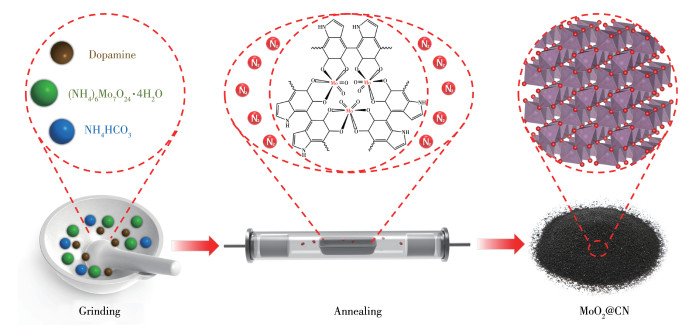
Scheme 1.
Schematic preparation process of MoO2@CN

工业废水中有毒有害有机污染物具有毒性大难降解等特点,严重威胁了生态环境和人体健康。因此,高效处理废水中的有机污染物是环境修复领域研究的热点和难点[1]。基于过一硫酸盐(PMS) 的高级氧化技术由于产生的强氧化性活性氧物种硫酸根自由基(SO4·-)能够彻底分解有机污染物,被认为是环境修复领域中最有前景的技术之一[2-3]。活化PMS是产生SO4·-的关键,目前活化PMS的方法主要有热[4]、紫外光、微波[5]、碱、过渡金属[6]等。其中,过渡金属活化PMS具有反应条件温和、活化速度快等优点,而且无需额外添加能源,是活化PMS理想途径。然而,目前活化PMS主要局限于钴、铁、铜镍、锰等常见的过渡金属,而钼在高级氧化技术中的探索很少。
钼作为人体及动植物所必需的微量元素之一,具有毒性低、化学稳定性好等特点。同时钼极易发生电子转移导致价态改变,价态可从Mo2-递增到Mo6+,因此在催化领域逐渐被关注[7]。在钼基化合物中,MoO2因具有合成方便、电导率高、化学稳定性好等优点逐渐成为高级氧化技术领域的研究热点[8]。Shen等[9]发现商业MoO2可以作为助催化剂促进Fe3+到Fe2+的循环转化,Fe2+/MoO2/H2O2体系可以在1 min内100% 降解罗丹明B(RhB),其降解效率是传统Fenton反应的2倍。Xing等[10]以商用MoO2为助催化剂添加到Fe2+/PMS体系中,大大加快了Fe3+/Fe2+转化速率,10 min内对RhB的去除率可达96.0%,其kobs是Fe2+/PMS体系的50.0倍。尽管MoO2是均相芬顿体系优异的助催化剂,但是由于均相芬顿体系中存在大量铁离子,容易带来二次污染。为此,Chen等[11]使用商用纳米MoO2作为催化剂直接活化PMS,无需铁离子存在,反应180 min后可有效降解萘衍生物。然而,虽然纳米MoO2已被发现能直接活化PMS降解有机污染物,但仍存在催化效率低的问题。因此,如何提高MoO2催化活性是环境催化领域亟待解决的关键难题。
多巴胺是一种富含碳、氮元素的化合物,且含有丰富的邻苯二酚基团、氨基官能团[12]。同时,多巴胺的邻苯二酚基团可与金属配位达到锚定金属的目的[13]。此外,多巴胺可在碱性环境中氧化自聚生成聚多巴胺(PDA),其在煅烧过程中形成的碳材料既可作为导电体,又可作为结构支撑体。它还具有较强的尺寸限制效应,避免了MoO2颗粒的聚集,为制备高活性钼基催化剂提供了可能。为此,我们以四水合钼酸铵、多巴胺、碳酸氢铵为主要原料,经研磨、煅烧制得一种MoO2@氮掺杂碳(MoO2@CN)复合物催化剂,其制备路线图见方案1。研究发现,MoO2@CN/PMS在12 min内对卡马西平(CBZ)的去除率高达99.2%,而且与商用MoO2相比,表观速率常数kobs(0.393 min-1)是商用MoO2(0.016 4 min-1)的24.0倍。将MoO2@CN引入Fe2+/PMS后,其催化降解CBZ的性能显著增强,kobs(1.25 min-1)是单独Fe2+/PMS体系(0.079 7 min-1)的15.7倍。制备的MoO2@CN能够直接高效降解多种有机污染物,拓宽了钼基催化剂在废水处理中的应用。
多巴胺(DA)、四水合钼酸铵((NH4)6Mo7O24· 4H2O)、碳酸氢铵、过一硫酸氢钾、CBZ、5,5-二甲基- 1-氧化吡咯啉氮氧化物(5,5-dimethyl-pyrroline Noxide,DMPO)、2,2,6,6-四甲基-4-哌啶酮(2,2,6,6- tetramethyl-4-oxopiperidine,TEMP)、甲醇(CH3OH,MeOH)、叔丁醇(C4H10O,TBA)均为分析纯,购于上海阿拉丁有限公司。
取0.20 g DA、2.00 g碳酸氢铵、0.74 g四水合钼酸铵置于研钵中,研磨30 min,得砖红色流体状中间物;然后将其放置于管式炉中,在氮气保护下,以2 ℃·min-1的升温速率加热至750 ℃并保温3 h,自然冷却至室温得MoO2@CN。以0.74 g四水合钼酸铵为原料,直接研磨煅烧制得MoOx。
实验在恒温水浴振荡器中进行。首先,将制备好的催化剂加入到40 mL污染物中,然后将锥形瓶置于恒温振荡器中进行反应,间隔一定时间取1 mL溶液,溶液经0.45 μm聚四氟乙烯滤头过滤后注入2 mL液相瓶中,进行高效液相色谱分析(UPLC型高效液相色谱仪,美国Waters公司)。有机染料采用紫外可见光分光光度计(1800型可见分光光度计(UV- 1800),美国尤尼柯上海有限公司)检测,其它污染物的具体检测条件见表S1(Supporting information)。反应初始条件:催化剂量为0.1 g·L-1,反应温度为25 ℃,污染物浓度为20 μmol·L-1,pH为6.5,振荡速度为150 r·min-1,条件可根据测试需求相应变化。实验用1 mol·L-1 HCl或NaOH调节初始溶液pH (6.5~10.5)。
