运动训练的PD患者姿势控制得到明显改善。该研究提示跑步训练可能涉及前额皮质回路,促进多巴胺D2R的恢复并因此通过涉及运动技能学习的回路改善患者姿势控制,对PD患者护理具有重要意义[62]。11C标记的显像剂[11C] fallypride也有相同的效果。11C核素标记的示踪剂在60~90 min内可以获得良好成像数据,而[11C] fallypride在90 min左右可以在外纹状体区域(丘脑、杏仁核、皮层等)实现最佳结合动力学,因此可以对外纹状体区域在1 d内对个体进行重复扫描以研究药物效应,并且短的扫描时间有效避免了被试者移动的干扰[63]。[11C]FLB 457同样广泛用于人体PET成像,研究抗精神病药物对多巴胺D2样受体占用情况,它对D2R和D3R有同等的高亲和力(Ki=0.02 nmol·L-1),是非选择性D2R/D3R显像剂。
多巴胺受体成像的另一种方法是对激动剂分子放射性标记,相比于拮抗剂,放射性标记的激动剂对内源多巴胺变化更加敏感,并且可以在体内区分高亲和力受体结合位点和低亲和力受体结合位点。四氢萘类化合物[11C]5-OH-DPAT在四氢萘的苯环上进行羟基化修饰,在四氢萘的2位连接N,N-二丙基链。它具有激动剂活性,其在纹状体的结合能被D2R拮抗剂舒必利取代说明具有D2R选择性,与D2R的选择性结合能被三磷酸鸟苷(guanosine triphosphate,GTP)类似物鸟苷5-[β,γ-亚氨基]三磷酸三钠盐阻断,表明[11C]5-OH-DPAT可以探测D2R的高亲和力状态[64]。非选择性D2R/D3R显像剂[11C]-(+)-PHNO为D3R偏好性显像剂,体内结合实验显示在D3R分布区域苍白球有高特异性摄取,在丘脑、脑干也有一定的特异性摄取,其也是激动剂,它对安非他命刺激释放的多巴胺有很好的敏感性。活体成像显示狒狒给药安非他命后D2R结合显著减少,与多巴胺释放占据靶点一致,因而临床上可用于评价突触多巴胺浓度的急性变化[65]。但由于[11C]-(+)-PHNO对D3R/D2R的选择性只有53,因此在D2R与D3R均表达的脑区如纹状体等,D2R的存在仍是干扰,需要用非标记的D3R拮抗剂消除D3R PET信号再反推出D3R存在区域的摄取值[66]。
11C标记的吗啡类似物[11C]NPA也是激动剂,阿扑吗啡的酚羟基结构是D2R受体结合的关键,[11C] NPA保留了该结构并用丙基取代阿扑吗啡叔胺上的甲基以提高D2R亲和力和降低5-HT1AR的亲和力[1, 67]。[11C]NPA具有良好的脑组织渗透能力以及适当的区域选择性,可对高亲和状态D2受体成像,对内源多巴胺敏感性良好。对狒狒进行不同浓度的安非他命(0.3、0.5、1.0 mg·kg-1)刺激,[11C]NPA在纹状体的摄取呈剂量依赖式下降(分别下降32%±2%、45%±3%、53%±9%),相比于[11C]raclopride(分别下降24%±10%、32%±6%、44%±9%),[11C]NPA的下降程度更多说明内源性DA更有效地与[11C]NPA竞争D2R结合位点,使得[11C]NPA对多巴胺变化得更为敏感[68]。
D3R在中脑边缘区域高水平的表达的这一特殊性使其越来越成为引人注目的靶点,大量证据表明D3R与认知、奖励、行为强化和渴望等情感行为及PD、SZ、药物成瘾等脑疾病息息相关[69-70]。但D3R与D2R在跨膜结合域有高达80% 的同源性,更重要的是,这些受体结合口袋内的残基几乎相同[71-72],并且D3R数量很少(约为D2R的1%)[73],选择性D3R探针的开发相当困难,直到目前仍没有选择性D3R PET显像剂被开发出来。
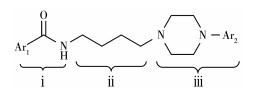
早期开发的D3R探针多为D2R/D3R探针,无法针对D3R成像,并且D3R与内源多巴胺的结合亲和力是所有多巴胺受体中最高的,内源多巴胺极易占用D3R对成像造成干扰,种种因素使得研究D3R与疾病关系的体内PET定量实验结果往往与一些体外实验结论相悖。因此开发具有高选择性高亲和性的D3R显像剂极具意义与挑战。得益于CADD技术,研究者们对D3R与配体的构效关系总结出了一定的规律,如图 9所示,即一个芳基或芳基取代酰胺(ⅰ),通过一个功能化的烃基部分(ⅱ)连接一个含芳基末端的结构(ⅲ)。ⅰ部分为较大芳环的苯甲酰胺类对维持共平面结构有重要作用,部分小环结构也具有良好选择性,如苯乙烯类。因为D3R与D2R结合袋内的残基本质上是相同的,为了实现靶向选择性,配体必须向结合口袋的细胞外开口延伸,因此配体要以一种更线性、延伸的结构与D3R结合[72],研究发现ⅱ部分为4个碳的烃基时对维持ⅰ与ⅲ之间的几何距离和空间构象最佳,表现出的亲和力效果最好[74]。ⅲ部分为含氮取代基的芳环,在这部分结构上进行改性对提高选择性有关键影响,近年来通过计算机模型研究此部分与D3R正构结合位点(OBS)、次级结合位点(SBS)的相互作用发现,苯基哌嗪药效团具有最强大的选择性[75]。因此,近年来设计的D3R PET探针大多为苯基哌嗪类衍生物。目前为止,效果最好的是Robert等设计的[18F]FTP。
Robert等设计、合成了一系列具有柔性构象的苯基哌嗪类苯甲酰胺分子,其中化合物5在苯基哌嗪芳环的2位上用氟乙氧基进行修饰,并在苯甲酰胺的4位用3-噻吩环取代,发现该化合物对D3R的亲和力以及D3R/D2R选择性最优,对D3R的亲和力达到0.1 nmol·L-1量级,D3R/D2R选择性高达160倍(D2R:Ki=27.7 nmol·L-1,D3R:Ki=0.17 nmol·L-1)[76]。而后该团队以该化合物为目标通过18F亲核取代标记法合成了[18F]FTP。恒河猴脑部PET成像显示其在纹状体、丘脑等D3R富集区域有高摄取,在其它非D3R区域未见明显摄取[77]。但由于异氟烷麻醉导致的DAT构象发生变化,使得突触间隙多巴胺浓度增加,从而大量占据突触后膜的多巴胺受体,阻碍了[18F]FTP与D3R的结合。安定药物劳拉西泮(lorazepam) 可以降低内源多巴胺水平,在未使用lorazepam抑制多巴胺分泌时,恒河猴脑部PET结果显示示踪剂的脑内分布在个体间的差异较大,但在Lorazepam抑制后不同个体间[18F]FTP在脑内分布的情况一致,即在纹状体、丘脑等D3R所在区域有明显摄取。该探针也是唯一进入临床试验的选择性D3R显像剂,然而临床结果显示D3R抑制剂奋乃静抑制前后[18F]FTP在D3R富集区域无显著差异,仍需对探针进一步优化和筛选[78]。图 8所示为上文介绍的D2R与D3R显像剂的结构式。
D4R是多巴胺D2样受体家族中含量较少的成员,信使RNA(messenger RNA,mRNA)和免疫组化研究显示其主要分布在外纹状体区域(皮层区、边缘区),D4R在各种神经行为和精神疾病中发挥着重要作用,如SZ、注意力缺陷多动障碍(ADHD)和勃起功能障碍(ED)等。
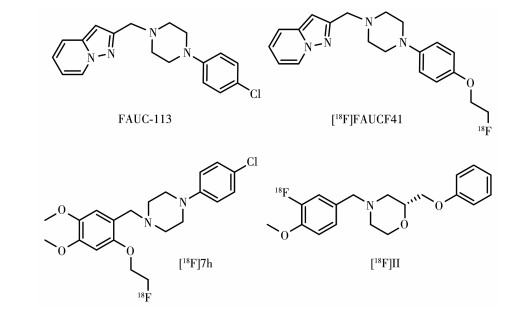
多年来,研究者们一直努力寻找具有高D4R亲和力的配体,研究发现N-芳基哌嗪的第二个N原子优先连接到杂芳烯部分苄基CH2位置时,配体具有高D4R亲和力,如图 10所示的FAUC113。用对(2-氟乙氧基)取代FAUC113邻甲氧基的苯基哌嗪衍生物FAUCF41对D4R有纳摩尔级的高亲和力以及对D2R和D3R都有1 000倍以上的选择性,[18F] FAUCF41体外稳定性高,脂水系数lg P为1.99。体外大鼠脑放射自显影显示,它选择性地结合在不同的脑区,在包括海马齿状回和其他富含D4R的区域的结合可被D4R拮抗剂FAUC113抑制78%~93%,这些发现支持[18F]FAUCF41作为D4R的潜在PET放射性配体[79]。18F标记的另一个苯基哌嗪类化合物[18F]7h,用间甲氧基苯生物电子等排体取代吡啶氮杂二烯,并在苯环对位引入额外的甲氧基,标记位点的氟乙氧基在临位,该结构对D4R的纳摩尔级高亲和力以及对D2R和D3R都有高选择性(D4/D2:450,D4/D3:2 300),lg P为2.8,在人血清的体外稳定性良好(大于95%,90 min),也是一种很有前途的PET候选物[80]。