实验中所用仪器:美国Thermo Scientific KAlpha公司K-Alpha型X射线光电子能谱仪(XPS,Al Kα射线,hν=1 486.6 eV);德国布鲁克AXS公司D8 Advance型X射线衍射仪(XRD,扫描速度为5 (°)· min-1,Cu Kα辐射,λ=0.154 06 nm,电压30 kV,电流200 mA,扫描范围10°~80°);司捷克TESCAN MIRA LMS扫描电子显微镜(SEM,铂金靶材,工作电压5 kV);日本电子株式会社JEOL JEM 200PLUS透射电子显微镜(TEM,电压200 kV);日本岛津公司TOC-L型总有机碳(TOC)分析仪;麦克默瑞提克(上海)仪器有限公司麦克ASAP2460型全自动比表面及孔隙度分析仪(200 ℃下真空脱气480 min,77 K液氮温度);德国Bruker公司A300型电子顺磁共振波谱仪(EPR);上海辰华仪器有限公司CHI 660E电化学工作站(银电极作为对电极,Hg/Hg2Cl2(饱和KCl)电极作为参比电极,在40 mL 1 mol·L-1 KOH溶液中进行线性扫描伏安法(LSV)测定,采集电流的扫描速率为20 mV·s-1)。
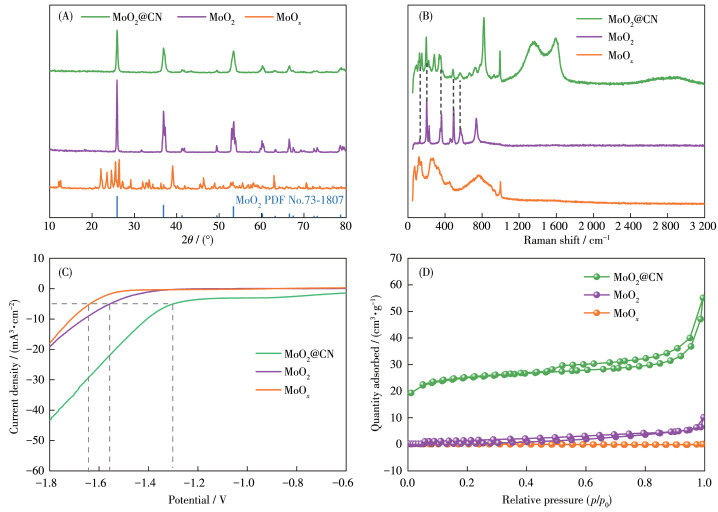
采用SEM对MoO2@CN、MoOx的表面形貌进行分析,结果如图 1所示。由图 1A~1C可见,MoO2@CN的颗粒状结构尺寸较小、分布较均匀,而MoOx呈现一种紧密堆积的光滑片状结构。这是由于制备过程中多巴胺的引入,其上的邻苯二酚基团对金属有很强的配位作用,且聚多巴胺在高温下具有不挥发的特点,这种双重限制策略有效地减少了MoO2团聚现象,提高了其分散性。图 1D显示催化剂MoO2@CN中Mo、C、N、O元素分布较均匀。TEM图(图 1E、1F)显示MoO2分散较均匀,而且在其表面包覆了一层碳。高分辨TEM(HRTEM)图(图 1G)呈现出连续的晶格条纹,其晶格间距为0.344和0.242 nm,分别对应MoO2的(011)和(211)面。上述分析初步表明催化剂是由含N的碳材料与MoO2复合而成。

采用XRD进一步分析MoO2@CN的晶体结构,结果如图 2A所示。由图可见,MoO2@CN在2θ为25.9°、36.9°、53.4°、60.2°和66.6°处有明显的特征衍射峰,这与商用MoO2特征峰基本一致,而MoOx没有MoO2的特征峰。经与MoO2标准卡片(PDF No.73- 1807)对比,这些特征峰分别对应MoO2的(011)、(211)、(311)、(031)、(404)晶面,进一步证实MoO2@CN中MoO2的存在。通过用XRD半峰宽计算得出MoO2@CN、商用MoO2和MoOx的主要粒径分别为26.3、44.1、68.3 nm,这是因为碳的引入限制了MoO2粒径的增长,且破坏了MoO2@CN的结晶度。为了进一步分析MoO2@CN中碳的结构,利用拉曼光谱对MoO2@CN进行表征,结果如图 2B所示。从图可见,与商用MoO2和MoOx相比,MoO2@CN在1 365和1 598 cm-1附近有明显的特征峰,这属于非晶态碳的D带和G带,其中D带是由无序碳拉伸振动造成的,G带属于石墨碳的拉伸振动[14],其D带和G带强度比值(ID/IG)为0.95,这表明MoO2@CN与MoO2相比具有较高的石墨化程度,更有利于催化反应过程中的电子转移,为增强MoO2@CN催化活性提供可能[15]。此外,商用MoO2和MoO2@CN在125、202、361、490、563 cm-1均出现了特征峰,这属于MoO2的拉伸振动[16],进一步证实MoO2@CN中MoO2的存在。为进一步探究MoO2@CN的导电性,采用LSV测试了MoO2@CN、商用MoO2和MoOx的电化学性能,结果如图 2C所示。由图可见,MoO2@CN达到5 mA·cm-2的电流密度时,其对应的过电位仅1.31 V,而商用MoO2和MoOx的过电位为1.53和1.64 V,证实碳、氮的引入提高了MoO2@CN的导电性能,导电性能的提高有利于加快催化反应过程中电子转移速率。MoO2@CN、MoO2和MoOx的N2吸附-脱附等温线如图 2D所示,为典型的Ⅳ型等温线[17],表明MoO2@CN形成了介孔结构[18],平均孔径大小为9.5 nm(图S1)。MoO2@CN的比表面积(70 m2·g-1)与MoOx(0 m2·g-1)和MoO2(2 m2·g-1)相比明显提高,较大的比表面积有利于暴露更多的活性位点[19]。