虽然上述配体对D4R具有亚纳摩尔级亲和力和高D4R/D2R选择性,但这种基于芳基哌嗪结构的D4R配体结构与许多GPCR配体相同,非D4R的特异性结合过多,D4R PET配体的开发仍需通过结构改进克服这一缺点。
最近,Willmann等合成的18F标记吗啉衍生物(S)-4-(3-氟-4-甲氧苄基)-2-(苯氧甲基)吗啉([18F]Ⅱ),对D4R亲和力为13 nmol·L-1,对五羟色胺受体和其他多巴胺受体亚型都有高选择性,lg P=3.38,大鼠体外自显影显示在整个大脑显示特异性结合,非特异性摄取较少,纹状体无摄取,在小脑的丘核和内侧核中有明显的高积聚,但在D4R聚集的前额皮质和海马区域仅观察到少量结合。该探针将进一步进行临床前动物模型的脑摄取与代谢功能等方面的研究[81]。
虽然D4R PET配体在亚型选择性上已经有了很大进展,但由于D4R在脑中含量低,极其容易受到非特异性信号干扰从而掩盖其特异性结合,目前开发的D4R PET显像剂都未能成功进入人体研究。
一些动物实验显示D5R在血压调控中起到重要作用[82],而腹侧被盖区神经元D5R的激活可能有助于成瘾性的形成[83]。但由于D1R与D5R在高度保守的7个跨膜结构域的序列同源性达80%,目前开发的D5R配体都无法克服对D1R的高亲和力。D5R在大脑的几个皮层和纹状体外区域低水平表达,其在脑内的密度远少于D1R,这对选择性D5R配体的开发更增加了难度[84]。目前尚无D5R选择性或特异性放射配体的报道。
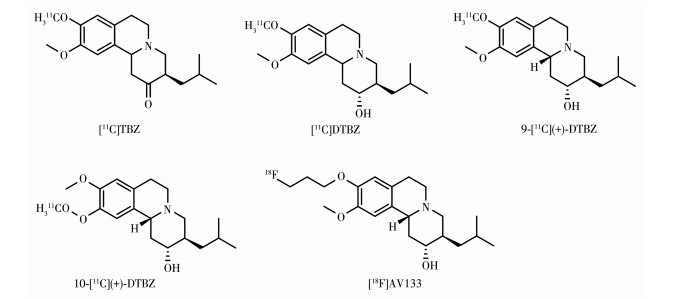
VMAT2是一种膜结合的突触前蛋白,负责将多巴胺和其他单胺类神经递质从细胞质转移到储存囊泡中。VMAT2主要存在于中枢神经系统的单胺能神经元中,包括多巴胺能、5-羟色胺能、去甲肾上腺素能和组胺能神经元,在尾壳核、伏隔核、黑质、被盖腹侧区、蓝斑、中缝核群和孤束核有高表达,纹状体中95% 以上的VMAT2结合位点都在多巴胺能神经元末端,因此纹状体VMAT2结合位点密度与中脑黑质纹状体神经元数量线性相关[85]。各种精神兴奋剂的神经毒性和成瘾性至少部分归因于它们对VMAT2功能的干扰。通过PET对VMAT2成像能够评估突触前多巴胺能通路的完整性。在临床上对PD和阿尔茨海默病的早期诊断和监测进展以及药物滥用机制的研究具有重要意义。VMAT2 PET显像剂起源于VMAT2抑制剂丁苯那嗪(tetrabenazine,TBZ)。TBZ及其衍生物对VMAT2有纳摩尔级高亲和性并且特异性识别VMAT2。自1993年[11C]TBZ的制备和成功的人体成像开始,一系列11C和18F标记的丁苯那嗪类似物被制备并在人体成像研究中进行了评估,如图 11所示。
[11C]TBZ是包含2种异构体的外消旋混合物(3R、11bR和3S、11bS),其标记位点为TBZ的9-[11C] 甲氧基。[11C]TBZ的分子量(molecular weight,MW=315)与亲脂性(lg P=2.7)使之易于入脑,体内实验显示其入脑迅速,在富含VMAT2的纹状体和下丘脑聚集并迅速从大脑其他大部分区域快速消失,在短时间内可获得高信噪比的清晰图像,TBZ抑制实验显示[11C]TBZ与VMAT2的结合具有特异性,适合人体PET成像,但是[11C]TBZ的酮羰基在肝脏中羰基还原酶的作用下快速代谢成仲醇,产生二氢丁苯那嗪(dihydrotetrabenazine,DTBZ) 代谢物,α和β构型的[11C]DTBZ可穿过BBB再次与VMAT2结合,对[11C] TBZ放射性分布的定量药代动力学建模造成困难[86-88],并且早期对[11C]TBZ的PET研究很可能代表了[11C]DTBZ的α-和β-异构体的大脑分布。之后对TBZ的α构型代谢物DTBZ的9-[11C]甲氧基位点标记得到[11C]DTBZ,标记位点选在9-[11C]甲氧基是因为在该位置的甲氧基代谢为羟基,产生的放射性代谢产物从脑内快速清除,不对脑内放射性信号造成干扰[89]。仲醇取代TBZ的羰基后其代谢稳定良好,具有可逆性结合和2 nmol·L-1的平衡结合亲和力,对DAT、SERT和NET等转运体以及其他受体和酶的亲和力都很低。与健康被试相比,MSA患者尾状核壳核中[11C]DTBZ的特异性结合明显减少,另一项研究中PD患者壳核和尾状核中[11C]DTBZ的特异性结合也明显减少[90]。具有临床应用价值,但相比于[11C]TBZ(BPND:1.49),[11C]DTBZ在人体体内成像结果(BPND:0.96)方面只有相对较小的改善。之后对[11C] DTBZ外消旋体进行拆分发现只有(+)对映体与VMAT2特异性结合,(-)对映体亲和性是(+)对映体的0.1%,使用外消旋的[11C]DTBZ降低了特异性信号并降低了图像对比度。将DTBZ对映异构体拆分得到更具特异性的示踪剂9-[11C](+)-DTBZ,相比于[11C] TBZ(BPND:2.56),人体体内成像结果(BPND:0.94)得到了显著改善。Bohnen等用9-[11C](+)-DTBZ评估正常被试与PD患者的VMAT2的结合密度,正常被试的纹状体特异性结合呈年龄相关性下降,每年下降约0.5%,在PD患者中,尾状核、前壳核、后壳核和黑质中的特异性结合呈显著下降,与临床上PD患者的诊断评分(UPDRSⅢ)结果呈现显著的线性相关性,9- [11C](+)-DTBZ已广泛用于人类神经系统疾病的临床研究[91]。9-[11C](+)-DTBZ与囊泡内的多巴胺竞争结合VMAT2,这一特性也使之用于甲基苯丙胺(俗称“冰毒”)作用机制的相关研究,研究显示甲基苯丙胺戒断早期(平均戒断2.6 d),9-[11C](+)-DTBZ纹状体结合升高(增加28%),并且升高程度与药物使用的严重程度和以及认知障碍和戒断症状相关,但对于戒断10 d的人群,9-[11C](+)-DTBZ纹状体结合与正常者接近,该成像结果支持了甲基苯丙胺吸食者死后多巴胺水平降低的报道,并表明早期戒断期脑内多巴胺水平较低,随着戒断时间增长,多巴胺水平逐渐恢复,这一研究为戒断治疗策略的制定提供思路。Johanson等发现9-[11C](+)-DTBZ在纹状体的结合水平在戒断甲基苯丙胺长达30年的人群中略有降低但降低程度与戒断年数无关,并且这种降低程度可能对黑质纹状体投射完整性影响的幅度足够小,提示人类长期高剂量甲基苯丙胺滥用并不一定会对黑纹状体神经末梢造成严重损伤[92-93]。国立台湾大学的Huang及其同事报道了在10-甲氧基位置进行11C标记的10-[11C](+)-DTBZ在啮齿动物脑内的合成和体内药代动力学,但由于10-位的标记在代谢过程中在脑内产生放射性代谢产物会对图像信号造成干扰,该放射性示踪剂尚未进入人体研究[94]。为了让更多不具备核素生产条件的医院或研