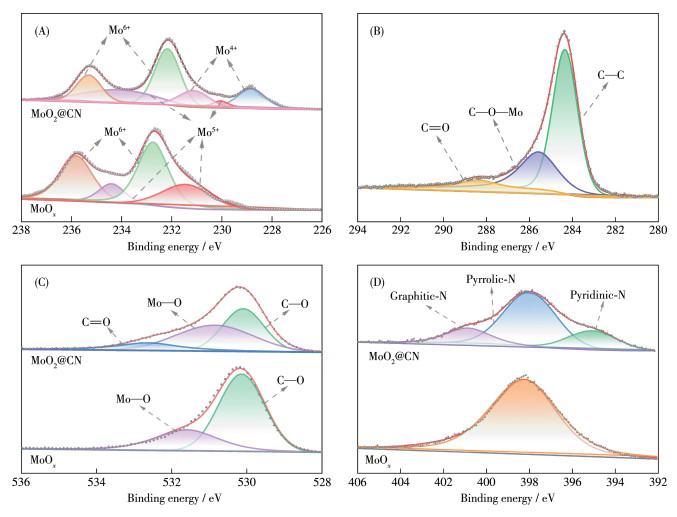
采用XPS对MoO2@CN的化学组成和元素价态进行了分析。图S2是MoO2@CN和MoOx的XPS全谱图,由图可见,与MoOx相比,MoO2@CN中C1s信号峰显著增强,MoOx的全谱图在284.4 eV处的微弱C1s信号来源于测试的碳基体。图 3A为Mo3d窄谱图,由图可知,与MoOx相比,MoO2@CN在228.8和231.1 eV处有新的特征峰,分别属于Mo4+的Mo3d3/2和Mo3d5/2特征峰[20-23],这可能归因于高温煅烧碳热还原将高价Mo5+、Mo6+变为低价Mo4+。C1s的窄谱图(图 3B)在284.3、285.6和288.4 eV处出现了特征峰,分别对应C—C、C—O—Mo和C=O[22],结合O1s窄谱图(图 3C)在530.86 eV的Mo—O特征峰,表明C掺杂MoO2后主要以C—C、C—O—Mo形式存在。N1s的窄谱图(图 3D)显示MoO2@CN在395.1、398.1和400.1 eV处出现了特征峰,分别归属于吡啶N、吡咯N和石墨N的特征峰,说明N已成功掺入碳层,其中吡啶N位点有利于PMS的吸附和活化,结合石墨N的导电特性,这有利于进一步提升催化剂的性能[24-25]。
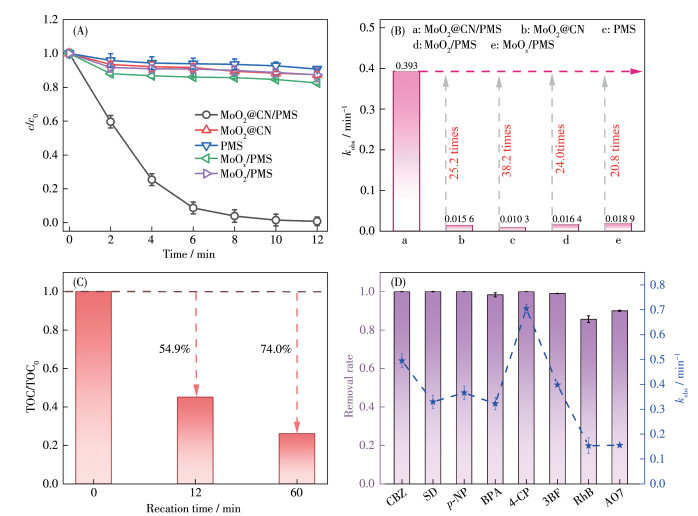
CBZ作为一种治疗癫痫、精神疾病的典型药物,近年来,因其大规模使用以及处理不完全等因素, 对生态环境造成严重危害。因此,选取CBZ为目标污染物,考察了不同体系对其降解性能,结果如图 4A所示。由图可知,反应12 min时,在仅有PMS存在的条件下,CBZ的去除率为9.30%,这是由部分PMS自分解引起的降解;单纯MoO2@CN对CBZ的去除率为11.2%,这归因于MoO2@CN吸附了部分CBZ[26];当MoO2@CN和PMS同时存在时,CBZ的去除率可达99.2%,远远高于MoO2/PMS体系(去除率13.6%)、MoOx/PMS体系(去除率17.3%),而且MoO2@CN/PMS体系kobs高达0.393 min-1,是MoOx/PMS体系(0.018 9 min-1)的20.8倍,MoO2/PMS体系(0.016 4 min-1)的24.0倍(图 4B)。这是因为MoO2@CN与商业MoO2和MoOx相比能暴露更多的活性位点,且具有更高的比表面积和更好的电子传输能力(图 2)。采用TOC分析仪进一步探究MoO2@CN对CBZ的矿化效果,结果如图 4C所示。由图可知,反应12 min时,TOC去除率为54.9%,反应60 min后TOC去除率达到74.0%,这说明MoO2@CN/PMS体系对CBZ具有良好的矿化效果。为了进一步探究MoO2@CN/PMS体系对有机污染物降解的普遍性,探究了其它常见污染物的降解效果(图 4D),在MoO2@CN/PMS体系反应12 min,磺胺嘧啶(SD)、对硝基苯酚(p -NP)、双酚A (BPA)、四氯苯酚(4-CP)及活性红(3BF)的降解率均能达到98.0% 以上,而且对难降解的RhB和酸性橙(AO7) 去除率也可高达85.6% 和90.0%,这表明MoO2@CN/PMS体系能够对大多数有机污染物有一个很好的去除效果。此外,重复性也是衡量催化剂性能的重要指标。如图S3所示,经过4次循环后,CBZ的去除率仍能达到70.0%,去除率降低可能是因为重复过程中催化剂损耗和吸附了部分污染物;与初始MoO2@CN相比,使用过后的MoO2@CN的XRD图变化可以忽略不计(图S4),证实MoO2@CN稳定性较好。
Conditions: cPMS,0=1 mmol·L-1, ccatalyst=0.1 g·L-1, ccontaminant,0=20 μmol·L-1, T=298 K, initial pH=6.5