究机构可开展VMAT2 PET显像剂的临床研究,需开发半衰期较长的18F标记的DTBZ衍生物。Goswami等报道的[18F]FP-(±)-DTBZ对VMAT2均具有与DTBZ相近的高亲和性,静脉注射后脑摄取快速,[18F]FP-(±)-DTBZ从非靶区清除快,具有更好的靶/背景比,注射后30 min,纹状体/小脑的摄取比值为3.0,其在体内脱氟代谢缓慢,并且代谢物18F烷基DTBZ仅占少量。相应手性拆分后的旋光异构体[18F]FP-(+)-DTBZ([18F]AV133)具有比[18F]FP-(±)-DTBZ更高的VMAT2亲和力和特异性,且代谢稳定性好,体内脱氟非常轻微且缓慢,是一种可用于人体研究的VMAT2 PET显像剂。VMAT2的放射性配体对内源性多巴胺的变化不太敏感,因此更准确地反映了活体黑质纹状体神经元末端丢失情况[95]。[18F] AV133对于早期PD患者的鉴别与诊断,以及预后和给药方案的制定都有很大帮助。Alexander等对临床不确定的PD患者进行[18F]AV133扫描,临床诊断专家在得到PET图像后对23% 的患者的诊断结果做出了改变,对53% 的患者的给药方案作出修订。相比于临床医生的临床经验判断,[18F]AV133成像将诊断正确率由11% 提高到了80%[96-97]。相比于PD患者的[123I]CIT DAT显像,[18F]AV133进行VMAT2成像具有潜在的优势,如更好的图像质量和定量、更短的示踪剂给药和扫描之间的时间、更短的扫描持续时间,并且相比于[123I]CIT,[18F]AV133不需要事先封锁甲状腺以防止放射性碘的摄入[97]。目前,[18F]AV133在美国已经进入Ⅲ期临床的研究。其他新结构的VMAT2 PET显像剂目前尚无报道。
脑多巴胺系统已被证实或显示出能在各种神经和精神疾病的诊疗中发挥关键作用,但多巴胺系统的生理功能相当复杂,是包括多巴胺的产生、转移、激活、再摄取及死亡等多个方面相互关联的结果,了解这些过程的内在变化关系对于研究疾病分子机理尤为重要。许多PET显像剂已被开发用于脑多巴胺系统的成像研究:靶向AADC的显像剂[18F]FDOPA等用于评估突触前多巴胺神经元合成多巴胺的能力,靶向VMAT2的显像剂[18F]AV133等用于评估从细胞质合成的多巴胺转移至多巴胺能神经末梢储存囊泡的能力,靶向DAT的显像剂[18F]FE-PE2I等用于评估突触间隙内多巴胺重摄入突触前细胞质的能力,靶向多巴胺受体的显像剂[18F] fallypride等则反映着多巴胺信号传递的功能,以及靶向单胺氧化酶A(MAO A)和单胺氧化酶B(MAO B) 的一些显像剂用于反映多巴胺代谢分解过程。这些显像剂为活体水平研究多巴胺的合成、突触囊泡的存储、释放及再吸收、受体结合、体内降解的全过程提供了重要帮助,反映了生物体真实的生理功能。目前针对多巴胺系统的探针仍存在缺陷,迫切需要新探针的开发,但新放射性探针的研发仍依赖于从现有小分子药物中筛选、改进、修饰,这种模式虽然产生了很多应用于临床的优秀探针,但却在一定程度上限制了新构型分子的开发。针对放射性药物的计算机辅助设计可以有效地发现和优化先导化合物,提高药物筛选效率,可以帮助我们实现更高效的新放射性药物的发现。
单一放射性探针只能反映多巴胺系统某一方面的功能变化,未来利用多巴胺系统不同靶点特异性探针的多探针PET成像方法,可以获取AADC、DAT、D1R-D5R、VMAT2等在脑内不同区域的分布、密度信息,从而获得整个多巴胺系统的功能变化情况,帮助更深入地理解多巴胺系统在PD、SZ等疾病发展过程中的变化规律,为疾病诊断与分子机制的研究以及新药开发提供依据。
Zhang A, Neumeyer J L, Baldessarini R J. Recent progress in development of dopamine receptor subtype-selective agents: Potential thera-peutics for neurological and psychiatric disorders[J]. Chem. Rev., 2007, 107(1): 274-302. doi: 10.1021/cr050263h
Sioka C, Fotopoulos A, Kyritsis A P. Recent advances in PET imaging for evaluation of Parkinson's disease[J]. Eur. J. Nucl. Med. Mol. Imag., 2010, 37(8): 1594-1603. doi: 10.1007/s00259-009-1357-9
Ametamey S M, Honer M, Schubiger P A. Molecular imaging with PET[J]. Chem. Rev., 2008, 108(5): 1501-1516. doi: 10.1021/cr0782426
Raviña E, Negreira J, Cid J, Masaguer C F, Rosa E, Rivas M E, Fontenla J A, Loza M I, Tristán H, Cadavid M I, Sanz F, Lozoya E, Carotti A, Carrieri A. Conformationally constrained butyrophenones with mixed dopaminergic (D2) and serotoninergic (5-HT2A, 5-HT2C) affinities: Synthesis, pharmacology, 3D-QSAR, and molecular modeling of (aminoalkyl) benzo-and-thienocycloalkanones as putative atypical antipsychotics[J]. J. Med. Chem., 2000, 43(6): 1250-1250. doi: 10.1021/jm9911837
De P, Roy K. QSAR modeling of PET imaging agents for the diagnosis of Parkinson's disease targeting dopamine receptor[J]. Theor. Chem. Acc., 2020, 139(12): 176. doi: 10.1007/s00214-020-02687-9
Kilbourn M R. 11C-and 18F-radiotracers for in vivo imaging of the dopamine system: Past, present and future[J]. Biomedicines, 2021, 9(2): 108. doi: 10.3390/biomedicines9020108
Libert L C, Franci X, Plenevaux A R, Ooi T, Maruoka K, Luxen A J, Lemaire C F. Production at the curie level of no-carrier-added 6-[18F] fluoro-L-DOP[J]. J. Nucl. Med., 2013, 54(7): 1154-1161. doi: 10.2967/jnumed.112.112284
Neves A C B, Hrynchak I, Fonseca I, Alves V H P, Pereira M M, Falcao A, Abrunhosa A J. Advances in the automated synthesis of 6-18F fluoro-L-DOPA[J]. EJNMMI Radiopharm. Chem., 2021, 6(1): 18. doi: 10.1186/s41181-021-00132-1
左传涛. 18F-多巴PET在帕金森病中的应用[J]. 国际放射医学核医学, 2000,24,(1): 4-7. ZUO C T. The Applications of 18F-DOPA PET in Parkinson's disease[J]. International Journal of Radiation Medicine and Nuclear Medicine, 2000, 24(1): 4-7.
Doudet D J, Mclellan C A, Carson R, Adams H R, Miyake H, Aigner T G, Finn R T, Cohen R M. Distribution and kinetics of 3-O-methyl-6-[18F]fluoro-L-DOPA in the rhesus monkey brain[J]. J. Cereb. Blood Flow Metab., 1991, 11(5): 726-734. doi: 10.1038/jcbfm.1991.129
Huang S C, Yu D C, Barrio J R, Grafton S, Melega W P, Hoffman J M, Satyamurthy N, Mazziotta J C, Phelps M E. Phelps M E. Kinetics and modeling of L-6-[18F]fluoro-DOPA in human positron emission tomographic studies[J]. J. Cereb. Blood Flow Metab., 1991, 11(6): 898-913. doi: 10.1038/jcbfm.1991.155
Morrish P K, Sawle G V, Brooks D J. Regional changes in[18F] DOPA metabolism in the striatum in Parkinson's disease[J]. Brain, 1996, 119(6): 2097-2103. doi: 10.1093/brain/119.6.2097
Morrish P K, Rakshi J S, Bailey D L, Sawle G V, Brooks D J. Measuring the rate of progression and estimating the preclinical period of Parkinson's disease with[18F]dopa PET[J]. J. Neurol. Neurosurg. Psy-chiatry, 1998, 64(3): 314-319. doi: 10.1136/jnnp.64.3.314
Gallagher C L, Holden J, Christian B, Harding S, Nickles R J, Johnson S. A within-subject comparison of 6-[18F]fluoro-m-tyrosine (FMT) and 6-[18F]fluoro-l-DOPA (FDOPA) in Parkinson disease (PD)[J]. Neuroimage, 2010, 52: S75-S75. doi: 10.1016/j.neuroimage.2010.04.059
Brooks D J. Molecular imaging of dopamine transporters[J]. Ageing Res. Rev., 2016, 30: 114-121. doi: 10.1016/j.arr.2015.12.009
Lee C S, Samii A, Sossi V, Ruth T J, Schulzer M, Holden J E, Wudel J, Pal P K, De La Fuente-Fernandez R, Calne D B, Stoessl A J. In vivo positron emission tomographic evidence for compensatory changes in presynaptic dopaminergic nerve terminals in Parkinson's disease[J]. Ann. Neurol., 2000, 47(4): 493-503. doi: 10.1002/1531-8249(200004)47:4<493::AID-ANA13>3.0.CO;2-4
Stehouwer J S, Goodman M M. Fluorine-18 radiolabeled PET tracers for imaging monoamine transporters: Dopamine, serotonin, and nor-epinephrine[J]. PET Clinics, 2009, 4(1): 101-128. doi: 10.1016/j.cpet.2009.05.006
Clarke R L, Daum S J, Gambino A J, Aceto M D, Pearl J, Levitt M, Cumiskey W R, Bogado E F. Compounds affecting the central nervous system. 4. 3 beta-phenyltropane-2-carboxylic esters and analogs[J]. J. Med. Chem., 1973, 16(11): 1260-1267. doi: 10.1021/jm00269a600
Jin C, Navarro H A, Carroll F I. Synthesis and structure-activity relationship of 3 beta-(4-alkylthio, -methylsulfinyl, and-methylsulfonyl-phenyl) tropane and 3 beta-(4-alkylthiophenyl) nortropane derivatives for monoamine transporters[J]. Biorg. Med. Chem., 2009, 17(14): 5126-5132. doi: 10.1016/j.bmc.2009.05.052
Carroll F I, Gao Y G, Rahman M A, Abraham P, Parham K, Lewin A H, Boja J W, Kuhar M J. Synthesis, ligand binding, QSAR, and CoMFA study of 3 beta-(p-substituted phenyl) tropane-2 beta-carboxylic acid methyl esters[J]. J. Med. Chem., 1991, 34(9): 2719-2725. doi: 10.1021/jm00113a008
Carroll F I. 2002 Medicinal chemistry division award address: Mono-amine transporters and opioid receptors. Targets for addiction therapy[J]. J. Med. Chem., 2003, 46(10): 1775-1794.