采用EPR分析催化过程中产生的活性氧物种,结果如图 5A所示。由图可见,与单独的PMS体系相比,MoO2@CN/PMS体系出现了强度比为1∶2∶2∶1的DMPO-·OH(羟基自由基)较强信号峰,而且还出现DMPO-SO4·-特征信号峰[27],这表明MoO2@CN的加入促进了PMS体系中·OH和SO4·-的生成。此外,我们还采用TEMP、DMPO检测体系中是否有单线态氧(1O2)和超氧阴离子自由基(·O2-)生成,结果如图S5所示。结果显示MoO2@CN/PMS出现较微弱的TEMP-1O2、DMPO-·O2-信号峰,但其峰强与单独PMS体系相比基本相同,说明MoO2@CN的加入对生成1O2和·O2-无影响。由此可见,MoO2@CN/PMS体系主要以·OH、SO4·-为主。由于MeOH与SO4·-反应(标准反应速率常数k=1.0×107 mol·L-1·s-1)和·OH反应(k=9.7×108 mol·L-1·s-1)的k均很大,因此MeOH通常被认为是SO4·-和·OH的捕获剂。然而,TBA与·OH反应(k=3.3×109 mol·L-1·s-1)的k远大于TBA与SO4·-反应(k=4.1×105~9.0×105 mol·L-1·s-1),因此TBA可作为·OH的捕获剂[26, 28]。为了进一步考察·OH、SO4·-对催化降解CBZ的贡献,分别采用MeOH和TBA进行捕获实验,结果如图 5B所示。当向体系中分别添加过量TBA(1 mol·L-1)和MeOH(1 mol·L-1)时,CBZ去除率分别下降至50.0% 和8.40%,这证实了·OH和SO4·-是MoO2@CN/PMS体系起主要作用的活性物质。
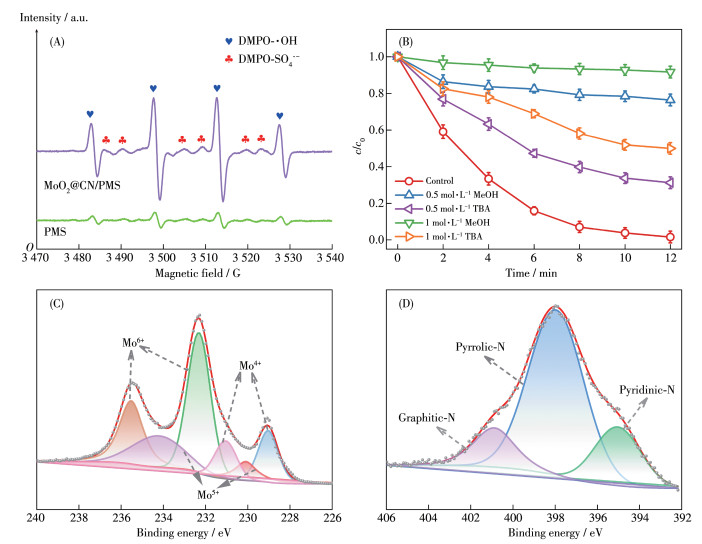
为了进一步分析其催化反应机理,利用XPS对反应后的MoO2@CN进行了测试,其结果如图 5C、5D所示。与反应前相比(表S2),反应后Mo4+的峰面积占比从26.6% 降低至20.9%,而Mo6+的峰面积从48.6%升高至55.5%,这是由于Mo4+与PMS发生反应被氧化成Mo6+ [1]。根据报道可知,乙二胺四乙酸(EDTA)可与钼配位从而失去活性[29-30],因此,为进一步证实Mo的作用,在MoO2@CN/PMS体系中添加EDTA,结果如图S6所示。从图中看出,CBZ的去除率从99.2% 大幅降低至35.1%,这表明Mo在MoO2@CN/PMS体系中起了重要作用。图 5D为反应后N1s窄谱图,与反应前相比,反应后吡啶N的峰面积占比从24.1%下降到了15.0%,而吡咯N和石墨N的比例有所增加,这可能是因为吡啶N在反应过程中与周围的Mo原子发生相互作用。Mo原子通过吡啶N位点传输电子,从而使吡啶N周围的电子密度增加,这更加利于PMS吸附和活化[31]。为了进一步明晰吡啶N位点在MoO2@CN催化剂中的作用,使用磷酸(H3PO4)作为质子化试剂来阻断吡啶N位点[32]。如图S7所示,在MoO2@CN/PMS体系中加入磷酸后,CBZ的降解率下降至65.9%,这说明在MoO2@CN/ PMS体系降解CBZ的过程中吡啶N发挥了重要作用。
基于上述分析,推测MoO2@CN/PMS体系去除CBZ的机理如下:首先,PMS吸附在CNx活性位点上形成亚稳态反应配合物CNx -PMS*,随后,Mo4+与CNx-PMS*发生反应生成SO4·-和·OH,部分SO4·-进一步与水、OH-反应生成·OH,在这个过程中Mo4+被氧化为Mo6+。活化PMS过程如公式1~4所示:
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
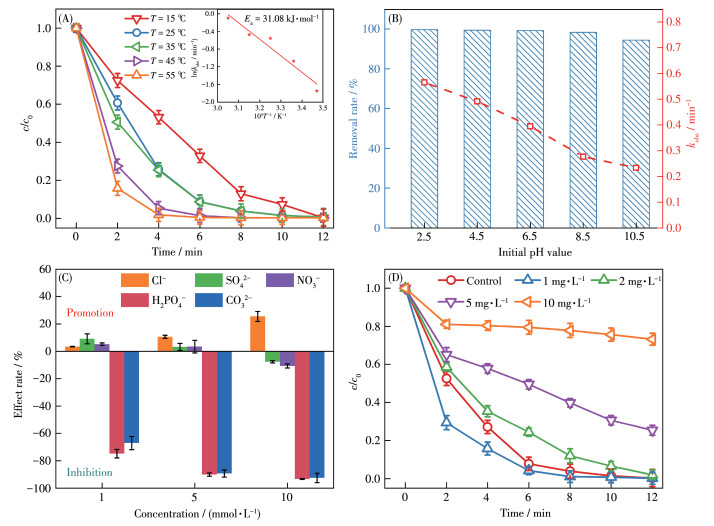
反应温度、初始pH是影响催化反应的重要因素。因此我们首先考察了温度、初始pH对催化剂性能的影响。图 6A为不同反应温度下,CBZ剩余率随时间变化的曲线,可见反应温度从15 ℃梯度上升至55 ℃,CBZ的去除速率随温度的升高而加快。这是因为反应温度越高,分子热运动越快,这增加了污染物与活性物质之间碰撞机率,提高了降解速率。通过Arrhenius方程进一步计算体系的反应活化能(Ea)为31.08 kJ·mol-1。图 6B显示MoO2@CN/ PMS体系初始pH在2.5~10.5之间均能有效去除CBZ,反应体系kobs随着pH升高呈下降趋势,这是因为PMS易与OH-发生反应生成低活性物质;但该体系对CBZ的去除率均能达到95.0% 以上,说明MoO2@CN/PMS体系具有较广的pH适应性,实验选择pH 6.5为CBZ溶解在去离子水中的pH值。此外,我们还研究PMS浓度和催化剂用量对降解CBZ的影响,结果如S8所示,由图可知CBZ的去除速率随着PMS浓度和催化剂用量的增加而加快。综合催化效率和成本考虑,选择PMS浓度1 mmol·L-1、催化剂剂量0.1 g·L-1作为后续探究的实验条件。
Conditions: cPMS, 0=1 mmol·L-1, ccatalyst=0.1 g·L-1, cCBZ, 0=20 μmol·L-1, T=298 K, initial pH=6.5