Meltzer P C, Liang A Y, Brownell A L, Elmaleh D R, Madras B K. Substituted 3-phenyltropane analogs of cocaine: Synthesis, inhibition of binding at cocaine recognition sites, and positron emission tomography imaging[J]. J. Med. Chem., 1993, 36(7): 855-862. doi: 10.1021/jm00059a010
Riss P J, Stockhofe K, Roesch F. Tropane-derived 11C-labelled and 18F-labelled DAT ligands[J]. J. Labelled Compd. Radiopharm., 2013, 56(3/4): 149-158.
Kerstens V S, Fazio P, Sundgren M, Matheson G J, Franzen E, Halldin C, Cervenka S, Svenningsson P, Varrone A. Reliability of dopamine transporter PET measurements with[18F]FE-PE2I in patients with Parkinson's disease[J]. EJNMMI Res., 2020, 10(1): 95. doi: 10.1186/s13550-020-00676-4
Cao S S, Tang J, Liu C Y, Fang Y, Ji L Y, Xu Y J, Chen Z P. Synthesis and biological evaluation of[18F]FECNT-d4 as a novel PET agent for dopamine transporter imaging[J]. Mol. Imaging Biol., 2021, 23(5): 733-744. doi: 10.1007/s11307-021-01603-2
Fan K L, Zhao H G, Li Y H, Du X X, Dai Y Y, Gao L L, Li Y, Sun Z H, Zhang Y. Characteristics and influencing factors of 11C-CFT PET imaging in patients with early and late onset Parkinson's disease[J]. Front. Neurol., 2023, 14: 1195577. doi: 10.3389/fneur.2023.1195577
Ouchi Y, Kanno T, Okada H, Yoshikawa E, Futatsubashi M, Nobezawa S, Torizuka T, Tanaka K. Changes in dopamine availability in the nigrostriatal and mesocortical dopaminergic systems by gait in Parkinson's disease[J]. Brain, 2001, 124(4): 784-792. doi: 10.1093/brain/124.4.784
Sun X, Liu F, Li Q Y, Gai Y K, Ruan W W, Wimalarathne D N, Hu F, Tan X B, Lan X L. Quantitative research of 11C-CFT and 18F-FDG PET in Parkinson's disease: A pilot study with NeuroQ software[J]. Front. Neurosci., 2019, 13: 299. doi: 10.3389/fnins.2019.00299
Nurmi E, Ruottinen H M, Kaasinen V, Bergman J, Haaparanta M, Solin O, Rinne J O. Progression in Parkinson's disease: A positron emission tomography study with a dopamine transporter ligand[18F] CF[J]. Ann. Neurol., 2000, 47(6): 804-808. doi: 10.1002/1531-8249(200006)47:6<804::AID-ANA14>3.0.CO;2-F
Park E, Hwang Y M, Lee C N, Kim S, Oh S Y, Kim Y C, Choe J G, Park K W. Differential diagnosis of patients with inconclusive Parkinsonian features using[18F]FP-CIT PET/CT[J]. Nucl. Med. Molec. Imag., 2014, 48(2): 106-113. doi: 10.1007/s13139-013-0253-1
Kerstens V S, Fazio P, Sundgren M, Halldin C, Svenningsson P, Varrone A. [18F]FE-PE2I DAT correlates with Parkinson's disease duration, stage, and rigidity/bradykinesia scores: A PET radioligand validation study[J]. EJNMMI Res., 2023, 13(1): 29. doi: 10.1186/s13550-023-00974-7
Fazio P, Svenningsson P, Cselenyi Z, Halldin C, Farde L, Varrone A. Nigrostriatal dopamine transporter availability in early Parkinson's disease[J]. Mov. Disord., 2018, 33(4): 592-599. doi: 10.1002/mds.27316
Mo S J, Axelsson J, Jonasson L, Larsson A, Ogren M J, Ogren M, Varrone A, Eriksson L, Backstrom D, af Bjerken S, Linder J, Riklund K. Dopamine transporter imaging with[18F]FE-PE2I PET and[123I]FP-CIT SPECTa clinical comparison[J]. EJNMMI Res., 2018, 8: 100. doi: 10.1186/s13550-018-0450-0
Marner L, Korsholm K, Anderberg L, Lonsdale M N, Jensen M R, Brodsgaard E, Denholt C L, Gillings N, Law I, Friberg L. [18F]FE-PE2I PET is a feasible alternative to[123I]FP-CIT SPECT for dopamine transporter imaging in clinically uncertain parkinsonism[J]. EJNMMI Res., 2022, 12(1): 56. doi: 10.1186/s13550-022-00930-x
Kerstens V S, Varrone A. Dopamine transporter imaging in neurode-generative movement disorders: PET vs. SPECT[J]. Clin. Transl. Imaging, 2020, 8(5): 349-356. doi: 10.1007/s40336-020-00386-w
Chalon S, Vercouillie J, Payoux P, Deloye J B, Malherbe C, Le Jeune F, Arlicot N, Salabert A S, Guilloteau D, Emond P, Ribeiro M J. The story of the dopamine transporter PET tracer LBT-999:From conception to clinical use[J]. Front. Med., 2019, 6: 90. doi: 10.3389/fmed.2019.00090
Niznik H B, Van Tol H H M. Dopamine receptor genes: New tools for molecular psychiatry[J]. J. Psychiatry Neurosci., 1992, 17(4): 158-180.
Banerjee A, Prante O. Subtype-selective dopamine receptor radioligands for PET imaging: Current status and recent developments[J]. Curr. Med. Chem., 2012, 19(23): 3957-3966. doi: 10.2174/092986712802002518
Halldin C, Stone-Elander S, Farde L, Ehrin E, Fasth K J, Långström B, Sedvall G. Preparation of 11C-labelled SCH 23390 for the in vivo study of dopamine D1 receptors using positron emission tomography[J]. Int. J. Rad. Appl. Instrum. A, 1986, 37(10): 1039-1043. doi: 10.1016/0883-2889(86)90044-4
Halldin C, Foged C, Farde L, Karlsson P, Hansen K, Grønvald F, Swahn C G, Hall H, Sedvall G. [11C]NNC 687 and[11C]NNC 756, dopamine D1 receptor ligands. Preparation, autoradiography and PET investigation in monkey[J]. Nucl. Med. Biol., 1993, 20(8): 945-953. doi: 10.1016/0969-8051(93)90095-C
Abi-Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y Y, Hwang D R, Keilp J, Kochan L, Van Heertum R, Gorman J M, Laruelle M. Prefrontal dopamine D1 receptors and working memory in schizophrenia[J]. J. Neurosci., 2002, 22(9): 3708-3719. doi: 10.1523/JNEUROSCI.22-09-03708.2002
Kosaka J, Takahashi H, Ito H, Takano A, Fujimura Y, Matsumoto R, Nozaki S, Yasuno F, Okubo Y, Kishimoto T, Suhara T. Decreased binding of[11C]NNC112 and[11C]SCH23390 in patients with chronic schizophrenia[J]. Life Sci., 2010, 86(21/22): 814-818.
Abi-Dargham A, Xu X, Thompson J L, Gil R, Kegeles L S, Urban N, Narendran R, Hwang D R, Laruelle M, Slifstein M. Increased prefrontal cortical D1 receptors in drug naive patients with schizophrenia: A PET study with[11C]NNC112[J]. J. Psychopharm., 2012, 26(6): 794-805. doi: 10.1177/0269881111409265
Besret L, Dollé F, Hérard A S, Guillermier M, Demphel S, Hinnen F, Coulon C, Ottaviani M, Bottlaender M, Hantraye P, Kassiou M. Dopamine D1 receptor imaging in the rodent and primate brain using the isoquinoline (+)-[11C]A-69024 and positron emission tomography[J]. J. Pharm. Sci., 2008, 97(7): 2811-2819. doi: 10.1002/jps.21168
Kassiou M, Scheffel U, Ravert H T, Mathews W B, Musachio J L, Lambrecht R M, Dannals R F. [11C]A-69024-A potent and selective non-benzazepine radiotracer for in-vivo studies of dopamine D1 receptors[J]. Nucl. Med. Biol., 1995, 22(2): 221-226. doi: 10.1016/0969-8051(94)00086-Y
Tang C, Tomkins D M, Sanci V, Houle S, Dasilva J N. Chronic ethanol increases binding of dopamine D1 agonist R-[11C]SKF 82957 in vivo in rat brain[J]. Society for Neuroscience Abstracts, 2000, 26(1/2): Abstract No.-290.295.
Dasilva J N, Schwartz R A, Greenwald E R, Lourenco C M, Wilson A A, Houle S. Dopamine D1 agonist R-[11C]SKF 82957: Synthesis and in vivo characterization in rats[J]. Nucl. Med. Biol., 1999, 26(5): 537-542. doi: 10.1016/S0969-8051(99)00015-3
Palner M, Mccormick P, Parkes J, Knudsen G M, Wilson A A. Systemic catechol-O-methyl transferase inhibition enables the D1 agonist radiotracer R-[11C]SKF 82957[J]. Nucl. Med. Biol., 2010, 37(7): 837-843. doi: 10.1016/j.nucmedbio.2010.04.193
Barret O, Zhang L, Alagille D, Constantinescu C C, Sandiego C, Papin C, Sullivan J M, Morley T, Carroll V M, Seibyl J, Chen J, Lee C, Villalobos A, Gray D, Mccarthy T J, Tamagnan G. Dopamine D1 receptor agonist PET tracer development: Assessment in nonhuman primates[J]. J. Nucl. Med., 2021, 62(9): 1307-1313. doi: 10.2967/jnumed.120.256008
Nishi A, Shuto T. Potential for targeting dopamine/DARPP-32 signaling in neuropsychiatric and neurodegenerative disorders[J]. Expert Opin. Ther. Targets., 2017, 21(3): 259-272. doi: 10.1080/14728222.2017.1279149
邓虞娇, 朱华, 杨志, 彭志平, 贾建华. 中枢神经系统临床用PET显像剂的研究进展[J]. 同位素, 2020,33,(4): 250-262. DENG Y J, ZHU H, YANG Z, PENG Z P, JIA J H. Recent progress of PET radio-tracer for clinical use in central nervous system[J]. Isotope, 2020, 33(4): 250-262.
Sikazwe D M N, Li S M, Mardenborough L, Cody V, Roth B L, Ablordeppey S Y. Haloperidol: Towards further understanding of the structural contributions of its pharmacophric elements at D2-like receptors[J]. Bioorg. Med. Chem. Lett., 2004, 14(23): 5739-5742. doi: 10.1016/j.bmcl.2004.09.046
Im D, Inoue A, Fujiwara T, Nakane T, Yamanaka Y, Uemura T, Mori C, Shiimura Y, Kimura K T, Asada H, Nomura N, Tanaka T, Yamashita A, Nango E, Tono K, Kadji F M N, Aoki J, Iwata S, Shimamura T. Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone[J]. Nat. Commun., 2020, 11(1): 6442. doi: 10.1038/s41467-020-20221-0
Wagner H N, Burns H D, Dannals R F, Wong D F, Langstrom B, Duelfer T, Frost J J, Ravert H T, Links J M, Rosenbloom S B, Lukas S E, Kramer A V, Kuhar M J. Imaging dopamine-receptors in the human-brain by positron tomography[J]. Science, 1983, 221(4617): 1264-1266. doi: 10.1126/science.6604315
Welch M J, Katzenellenbogen J A, Mathias C J, Brodack J W, Carlson K E, Chi D Y, Dence C S, Kilbourn M R, Perlmutter J S, Raichle M E, Terpogossian M M. N-(3-[18Ffluoropropyl)-spiperone: The preferred 18F labeled spiperone analog for positron emission tomographic studies of the dopamine receptor[J]. Nucl. Med. Biol., 1988, 15(1): 83-97.
De Paulis T. The discovery of epidepride and its analogs as high-affinity radioligands for imaging extrastriatal dopamine D2 receptors in human brain[J]. Curr. Pharm. Des., 2003, 9(8): 673-696. doi: 10.2174/1381612033391135
Sawle G V, Playford E D, Brooks D J, Quinn N, Frackowiak R S J. Asymmetrical presynaptic and postsynaptic changes in the striatal dopamine projection in dopa naive parkinsonism-diagnostic implications of the D2 receptor status[J]. Brain, 1993, 116: 853-867. doi: 10.1093/brain/116.4.853
Murakami S, Marubayashi N, Fukuda T, Takehara S, Tahara T. Anti-dopaminergic effects of the stereoisomers of N-(1-alkyl-2-pyrrolidinyl) methyl-5-sulfamoylbenzamides and 2, 3-dihydrobenzofuran-7-carboxamides[J]. J. Med. Chem., 1991, 34(1): 261-267. doi: 10.1021/jm00105a041
Mukherjee J, Yang Z Y, Das M K, Brown T. Fluorinated benzamide neuroleptics .3. development of (S)-N-(1-allyl-2-pyrrolidinyl) methyl-5-(3-[18Ffluoropropyl)-2, 3-dimethoxybenzamide as an improved dopamine D2 receptor tracer[J]. Nucl. Med. Biol., 1995, 22(3): 283-296. doi: 10.1016/0969-8051(94)00117-3
Buchsbaum M S, Christian B T, Lehrer D S, Narayanan T K, Shi B, Mantil J, Kemether E, Oakes T R, Mukherjee J. D2/D3 dopamine receptor binding with[F-18] fallypride in thalamus and cortex of patients with schizophrenia[J]. Schizophr. Res., 2006, 85(1): 232-244.