实际水体中不可避免会存在大量的阴离子和一些天然有机物质,实验首先探究了5种常见阴离子(Cl-、NO3-、SO42-、H2PO42-和CO32-)对去除CBZ的kobs的影响程度(图 6C),添加阴离子后CBZ的去除率见图S9~S13。由图可知,Cl-对降解CBZ有一定的促进作用,这可能是因为Cl-可以与PMS反应生成Cl2,Cl2可进一步与水反应生成具有较高还原电位的HOCl (1.16 V)[33],因此Cl-对MoO2@CN/PMS体系会有明显的促进作用(公式S1~S2)。当NO3-、SO42-以1、5 mmol·L-1存在时,kobs和CBZ的去除率影响均较小,而当其浓度增加为10、20 mmol·L-1时,kobs和CBZ的去除率均降低,这是由于高浓度NO3-与·OH和SO4·-反应生成了低活性的NO3- (公式S3~S4),高浓度SO42-使SO4·-/SO42-的氧化还原电位明显降低。H2PO42-和CO32-极大抑制了CBZ的降解,其kobs和CBZ的去除率均明显降低,这归因于H2PO42-和CO32-与·OH和SO4·-分别反应生成低活性物种H2PO4 ·和CO3·-(公式S5~S8)[34-35]。紧接着以腐殖酸(HA)为代表来探究天然有机物对MoO2@CN/PMS体系降解CBZ的影响。如图 6D所示,HA为1 mg·L-1时,其对CBZ降解速率有一定促进作用,这是由于HA富含羧基、羟基和酚羟基,在一定程度上可以诱导自由基的形成;当HA浓度继续增加,HA与污染物之间的竞争效应逐渐明显,当HA的浓度达到10 mg·L-1时,CBZ的降解率仅有27.8%,降解受到一定程度的抑制。
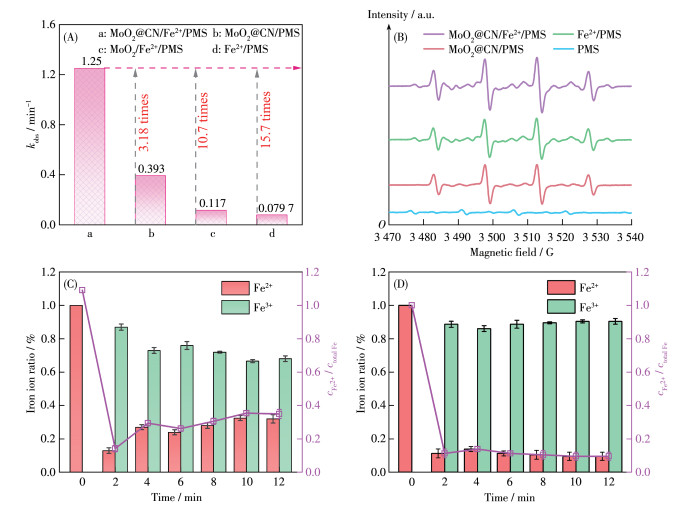
有趣的是,MoO2@CN引入铁离子浓度为5 μmol·L-1的类芬顿Fe2+/PMS体系(图 7A) 时,MoO2@CN/Fe2+/PMS体系降解CBZ的kobs(1.25 min-1) 分别是Fe2+/PMS(0.0797 min-1)、MoO2@CN/PMS (0.393 min-1)体系的15.7倍和3.18倍,而商用MoO2引入Fe2+/PMS体系时kobs(0.117 min-1)仅提高了0.47倍,表明引入MoO2@CN能显著提升Fe2+/PMS体系的催化性能,且MoO2@CN比商用MoO2更具优势。为进一步探讨MoO2@CN增强催化活性的原因,采用EPR测试不同反应体系中的活性物种(图 7B),与MoO2@CN/PMS和Fe2+/PMS体系相比,MoO2@CN/ Fe2+/PMS体系中DMPO-·OH加合物的信号明显增强,这说明MoO2@CN引入Fe2+/PMS体系生成了更多·OH。类芬顿反应体系中Fe3+向Fe2+的转变是影响产生·OH的关键,为此,进一步测试了MoO2@CN/ Fe2+/PMS反应体系中Fe2+和Fe3+浓度变化。结果如图 7C所示,可见溶液中Fe2+在反应开始后迅速被氧化成Fe3+并在2 min内达到最低值;在PMS几乎完全消耗后,Fe3+逐渐被MoO2@CN还原为Fe2+,最终Fe2+浓度保持在一个较高的水平[10]。在Fe2+/PMS体系中,Fe3+被还原为Fe2+的速度极慢,Fe2+浓度始终处于较低值(图 7D)。证实了MoO2@CN加快了Fe2+/PMS体系中Fe3+向Fe2+的转变,促进体系中·OH的生成,达到增强去除有机污染物的目的。
Conditions: cPMS, 0=1 mmol·L-1, cFe, 0=5 μmol·L-1, ccatalyst=0.1 g·L-1, cCBZ, 0=20 μmol·L-1, T=298 K, initial pH=6.5
以四水合钼酸铵、碳酸氢铵为主要原料,引入多巴胺,经研磨、煅烧制得MoO2@氮掺杂碳复合物(MoO2@CN)。表征发现MoO2被碳材料包覆,且复合物中存在吡啶N、吡咯N和石墨N,说明N已成功掺入碳层中,表明MoO2成功和氮掺杂碳材料复合。实验探究了MoO2@CN对废水中有机污染物的去除能力,发现在温度为25 ℃、pH为6.5的条件下,MoO2@CN/PMS可在12 min内降解99.2%的CBZ,且其kobs(0.393 min-1)是商用MoO2(0.016 4 min-1)的24.0倍,这归因于碳的引入可限制MoO2生长,同时提高MoO2@CN的比表面积和电子传输能力。MoO2@CN在较宽的pH范围内(2.5~10.5)均能有效降解CBZ,且对多种有机污染物和染料均具有良好的降解性能。反应60 min后,MoO2@CN/PMS体系中CBZ的TOC去除率高达74.0%。催化机理显示吡啶N在反应过程中与周围的C原子反应构成CNx活性位点,可吸附PMS形成亚稳态反应配合物CNx -PMS*,随后,Mo4+与CNx-PMS*发生反应生成SO4·-和·OH。在Fe2+浓度极低(5 μmol·L-1)的类芬顿Fe2+/PMS体系中引入MoO2@CN,其kobs(1.25 min-1)是单独Fe2+/PMS体系(0.079 7 min-1)的15.7倍,表明MoO2@CN显著增强了类芬顿Fe2+/PMS降解CBZ的性能,这是由于MoO2@CN加快了Fe3+到Fe2+的转变,使Fe2+保持在相对较高的浓度,导致更多·OH生成。制备的碳氮改性MoO2能够直接高效降解有机污染物,为钼基催化剂在废水治理领域的应用提供了新选择。
Zhao Y, An H Z, Feng J, Ren Y M, Ma J. Impact of crystal types of AgFeO2 nanoparticles on the peroxymonosulfate activation in the water[J]. Environ. Sci. Technol., 2019, 53(8): 4500-4510. doi: 10.1021/acs.est.9b00658
Li X N, Huang X, Xi S B, Miao S, Ding J, Cai W Z, Liu S, Yang X L, Yang H B, Gao J J, Wang J H, Hang Y Q, Zhang T, Liu B. Single cobalt atoms anchored on porous N-doped graphene with dual reaction sites for efficient Fenton-like catalysis[J]. J. Am. Chem. Soc., 2018, 140(39): 12469-12475. doi: 10.1021/jacs.8b05992
Su C, Duan X G, Miao J, Zhou Y J, Zhou W, Wang S B, Shao Z P. Mixed conducting perovskite materials as superior catalysts for fast aqueous-phase advanced oxidation: a mechanistic study[J]. ACS Catal., 2017, 7(1): 388-397. doi: 10.1021/acscatal.6b02303
Waldemer R H, Tratnyek P G, Johnson R L, Nurmi J T. Oxidation of chlorinated ethenes by heat-activated persulfate: Kinetics and products[J]. Environ. Sci. Technol., 2007, 41(3): 1010-1015. doi: 10.1021/es062237m
Zhang L, Guo X J, Yan F, Su M M, Li Y. Study of the degradation behaviour of dimethoate under microwave irradiation[J]. J. Hazard. Mater., 2007, 149(3): 675-679. doi: 10.1016/j.jhazmat.2007.04.039
Anipsitakis G P, Dionysiou D D. Degradation of organic contaminants in water with sulfate radicals generated by the conjunction of peroxymonosulfate with cobalt[J]. Environ. Sci. Technol., 2003, 37(20): 4790-4797. doi: 10.1021/es0263792
Smedley P L, Kinniburgh D G. Molybdenum in natural waters: A review of occurrence, distributions and controls[J]. Appl. Geochem., 2017, 84: 387-432. doi: 10.1016/j.apgeochem.2017.05.008
Ji J H, Bao Y, Liu X Y, Zhang J L, Xing M Y. Molybdenum-based heterogeneous catalysts for the control of environmental pollutants[J]. EcoMat, 2021, 3(6): e12155.