Grunder G, Landvogt C, Vernaleken I, Buchholz H G, Ondracek J, Siessmeier T, Hartter S, Schreckenberger M, Stoeter P, Hiemke C, Rosch F, Wong D F, Bartenstein P. The striatal and extrastriatal D2/D3 receptor-binding profile of clozapine in patients with schizophrenia[J]. Neuropsychopharmacology, 2006, 31(5): 1027-1035. doi: 10.1038/sj.npp.1300931
Fisher B E, Li Q, Nacca A, Salema G J, Song J, Yip J, Hui J S, Jakowec M W, Petzinger G M. Treadmill exercise elevates striatal dopamine D2 receptor binding potential in patients with early Parkinson's disease[J]. Neuroreport, 2013, 24(10): 509-514. doi: 10.1097/WNR.0b013e328361dc13
Mukherjee J, Shi B, Christian B T, Chattopadhyay S, Narayanan T K. 11C-fallypride: Radiosynthesis and preliminary evaluation of a novel dopamine D2/D3 receptor PET radiotracer in non-human primate brain[J]. Biorg. Med. Chem., 2004, 12(1): 95-102. doi: 10.1016/j.bmc.2003.10.020
Mukherjee J, Narayanan T K, Christian B T, Shi B Z, Dunigan K A, Mantil J. In vitro and in vivo evaluation of the binding of the dopamine D2 receptor agonist C-11-(R,S)-5-hydroxy-2-(di-n-propylamino) tetralin in rodents and nonhuman primate[J]. Synapse, 2000, 37(1): 64-70. doi: 10.1002/(SICI)1098-2396(200007)37:1<64::AID-SYN7>3.0.CO;2-F
Narendran R, Slifstein M, Guillin O, Hwang Y Y, Hwang D R, Scher E, Reeder S, Rabiner E, Laruelle M. Dopamine D2/3 receptor agonist positron emission tomography radiotracer[11C]-(+)-PHNO is a D3 receptor preferring agonist in vivo[J]. Synapse, 2006, 60(7): 485-495. doi: 10.1002/syn.20325
Tziortzi A C, Searle G E, Tzimopoulou S, Salinas C, Beaver J D, Jenkinson M, Laruelle M, Rabiner E A, Gunn R N. Imaging dopamine receptors in humans with[11C]-(+)-PHNO: Dissection of D3 signal and anatomy[J]. Neuroimage, 2011, 54(1): 264-277. doi: 10.1016/j.neuroimage.2010.06.044
Zhang A, Zhang Y, Branfman A R, Baldessarini R J, Neumeyer J L. Advances in development of dopaminergic aporphinoids[J]. J. Med. Chem., 2007, 50(2): 171-181. doi: 10.1021/jm060959i
Narendran R, Hwang D R, Slifstein M, Talbot P S, Erritzoe D, Huang Y Y, Cooper T B, Martinez D, Kegeles L S, Abi-Dargham A, Laruelle M. In vivo vulnerability to competition by endogenous dopamine: Comparison of the D2 receptor agonist radiotracer (-)-N-[11C] propyl-norapomorphine ([11] NPA) with the D2 receptor antagonist radiotracer[11C]raclopride[J]. Synapse, 2004, 52(3): 188-208. doi: 10.1002/syn.20013
Joyce J N, Milian M J. Dopamine D3 receptor antagonists as therapeutic agents[J]. Drug Discovery Today, 2005, 10(13): 917-925. doi: 10.1016/S1359-6446(05)03491-4
Rubi B, Ljubicic S, Pournourmohammadi S, Carobbio S, Armanet M, Bartley C, Maechler P. Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion[J]. J. Biol. Chem., 2005, 280(44): 36824-36832. doi: 10.1074/jbc.M505560200
Sokoloff P, Giros B, Martres M P, Bouthenet M L, Schwartz J C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics[J]. Nature, 1990, 347(6289): 146-151. doi: 10.1038/347146a0
Chien E Y T, Liu W, Zhao Q, Katritch V, Han G W, Hanson M A, Shi L, Newman A H, Javitch J A, Cherezov V, Stevens R C. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist[J]. Science, 2010, 330(6007): 1091-1095. doi: 10.1126/science.1197410
Xu J B, Hassanzadeh B, Chu W H, Tu Z D, Jones L A, Luedtke R R, Perlmutter J S, Mintun M A, Mach R H. [3H]4-(dimethylamino)-N-(4-(4-(2-methoxyphenyl) piperazin-1-yl) butyl) benzamide: A selective radioligand for dopamine D3 receptors. Ⅱ. Quantitative analysis of dopamine D3 and D2 receptor density ratio in the caudate-putamen[J]. Synapse, 2010, 64(6): 449-459. doi: 10.1002/syn.20748
Wang Q, Mach R H, Luedtke R R, Reichert D E. Subtype selectivity of dopamine receptor ligands: Insights from structure and ligand-based methods[J]. J. Chem. Inf. Model., 2010, 50(11): 1970-1985. doi: 10.1021/ci1002747
Newman A H, Beuming T, Banala A K, Donthamsett P, Pongetti K, Labounty A, Levy B, Cao J J, Michino M, Luedtke R R, Javitch J A, Shi L. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor[J]. J. Med. Chem., 2012, 55(15): 6689-6699. doi: 10.1021/jm300482h
Tu Z, Li S, Cui J, Xu J, Taylor M, Ho D, Luedtke R R, Mach R H. Synthesis and pharmacological evaluation of fluorine-containing D3 dopamine receptor ligands[J]. J. Med. Chem., 2011, 54(6): 1555-1564. doi: 10.1021/jm101323b
Mach R H, Tu Z D, Xu J B, Li S H, Jones L A, Taylor M, Luedtke R R, Derdeyn C P, Perlmutter J S, Mintun M A. Endogenous dopamine (DA) competes with the binding of a radiolabeled D3 receptor partial agonist in vivo: A positron emission tomography study[J]. Synapse, 2011, 65(8): 724-732. doi: 10.1002/syn.20891
Doot R K, Young A J, Dominguez T L, Ward C G, Li S, Helili Z, Sheffer R, Lee H, Schubert E K, Mach R H, Dubroff J G. Human blocking study to assess selectivity of [18FFTP PET for dopamine D3 receptors[J]. J. Cereb. Blood Flow Metab., 2021, 41(Suppl1): 244-245.
Prante O, Tietze R, Hocke C, Loeber S, Huebner H, Kuwert T, Gmeiner P. Synthesis, radiofluorination, and in vitro evaluation of pyrazolo[1, 5-a] pyridine-based dopamine D4 receptor ligands: Discovery of an inverse agonist radioligand for PET[J]. J. Med. Chem., 2008, 51(6): 1800-1810. doi: 10.1021/jm701375u
Tietze R, Loeber S, Huebner H, Gmeiner P, Kuwert T, Prante O. Discovery of a dopamine D4 selective PET ligand candidate taking advantage of a click chemistry based REM linker[J]. Bioorg. Med. Chem. Lett., 2008, 18(3): 983-988. doi: 10.1016/j.bmcl.2007.12.026
Willmann M, Ermert J, Prante O, Huebner H, Gmeiner P, Neumaier B. Radiosynthesis and evaluation of 18F-labeled dopamine D4-receptor ligands[J]. Nucl. Med. Biol., 2021, 92: 43-52. doi: 10.1016/j.nucmedbio.2020.07.004
于长青, 曾春雨, 杨志伟. 多巴胺D5受体在血压调控中的作用[J]. 中华高血压杂志, 2007,14,(2): 98-100. YU C C, ZENG C Y, YANG Z W. The role of dopamine D5 receptor in blood pressure regulation[J]. Chinese Journal of Hypertension, 2007, 14(2): 98-100.