Shen B, Dong C C, Ji J H, Xing M Y, Zhang J L. Efficient Fe(Ⅲ)/Fe(Ⅱ) cycling triggered by MoO2 in Fenton reaction for the degradation of dye molecules and the reduction of Cr(Ⅵ)[J]. Chin. Chem. Lett., 2019, 30(12): 2205-2210. doi: 10.1016/j.cclet.2019.09.052
Ji J, Aleisa R M, Duan H, Zhang J L, Yin Y D, Xing M Y. Metallic active sites on MoO2(110) surface to catalyze advanced oxidation processes for efficient pollutant removal[J]. iScience, 2020, 23(2): 100861. doi: 10.1016/j.isci.2020.100861
Chen X W, Vione D, Borch T, Wang J, Gao Y. Nano-MoO2 activates peroxymonosulfate for the degradation of PAH derivatives[J]. Water Res., 2021, 192: 116834. doi: 10.1016/j.watres.2021.116834
Wang Y W, Yu L, Lou X W. Formation of triple-shelled molybdenumpolydopamine hollow spheres and their conversion into MoO2/carbon composite hollow spheres for lithium-ion batteries[J]. Angew. Chem. Int. Ed., 2016, 55(47): 14668-14672. doi: 10.1002/anie.201608410
Zhao C Y, Kong J H, Yang L P, Yao X Y, Phuaa S L, Lu X H. The dopamine-Mo-Ⅵ complexation-assisted large-scale aqueous synthesis of a single-layer MoS2/carbon sandwich structure for ultrafast, long-life lithium-ion batteries[J]. Chem. Commun., 2014, 50(68): 9672-9675. doi: 10.1039/C4CC04099F
Wu C, Chen Z F, Wang M L, Cao X, Zhang Y, Song P, Zhang T Y, Ye X L, Yang Y, Gu W H, Zhou J D, Huang Y Z. Confining tiny MoO2 clusters into reduced graphene oxide for highly efficient low frequency microwave absorption[J]. Small, 2020, 16(30): 2001686. doi: 10.1002/smll.202001686
Zhang H K, Song X, Zhang J, Liu Y D, Zhao H L, Hu J K, Zhao J H. Performance and mechanism of sycamore flock based biochar in removing oxytetracycline hydrochloride[J]. Bioresour. Technol., 2022, 350: 126884. doi: 10.1016/j.biortech.2022.126884
Liu Z, Zhou S F, Xue S J, Guo Z, Li J, Qu K G, Cai W W. Heterointerface-rich Mo2C/MoO2 porous nanorod enables superior alkaline hydrogen evolution[J]. Chem. Eng. J., 2021, 421: 127807. doi: 10.1016/j.cej.2020.127807
王静羚, 董曼茹, 张起程, 林文松, 邢跃. 镂空Bi2MoO6微球的制备及其对氧氟沙星抗生素的降解性能[J]. 无机化学学报, 2020,36,(5): 827-834.
Foo K Y, Hameed B H. Insights into the modeling of adsorption isotherm systems[J]. Chem. Eng. J., 2010, 156(1): 2-10. doi: 10.1016/j.cej.2009.09.013
Huang Y, Gong Q F, Song X N, Feng K, Nie K Q, Zhao F P, Wang Y Y, Zeng M, Zhong J, Li Y G. Mo2C nanoparticles dispersed on hierarchical carbon microflowers for efficient electrocatalytic hydrogen evolution[J]. ACS Nano, 2016, 10(12): 11337-11343. doi: 10.1021/acsnano.6b06580
Yang L, Chen H, Jia F F, Peng W J, Tian X, Xia L, Wu X Y, Song S X. Emerging hexagonal Mo2C nanosheet with (002) facet exposure and Cu incorporation for peroxymonosulfate activation toward antibiotic degradation[J]. ACS Appl. Mater. Interfaces, 2021, 13(12): 14342-14354. doi: 10.1021/acsami.1c03601
邱灵芳, 马梦帆, 刘哲媛, 陈建, 李平, 陈祥树, 喜多英敏, 多树旺. 碳化MoS2/掺硫g-C3N4异质结的合成及其在可见光下催化降解罗丹明B机理[J]. 无机化学学报, 2014,2,7281-7287.
Yao Y, Chen Z A, Yu R H, Chen Q, Zhu J X, Hong X F, Zhou L, Wu J S, Mai L Q. Confining ultrafine MoO2 in a carbon matrix enables hybrid Li ion and Li metal storage[J]. ACS Appl. Mater. Interfaces, 2020, 12(36): 40648-40654. doi: 10.1021/acsami.0c10833
Han X, Gerke C S, Banerjee S, Zubair M, Jiang J J, Bedford N M, Miller E M, Thoi V S. Strategic design of MoO2 nanoparticles supported by carbon nanowires for enhanced electrocatalytic nitrogen reduction[J]. ACS Energy Lett., 2020, 5(10): 3237-3243. doi: 10.1021/acsenergylett.0c01857
Duan X G, Ao Z M, Sun H Q, Indrawirawan S, Wang Y X, Lang J, Liang F L, Zhu Z H, Wang S B. Nitrogen-doped graphene for generation and evolution of reactive radicals by metal-free catalysis[J]. ACS Appl. Mater. Interfaces, 2015, 7(7): 4169-4178. doi: 10.1021/am508416n
Miao J, Geng W, Alvarez P J J, Long M C. 2D N-doped porous carbon derived from polydopamine-coated graphitic carbon nitride for efficient nonradical activation of peroxymonosulfate[J]. Environ. Sci. Technol., 2020, 54(13): 8473-8481. doi: 10.1021/acs.est.0c03207
Wu Z L, Song W K, Xu X W, Yuan J N, Lv W Y, Yao Y Y. High 1T phase and sulfur vacancies in C-MoS2@Fe induced by ascorbic acid for synergistically enhanced contaminants degradation[J]. Sep. Purif. Technol., 2022, 286: 120511. doi: 10.1016/j.seppur.2022.120511
崔金萍, 陈温贤, 郁非繁, 曹诗雨, 吕维扬, 姚玉元. 碳掺杂六方氮化硼/二硫化钼吸附还原六价铬和助催化降解有机污染物[J]. 高等学校化学学报, 2021,42,(10): 3125-3134. CUI J P, CHEN W X, YU F F, CAO S Y, LÜ W Y, YAO Y Y. Adsorption reduction of hexavalent chromium and co-catalytic degradation of organic pollutants by carbon doped hexagonal boron nitride supported MoS2[J]. Chem. J. Chinese Universities, 2021, 42(10): 3125-3134.