Giorgioni G, Piergentili A, Ruggieri S, Quaglia W. Dopamine D5 receptors: A challenge to medicinal chemists[J]. Mini-Rev. Med. Chem., 2008, 8(10): 976-995. doi: 10.2174/138955708785740661
乔晋萍, 乔洪文, 武仙英, 邓艾芳, 朱霖. 多巴胺神经系统显像分子探针研究[J]. 生命的化学, 2014,34,(2): 154-165. QIAO J P, QIAO H W, WU X Y, DENG A F, ZHU L. Recent advances in PET tracers for imaging of the dopaminergic system[J]. Chemistry of Life, 2014, 34(2): 154-165.
Kish S J, Robitaille Y, Elawar M, Clark B, Schut L, Ball M J, Young L T, Currier R, Shannak K. Striatal monoamine neurotransmitters and metabolites in dominantly inherited olivopontocerebellar atrophy[J]. Neurology, 1992, 42(8): 1573-1577. doi: 10.1212/WNL.42.8.1573
Dasilva J N, Kilbourn M R, Mangner T J. Synthesis of[11C] tetrabenazine, a vesicular monoamine uptake inhibitor, for PET imaging studies[J]. Appl. Radiat. Isot., 1993, 44(4): 673-676. doi: 10.1016/0969-8043(93)90130-3
Kilbourn M R, Dasilva J N, Frey K A, Koeppe R A, Kuhl D E. In vivo imaging of vesicular monoamine transporters in human brain using[11C] tetrabenazine and positron emission tomography[J]. J. Neurochem., 1993, 60(6): 2315-2318. doi: 10.1111/j.1471-4159.1993.tb03521.x
Dasilva J N, Carey J E, Sherman P S, Pisani T J, Kilbourn M R. Characterization of 11C tetrabenazine as an in-vivo radioligand for the vesicular monoamine transporter[J]. Nucl. Med. Biol., 1994, 21(2): 151-156. doi: 10.1016/0969-8051(94)90003-5
Schwartz D E, Bruderer H, Rieder J, Brossi A. Metabolic studies of tetrabenazine, a psychotropic drug in animals and man[J]. Biochem. Pharmacol., 1966, 15(5): 645-655. doi: 10.1016/0006-2952(66)90031-1
Frey K A, Koeppe R A, Kilbourn M R, Vanderborght T M, Albin R L, Gilman S, Kuhl D E. Presynaptic monoaminergic vesicles in Parkinson's disease and normal aging[J]. Ann. Neurol., 1996, 40(6): 873-884. doi: 10.1002/ana.410400609
Bohnen N I, Albin R L, Koeppe R A, Wernette K A, Kilbourn M R, Minoshima S, Frey K A. Positron emission tomography of monoami-nergic vesicular binding in aging and Parkinson disease[J]. J. Cereb. Blood Flow Metab., 2006, 26(9): 1198-1212. doi: 10.1038/sj.jcbfm.9600276
Johanson C E, Frey K A, Lundahl L H, Keenan P, Lockhart N, Roll J, Galloway G P, Koeppe R A, Kilbourn M R, Robbins T, Schuster C R. Cognitive function and nigrostriatal markers in abstinent metham-phetamine abusers[J]. Psychopharmacology, 2006, 185(3): 327-338. doi: 10.1007/s00213-006-0330-6
Boileau I, Mccluskey T, Tong J, Furukawa Y, Houle S, Kish S J. Rapid recovery of vesicular dopamine levels in methamphetamine users in early abstinence[J]. Neuropsychopharmacology, 2016, 41(4): 1179-1187. doi: 10.1038/npp.2015.267
Kilbourn M R, Koeppe R A. Classics in neuroimaging: Radioligands for the vesicular monoamine transporter 2[J]. ACS Chem. Neurosci., 2019, 10(1): 25-29. doi: 10.1021/acschemneuro.8b00429
Kilbourn M R, Frey K A, Borght T V, Sherman P S. Effects of dopaminergic drug treatments on in vivo radioligand binding to brain vesicular monoamine transporters[J]. Nucl. Med. Biol., 1996, 23(4): 467-471. doi: 10.1016/0969-8051(96)00023-6
Lee C S, Schulzer M, De La Fuente-Fernández R, Mak E, Kuramoto L, Sossi V, Ruth T J, Calne D B, Stoessl A J. Lack of regional selectivity during the progression of parkinson disease: Implications for pathogenesis[J]. Arch. Neurol., 2004, 61(12): 1920-1925.
Alexander P K, Lie Y, Jones G, Sivaratnam C, Bozinvski S, Mulligan R S, Young K, Villemagne V L, Rowe C C. Management impact of imaging brain vesicular monoamine transporter type 2 in clinically uncertain parkinsonian syndrome with 18F-AV133 and PET[J]. J. Nucl. Med., 2017, 58(11): 1815-1820. doi: 10.2967/jnumed.116.189019

图 4 DAT抑制剂GBR129090与二苯甲基类DAT PET显像剂的结构示意图
Figure 4 Structure diagrams of DAT inhibitor GBR129090 and benzhydryl DAT PET radiotracer

图 5 可卡因与3β-苯基托品烷类DAT PET显像剂的结构示意图
Figure 5 Structure diagrams of cocaine and 3β-phenyltropanes DAT PET radiotracers

图 7 基于SCH23390的苯基苯并吖庚因结构骨架的结构示意图
Figure 7 Sructure diagrams of phenylbenzazepine structural framework based on SCH23390
表 1 DAT PET显像剂优缺点比较
Table 1. Comparison of advantages and disadvantages of DAT PET radiotracers
| Radiotracer | Advantage | Disadvantage |
| [18F]GBR13119 | Rapid kinetics, the metabolite not penetrating the blood-brain barrier, low affinity for the norepinephrine transporter | High lipophilicity, high non-specific uptake |
| [11C]cocaine | Be used to study the pharmacological properties related to cocaine | Unstable metabolism, rapid brain clearance, poor selectivity, BBB-penetrating metabolites |
| [11C]CFT | Good in-vivo stability, selective for other monoamine transporters | Slow kinetics, difficultly in reaching dynamic equilibrium |
| [11C]β-CIT | Good metabolic stability | High affinity for other monoamine transporters, the slow kinetics make it difficult to reach dynamic equilibrium |
| [11C]PE2I | High specificity, selective for other monoaminetransporters (above 30), stable brain metabolism | Slow kinetics, BBB-penetrating metabolites, long time to peak concentration |
| [18F]-β-CFT | Good in-vivo stability, selective for other monoamine transporters | Low kinetics, slow radio-uptake ratio, long time to peak concentration (225 min for striatum) |
| [18F]-FP-CIT | High specificity, fast kinetics, no lipophilic metabolites | Poor selectivity for other monoamine transporters, slight in-vivo defluorination |
| [18F]LBT-999 | High selectivity for other monoamine transporters, high specificity, high signal to noise ratio | Slight in-vivo defluorination, high affinity between radioactive metabolite and DAT which causing difficulties in quantification |
| [18F]FECNT | High selectivity for other monoamine transporters, high specificity, high signal to noise ratio | BBB-penetrating metabolites |
| [18F]FE-PE2I | High specificity, fast kinetics, high selectivity for serotonin transporters, fewer brain metabolites | — |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们