Li H C, Shan C, Pan B C. Fe(Ⅲ)-doped g-C3N4 mediated peroxymonosulfate activation for selective degradation of phenolic compounds via high-valent iron-oxo species[J]. Environ. Sci. Technol., 2018, 52(4): 2197-2205. doi: 10.1021/acs.est.7b05563
Kumar D, Singh M, Ramanan A. Crystallization of Mo-EDTA complex based solids: Molecular insights[J]. J. Mol. Struct., 2012, 1030: 89-94. doi: 10.1016/j.molstruc.2012.07.024
Al-Dalama K, Stanislaus A. Temperature programmed reduction of SiO2-Al2O3 supported Ni, Mo and NiMo catalysts prepared with EDTA[J]. Thermochim. Acta, 2011, 520(1/2): 67-74.
Mamtani K, Jain D, Zemlyanov D, Celik G, Luthman J, Renkes G, Co A C, Ozkan U S. Probing the oxygen reduction reaction active sites over nitrogen-doped carbon nanostructures (CNx) in acidic media using phosphate anion[J]. ACS Catal., 2016, 6(10): 7249-7259. doi: 10.1021/acscatal.6b01786
Liang J, Duan X G, Xu X Y, Chen K X, Zhang Y, Zhao L, Qiu H, Wang S B, Cao X D. Persulfate oxidation of sulfamethoxazole by magnetic iron-char composites via nonradical pathways: Fe? versus surface-mediated electron transfer[J]. Environ. Sci. Technol., 2021, 55(14): 10077-10086. doi: 10.1021/acs.est.1c01618
Nikravesh B, Shomalnasab A, Nayyer A, Aghababaei N, Zarebi R, Ghanbari F. UV/Chlorine process for dye degradation in aqueous solution: mechanism, affecting factors and toxicity evaluation for textile wastewater[J]. J. Environ. Chem. Eng., 2020, 8(5): 104244. doi: 10.1016/j.jece.2020.104244
Hu P D, Long M C. Cobalt-catalyzed sulfate radical-based advanced oxidation: a review on heterogeneous catalysts and applications[J]. Appl. Catal. B-Environ., 2016, 181: 103-117. doi: 10.1016/j.apcatb.2015.07.024
Madihi-Bidgoli S, Asadnezhad S, Yaghoot-Nezhad A, Hassani A. Azurobine degradation using Fe2O3@multi-walled carbon nanotube activated peroxymonosulfate (PMS) under UVA-LED irradiation: Performance, mechanism and environmental application[J]. J. Environ. Chem. Eng., 2021, 9(6): 106660. doi: 10.1016/j.jece.2021.106660

图 1 (A) MoOx的SEM图; (B、C) MoO2@CN的SEM图; (D) MoO2@CN对应的Mo、C、N和O的元素分布图; (E~G) MoO2@CN的HRTEM图
Figure 1 (A) SEM images of MoOx; (B, C) SEM images of MoO2@CN; (D) MoO2@CN corresponding element mappings of Mo, C, N, and O; (E-G) HRTEM image of MoO2@CN

图 2 MoO2@CN、MoO2和MoOx的(A) XRD图、(B) 拉曼谱图、(C) LSV极化曲线和(D) N2吸附-脱附等温线
Figure 2 (A) XRD patterns, (B) Raman spectra, (C) LSV polarization curves, and (D) N2 adsorption-desorption isotherms of MoO2@CN, MoO2, and MoOx

图 3 MoO2@CN和MoOx的(A) Mo3d、(B) C1s、(C) O1s和(D) N1s XPS窄谱图
Figure 3 High-resolution XPS spectra of (A) Mo3d, (B) C1s, (C) O1s, and (D) N1s for MoO2@CN and MoOx

图 4 (A) 12 min内CBZ在不同体系中的降解曲线和(B) 对应的kobs; (C) TOC去除率; (D) 多种有机污染物的去除率及相应的kobs
Figure 4 (A) CBZ degradation curves in various systems in 12 min and (B) corresponding kobs; (C) TOC removal rate; (D) Removal rate and corresponding kobs for multiple organic contaminants
Conditions: cPMS,0=1 mmol·L-1, ccatalyst=0.1 g·L-1, ccontaminant,0=20 μmol·L-1, T=298 K, initial pH=6.5

图 5 (A) 不同体系中的DMPO/H2O中的EPR谱图; (B) MeOH、TBA对体系降解CBZ的影响; 反应后MoO2@CN的(C) Mo3d和(D) N1s XPS窄谱图
Figure 5 (A) EPR spectra of DMPO/H2O in different systems; (B) Effect of TBA and MeOH on the CBZ removal rate; High-resolution XPS spectra of (C) Mo3d and (D) N1s after reaction

图 6 (A) 反应温度(插图为反应活化能)、(B) 初始pH、(C) 不同浓度阴离子和(D) HA对MoO2@CN/PMS体系影响
Figure 6 Influence of (A) reaction temperature (Inset: activation energy), (B) initial pH values, different concentrations of (C) organic anions and (D) HA on MoO2@CN/PMS system
Conditions: cPMS, 0=1 mmol·L-1, ccatalyst=0.1 g·L-1, cCBZ, 0=20 μmol·L-1, T=298 K, initial pH=6.5

图 7 (A) 不同体系的kobs; (B) 不同体系在DMPO/H2O中的EPR谱图; (C) MoO2@CN/Fe2+/PMS体系和(D) Fe2+/PMS体系中Fe2+和Fe3+浓度的变化
Figure 7 (A) kobs of different systems; (B) EPR spectra of DMPO/H2O in different systems; Variation of Fe2+ and Fe3+ concentrations in the (C) MoO2@CN/Fe2+/PMS system and (D) Fe2+/PMS system
Conditions: cPMS, 0=1 mmol·L-1, cFe, 0=5 μmol·L-1, ccatalyst=0.1 g·L-1, cCBZ, 0=20 μmol·L-1, T=298 K, initial pH=6.5

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载: