Table 1.
Crystal data and structure refinement for complexes 1⁃4
Citation:
Yong-Lan FENG, Wu-Jiu JIANG, Fu-Xing ZHANG, Dai-Zhi KUANG. Solvothermal Synthesis, Structure, and Fluorescence Properties of Four Organotin Complexes Based on m-Phthaloyl Bis(substituted salicylaldehyde acylhydrazone)[J]. Chinese Journal of Inorganic Chemistry,
2022, 38(6): 1171-1179.
doi:
10.11862/CJIC.2022.105

四个基于间苯二(取代水杨醛酰腙)的有机锡配合物的溶剂热合成、结构和荧光性能
摘要:
采用间苯二(取代水杨醛酰腙)(H4L)与R32SnOH溶剂热反应,或间苯二甲酰肼、3-叔丁基水杨醛和三环己基氢氧化锡一锅溶剂热法反应,合成了4个新的有机锡配合物(SnR2)2L(1~4),其中,H4L=m-Ph(CONH—N=CH(o-OH)PhR1)2;R1=NEt2,R2=Ph(1);R1=3,5-di-tert-butyl=3,5-t-2Bu,R2=Ph(2);R1=3,5-t-2Bu,R2=Cy(3);R1=3-tert-butyl=3-t-Bu,R2=Cy(4)。经元素分析、红外光谱和(1H、13C、119Sn)核磁共振谱表征,并用X射线衍射方法确证配合物1~4的结构。配体H4L的2个取代水杨醛酰腙链向内取向并与锡原子配位形成3个内向E型配合物1~3,取代水杨醛酰腙链向外取向并与锡原子配位形成外向E型配合物4。配合物1、2和4属于三斜晶系P1空间群,配合物3属于单斜晶系P21/c空间群。中心锡与配位原子构成畸形双角三锥构型。配体、配合物-三氯甲烷溶液的荧光性能表明,当具有弱荧光的配体m-Ph(CONH—N=CH(o-OH)PhNEt2)2(H4L1)和无荧光的配体m-Ph(CONH—N=CH(o-OH)Ph(3,5-t-2Bu))2(H4L2)分别与苯基锡、环己基锡配位后,配合物-三氯甲烷溶液发出强荧光。
-
关键词:
- 间苯二(取代水杨醛酰腙)
- / 有机锡配合物
- / 溶剂热合成
- / 晶体结构
- / 荧光性质
English
Solvothermal Synthesis, Structure, and Fluorescence Properties of Four Organotin Complexes Based on m-Phthaloyl Bis(substituted salicylaldehyde acylhydrazone)
Abstract:
Four new organotin complexes based on m-phthaloyl bis(substituted salicylaldehyde acylhydrazone) (H4L), (SnR2)2L (1-4), were synthesized by solvothermal reaction of H4L with R32SnOH, or one-pot solvothermal reaction of m-phthaloyl hydrazide, 3-tert-butyl salicylaldehyde, and tricyclohexyltin hydroxide, where H4L=m-Ph (CONH—N=CH(o-OH)PhR1)2; R1=NEt2, R2=Ph (1); R1=3, 5-di-tert-butyl=3, 5-t-2Bu, R2=Ph (2); R1=3, 5-t-2Bu, R2=Cy (3); R1=3-tert-butyl=3-t-Bu, R2=Cy (4). And they were characterized by elemental analysis, IR, and (1H, 13C, and 119Sn) NMR. The structures of complexes 1-4 were confirmed by X-ray diffraction. Three"inward E-type"complexes 1-3 were formed by the inward orientation of two substituted salicylaldehyde acylhydrazone chains of H4L and coordination with tin atoms. And two substituted salicylaldehyde acylhydrazone chains were oriented outward and coordinated with tin atoms to form an"outward E-type"complex 4. Complexes 1, 2, and 4 belong to the triclinic P1 space group and complex 3 belongs to the monoclinic P21/c space group. The central tin and the coordination atom form a five-coordinate distorted triangular bipyramids configuration. The fluorescence properties of the ligands and the complexes-chloroform solution showed that when free ligand m-Ph(CONH—N=CH(o-OH)PhNEt2)2 (H4L1) with weak fluorescence and ligand m-Ph(CONH—N=CH(o-OH)Ph(3, 5-t-2Bu))2 (H4L2) without fluorescence coordinated with phenyltin or cyclohexyltin, the chloroform solution of the complexes emitted strong fluorescence.
-
0. Introduction
Nucleophilic addition and dehydration of organic aldehydes or ketones with hydrazide result in acylhydrazone compounds containing amide Schiff base (R1C(O) —NH—N=CH(R) —R2). The acylhydrazone compounds not only have good bactericidal[1-2], antitumor[3-4], and optical properties[5-9] but also provide multiple coordination atoms such as oxygen and nitrogen. These —CH=N—, >NH and —CONH— active groups have their respective chemical reactivity and cooperation interaction effect. The lone pair electrons of amino-group (>NH) are p-π conjugated with adjacent carbonyl and double bond imine- groups, and the double bond produces Z-, E-isomerization[10]. Therefore, acylhydrazone compounds have a variety of coordination modes with metals, providing a broad space for the synthesis of complexes with diverse structures and functions[11-13]. Therefore, people are inspired to think that acylhydrazone compounds[14-15] containing bis (poly)-acylhydrazone X[NH—N=CHAr(OH)]2 (X can be carbonyl >C=O/S, aromatic polycarbonyl, and other bridge groups or atoms) have more active groups, more coordination modes than monoacylhydrazone, and are more prone to keto and enol conversion of acylhydrazone chain and deprotonation of amide nitrogen[16]. The coordination of acylhydrazone compounds with metals to assemble complexes with various structures and properties[17] has attracted more interest. In this work, m-phthaloyl bis(substituted salicylaldehyde acylhydrazone) compounds (H4L) were prepared by conden- sation of 4-diethylaminosalicylaldehyde, 3, 5-di-tert-butyl salicylaldehyde and 3-tert-butyl salicylaldehyde with m-phthaloyl hydrazine, respectively. The triphen- yltin hydroxide and tricyclohexyltin hydroxide reacted with H4L respectively to synthesize organotin diacylhy-drazone complexes (SnR2)2L (1-4), where H4L=m-Ph (CONH—N=CH(o-OH)PhR1)2; R1=NEt2, R2=Ph (1); R1=3, 5-di-tert-butyl=3, 5-t-2Bu, R2=Ph (2); R1=3, 5-t-2Bu, R2=Cy (3); R1=3-tert-butyl=3-t-Bu, R2=Cy (4). The fluorescence properties of the complexes were preliminarily evaluated.
1. Experimental
1.1 Material and instruments
FT-IR spectra were recorded on a Bruker TENSOR Ⅱ Fourier infrared spectrometer with the samples prepared as KBr (400-4 000 cm-1) pellets. C, H, and N analyses were carried out with a PE-2400 Ⅱ element analyzer. UV-Vis absorption spectra were recorded on a UV-2500PC spectrophotometer. 1H, 13C, and 119Sn NMR spectra were determined by Bruker Avance 500 NMR (TMS as internal standard and CDCl3 or DMSO-d6 as solvent). Melting points were measured by an X-4 microscopic melting point apparatus made by Beijing Tech Instrument Co., Ltd. and were uncorrected. Fluo-rescence spectra in the solution were recorded on a Hitachi F-7000 spectrometer.
4-Diethylaminosalicylaldehyde(99%) and 3-tert-butyl salicylaldehyde (CP) (Beijing, J&K Chemical Ltd.), m-phthaloyl hydrazide and 3, 5-di-tert-butyl salicylaldehyde (CP) (Shanghai Shaoyuan Co., Ltd.), tricyclohexyltin hydroxide (CP) (Hubei Jusheng Technology Co., Ltd.), triphenyltin hydroxide (CP) (Shanghai Civi Chemical Technology Co., Ltd.) were used without further purification.
1.2 Preparation of the ligands
A mixture of m-phthaloyl hydrazide (3.890 g, 0.020 mol), 4-diethylaminosalicylaldehyde (7.732 g, 0.040 mol), and 60 mL ethanol solution was placed in a flask. The reactant was stirred and refluxed until the solid was dissolved, and the reaction continued for 24 h. Then the mixture was cooled and filtered. The obtained solid was recrystallized and dried in a vacuum to obtain 8.420 g of orange powder of m-phthaloyl bis (4-diethylaminosalicylaldehyde acylhydrazone) (H4L1), with a yield of 75.0%. m. p. 300 ℃. Anal. Calcd. for C30H36N6O4(%): C, 66.16; H, 6.66; N, 15.43. Found(%): C, 66.20; H, 6.68; N, 15.38. FT-IR (KBr, cm-1): 3 448 (m, νO—H); 3 292, 3 239(m, νN—H); 3 072, 2 972, 2 930, 2 900(m, νAr—H and νC—H); 1 648, 1 628(vs, νC=O and νC=N); 1 585, 1 546 (m, benzene νC=C). 1H NMR (500 MHz, DMSO-d6): δ 12.02(s, 2H), 11.47(s, 2H), 8.50-6.15(m, 12H), 3.36(m, 8H), 1.13(s, 12H). 13C NMR (126 MHz, DMSO-d6): δ 162.14, 160.24(C=O); 150.71 (C=N); 133.99, 132.11, 131.01, 129.28, 127.17, 106.89, 104.17, 97.95 (spectral line of benzene carbon); 44.30, 40.48, 40.40, 40.31, 40.23, 40.15, 39.98, 39.81, 39.64, 39.48 (methylene carbon spectral line of ethylamine); 13.00 (methyl carbon of ethylamine).
m-Phthaloyl hydrazide (5.825 g, 0.030 mol), 3, 5-di-tert-butyl salicylaldehyde (14.059 g, 0.060 mol) and 50 mL of ethanol were added into the reaction flask. The reactant was stirred and refluxed for 48 h. Then the mixture was cooled and filter. The precipitate was dried in a vacuum. The yellow powder (8.961 g) of m-phthaloyl bis(3, 5-di-tert-butyl salicylaldehyde acylhydrazone) (H4L2) was obtained, with a yield of 69.1%. m.p. 114 ℃. Anal. Calcd. for C38H50N4O4(%): C, 72.81; H, 8.04; N, 8.94. Found(%): C, 72.85; H, 8.08;N, 8.76. FT-IR(KBr, cm-1): 3 476(m, νO—H); 3 252(m, νN—H); 3 062, 2 960, 2 909, 2 870(m, νAr—H and νC—H); 1 669, 1 646(vs, νC=O and νC=N); 1 622, 1 589 (m, benzene νC=C). 1H NMR (500 MHz, CDCl3): δ 11.91(s, 2H), 8.76 (s, 2H), 7.46-7.01(m, 10H), 1.57-1.20(m, 36H). 13C NMR (126 MHz): δ 165.29(C=O); 156.86(C=N); 141.29, 136.88, 128.24, 126.98, 116.70 (spectral line of benzene carbon); 35.15, 34.21, 31.45, 29.45 (tert-butyl carbon).
1.3 Synthesis of the complexes
H4L1 (0.545 g, 1 mmol), triphenyltin hydroxide (0.734 g, 2 mmol), and 8 mL mixed solvent (3 mL methanol and 5 mL DMF) were mixed and heated to 120 ℃ for 72 h, then the mixture was reduced to room temperature at 1 ℃·h-1 and filtered. Complex 1 was obtained by recrystallization.
Complex 2 was synthesized by replacing H4L1 with H4L2 (0.433 g, 1 mmol). Complex 3 was synthesized by replacing triphenyltin hydroxide with tricyclohexyltin hydroxide and reacting with H4L2.
One-pot solvothermal synthesis of complex 4: m-phthaloyl hydrazide (0.194 g, 1 mmol), 3-tert-butyl salicylaldehyde (0.356 g, 2 mmol), tricyclohexyltin hydroxide (0.771 g, 2 mmol) and 8 mL methanol were added to the polytetrafluoroethylene reactor. Then, following the synthesis steps of complex 1, complex 4 was obtained.
Complex 1, orange crystal, 0.869 g, Yield: 80.0%. m. p. 300 ℃. Anal. Calcd. for C54H52N6O4Sn2(%): C, 59.70; H, 4.82: N, 7.74. Found(%): C, 60.01; H, 4.78: N, 7.71. FT-IR (KBr, cm-1): 3 065, 3 047, 2 971, 2 928, 2 902, 2 809(m, νAr—H and νC—H); 1 607, 1 586(s, νC=O and νC=N); 1 505, 1 479 (m, benzene νC=C); 546(w, νSn—O); 500(w, νSn—N); 451(w, νSn—C). 1H NMR (500 MHz, CDCl3): δ 8.97(s, 2H), 8.57-6.16(m, 10H) 3.40(m, 8H), 1.25(s, 12H). 13C NMR (126 MHz, CDCl3): δ 169.16, 166.57(C=O); 160.05, 153.87(C=N); 140.02, 136.57, 136.40, 136.35, 136.14, 133.99, 130.12, 129.38, 129.00, 128.67, 128.32, 128.02, 126.30, 107.18, 103.99, 100.98 (carbon of benzene); 44.63 (methylene carbon of ethylamino); 12.86 (methyl carbon of ethylamino). 119Sn NMR (SnMe4, 187 MHz, CDCl3): δ -329.62.
Complex 2, orange red crystal, 0.594 g, Yield: 50.8%. m.p. 253-255 ℃. Anal. Calcd. for C62H67N4O4Sn2 (%): C, 63.72; H, 5.69; N, 4.79. Found(%): C, 63.68; H, 5.75; N, 4.75. FT-IR (KBr, cm-1): 3 059, 2 959, 2 906, 2867(s, νAr—H and νC—H); 1 611, 1 591(s, νC=O and νC=N); 1 552, 1 537, 1 509, 1 477, 1 460 (m, benzene νC=C); 694(w, νSn—O); 443(w, νSn—N); 407(w, νSn—C). 1H NMR (500 MHz, CDCl3): δ 9.00, 8.87(s, 2H), 8.75-7.02(m, 30H), 1.53, 1.47, 1.32(m, 36H). 13C NMR (126 MHz, CDCl3): δ 168.46, 165.27, 164.94, 163.11(C=O); 156.86 (C=N); 141.29, 140.58, 139.63, 139.21, 137.95, 137.24, 136.89, 136.46, 136.24, 136.14, 136.02, 133.53, 131.03, 130.35, 130.31, 129.13, 128.96, 128.79, 128.64, 128.44, 128.33, 128.22, 127.02, 126.97, 116.71, 116.29 (carbon of benzene); 35.48, 35.14, 34.19, 34.03, 31.44, 31.29, 30.09, 29.46 (carbon of tert-butyl). 119Sn NMR (SnMe4, 187 MHz, CDCl3): δ -324.04.
Complex 3, orange flake crystal, 0.476 g, Yield: 39.9%. m. p. 278 ℃. Anal. Calcd. for C62H92N4O4Sn2 (%): C, 62.32; H, 7.76; N, 4.69. Found(%): C, 62.45;H, 7.78; N, 4.65. FT-IR (KBr, cm-1): 3 014, 2 950, 2 920, 2 848 (s, νAr—H and νC—H); 1 611, 1 595(s, νC=O and νC=N); 1 548, 1 534, 1 518 (m, benzene νC=C); 525(w, νSn—O); 484(w, νSn—N); 459(w, νSn—C). 1H NMR (500 MHz, CDCl3): δ 9.07-8.63(m, 2H), 8.19-6.81(m, 8H), 1.96-1.30(m, 80H). 13C NMR (126 MHz, CDCl3): δ 168.89, 165.35, 162.47(C=O); 156.86(C=N); 140.07, 137.93, 133.98, 130.28, 129.87, 128.53, 128.02, 126.67, 116.08 (carbon of benzene); 39.94, 35.29, 33.95, 31.35, 31.13, 29.93, 29.91, 29.69, 28.82, 28.55, 28.53, 26.76, 26.61 (carbon of tert-butyl and cyclohexyl). 119Sn NMR (SnMe4, 187 MHz, CDCl3): δ -249.76.
Complex 4, yellow block crystal, 0.26 g, Yield: 24.1%. m. p. 226 ℃. Anal. Calcd. for C54H74N4O4Sn2 (%): C, 60.02; H, 6.90; N, 5.18. Found(%): C, 60.05; H, 6.92: N, 5.11. FT-IR (KBr, cm-1): 3 053, 3 017, 2 996 (s, νAr—H and νC—H); 1 606(s, νC=O and νC=N); 1 547, 1 516, 1 443(m, benzene νC=C); 534(w, νSn—O); 497(w, νSn—N); 455(w, νSn—C). 1H NMR (500 MHz, CDCl3): δ 8.79(m, 2H), 8.19-6.68(m, 10H), 2.13-1.21(m, 62H). 13C NMR (126 MHz, CDCl3): δ 168.46, 165.27, 164.94, 163.11 (C=O); 156.86, 141.29, 140.58, 139.63, 139.21, 137.95, 137.24, 136.89, 136.46, 136.24, 136.14, 136.02, 133.53, 131.03, 130.35, 130.31, 129.13, 128.96, 128.79, 128.64, 128.44, 128.33, 128.22, 127.02, 126.97, 116.71, 116.29 (carbon of benzene); 35.48, 35.14, 34.19, 34.03, 31.44, 31.29, 30.09, 29.46 (carbon of tert-butyl and cyclohexyl). 119Sn NMR (SnMe4, 187 MHz, CDCl3): δ -250.68.
1.4 X-ray data collection and refinement
The suitable crystals of the complexes were obtained by slow volatilization of methanol solvent. The single crystals of the complexes were mounted on a glass capillary of a Bruker SMART APEX Ⅱ CCD X-ray diffractometer. Intensity data for the crystals were measured on a diffractometer with graphite-monochro-matized Mo Kα radiation (λ =0.071 073 nm) by using the φ-ω scan technique and a certain θ range. Multiscan absorption correction was applied to the intensity data using the SAINT program. The structures were solved and refined by OLEX2 software[18] and SHELXS[19] program. All non-hydrogen atoms and their anisotropic thermal parameters were refined to convergence by the full-matrix least square method with the SHELXL program. Complexes 2 and 3 have a disorder of tert-butyl and cyclohexyl groups, respectively. After splitting and restraint, a chemically reasonable structural model and atomic displacement parameters were obtained. All hydrogen atoms were positioned geometrically and refined using a riding model. Details of the crystal data and structure refinement parameters for 1-4 are summarized in Table 1.
Table 1

 下载:
导出CSV
下载:
导出CSV
Parameter 1 2 3 4 Empirical formula C54H52N6O4Sn2 C62H66N4O4Sn2 C62H92N4O4Sn2 C54H74N4O4Sn2 Formula weight 1 086.39 1 168.70 1 194.77 1 080.55 Temperature/K 296.15 296.15 296.15 296.15 Crystal system Triclinic Triclinic Monoclinic Triclinic Space group P1 P1 P21/c P1 a/nm 1.018 44(7) 0.928 60(5) 2.121 75(13) 0.830 4(14) b/nm 1.384 59(9) 1.445 83(8) 1.019 40(6) 1.749(3) c/nm 1.854 86(12) 2.245 08(13) 2.885 28(17) 1.769(3) α/(°) 74.457 0(10) 99.703 0(10) 86.64(3) β/(°) 84.644 0(10) 90.020 0(10) 92.824 0(10) 79.44(2) γ/(°) 89.875 0(10) 91.683 0(10) 78.39(2) V/nm3 2.508 2(3) 2.969 8(3) 6.233 0(6) 2.473(7) Z 2 2 4 2 Dc/(Mg·m-3) 1.438 1.308 1.273 1.451 μ/mm-1 1.046 0.888 0.847 1.059 F(000) 1 100.0 1 198.0 2 496.0 1 116.0 Crystal size/mm 0.23×0.2×0.18 0.14×0.14×0.12 0.22×0.2×0.15 0.15×0.12×0.12 θ range/(°) 2.141-25.749 2.375-25.478 2.217-25.719 2.343-25.098 Limiting indices (h, k, l) -12-12,
-16-16,
-22-22-11-11,
-17-16,
-27-27-25-25,
-12-12,
-35-35-9-9,
-20-20,
-21-21Reflection collected 27 617 32 238 66 746 25 880 Unique reflection (Rint) 9 531 (0.016 3) 10 970 (0.019 0) 11 869 (0.031 6) 8 752 (0.020 8) Completeness to θ/% 99.7 99.6 99.8 99.5 Data, restraint, parameter 9 531, 156, 595 10 970, 1 671, 720 11 869, 2 865, 896 8 752, 24, 559 Goodness-of-fit on F2 1.024 1.049 1.020 1.090 Final R indices [I>2σ(I)] R1=0.039 3,
wR2=0.103 8R1=0.036 3,
wR2=0.089 9R1=0.074 1,
wR2=0.214 4R1=0.037 0,
wR2=0.098 1R indices (all data) R1=0.045 7,
wR2=0.110 6R1=0.045 5,
wR2=0.097 3R1=0.094 4,
wR2=0.238 5R1=0.046 1,
wR2=0.104 8Largest diff. peak and hole/(e·nm-3) 1 220 and -1 560 760 and -970 1 220 and -1 010 1 340 and -740 CCDC: 2111478, 1; 2111479, 2; 2111480, 3; 2111481, 4.
2. Results and discussion
2.1 Crystal structure of complexes
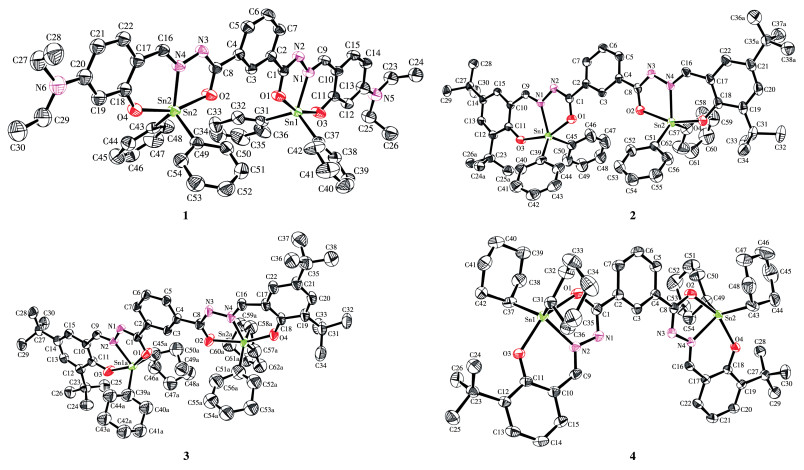
The ligands, m-phthaloyl bis(substituted salicylaldehyde acylhydrazone) with diacylhydrazone chains contain multiple nitrogen and oxygen atoms that can coordinate with metals. Due to the double bond C=N, the arrangement of phenyl and acyl hydrazone chains produces E-, Z-isomerism. In solids, due to hydrogen bonding, it often exists in an E-type configuration[20-21], In addition, the acylhydrazone chain contains a rotatable single bond. The C1—N—N=CPh(OH) and C8—N—N=CPh(OH) chains on the central benzene ring can be oriented inward and outward by C1—C2 or C4—C8 bond rotation. Theoretical calculations show that the coplanar structure of benzene and acylhydrazone chain of ligands H4L1 and H4L2 is assumed as the initial conformation (α =0°) and the C1—C2 bond rotates 360° around space. Other configurations remain unchanged, and the system energy (E) changes with the rotation (α) was explored. The difference between the maximum energy barrier and the lowest energy conformation of the curve was found (ΔE=Emax-Emin) to be 20.15 kJ (H4L1) and 19.78 kJ (H4L2) respectively. It can be seen that the C1—C2 bond of H4L1 and H4L2 can rotate freely, and the two acylhydrazone chains of the ligand can coordinate with the metal in an inward orientation or outward orientation to construct mono-, poly-metal complexes. X-ray crystal structure analysis shows that complexes 1-3 form an E-type internal orientation coordination structure. In complex 4, the C1—C2 and C4—C8 bond rotate, the configuration of the benzoylacylhydrazone chain is reversed, and the ligand atom coordinates with cyclohextin to form an E-type outward orientation structure. The selected bond length and bond angle data are shown in Table 2 and the molecular structure is shown in Fig. 1. Complexes 1, 2, and 4 belong to the triclinic P1 space group and complex 3 belongs to the monoclinic P21/c space group. These two kinds of internally and externally oriented coordination structures are produced by nitrogen, oxygen from ligand with tin, which coordinate with diphenyl (dicyclohexyl) tin to construct five coordinated diphenyl (dicyclohexyl) tin complexes, in which C1—O1 and C8—O2 bonds are 0.130 1(4) and 0.130 4(4) nm, respectively, and the bond length is between the normal carbon-oxygen single bond length (0.143 0 nm) and double bond length (0.122 4 nm)[22], indicating that the carbonyl group becomes enol type. Two phenyl groups(cyclohexyl) extend up and down the ligand "plane" to form ∠C—Sn—C: 119.39(15)° (1), 115.98(15)° (2), 134.7(6)° (3), and 127.97(16)° (4). It can be seen that cyclohexyl has a greater steric effect than phenyl. The bond parameters of the covalent bond composed of central tin and the nitrogen, oxygen, and carbon atoms of the ligand are different. O1 and O3 atoms are in the axial position at the top of the triangular bipyramid, the axial angles (∠O1—Sn1—O3) are 153°-159°, which deviates from the linear structure, and the cyclohexyltin of 3 and 4 deviates 180° more than that of 1 and 2 phenyltin complexes, which further supports the influence of space effect. The bond angle between the equatorial carbon and nitrogen atoms and the axial position, such as ∠O1—Sn1—C(N), varies from 72° to 98°. It can be seen that the central tin and the coordination atom form a distorted triangular bipyramids configuration.
Table 2
Table 2. Selected bond lengths (nm) and bond angles (°) of complexes 1⁃4下载:
导出CSV
Length or angle* 1 2 3 4 Sn1—O1 0.214 2(3) 0.214 4(2) 0.215 6(5) 0.210 4(3) Sn1—O3 0.208 0(3) 0.208 1(2) 0.210 4(5) 0.204 3(4) Sn1—Ny 0.212 4(3) 0.215 4(3) 0.214 7(5) 0.214 6(4) Sn1—Cx1 0.211 7(4) 0.211 8(4) 0.209 0(13) 0.211 7(5) Sn1—Cx2 0.212 4(4) 0.212 7(4) 0.220 6(16) 0.213 7(5) O1—Sn1—O3 158.83(11) 155.03(10) 153.72(19) 153.76(11) O1—Sn1—Ny 73.80(10) 73.18(9) 72.58(17) 71.81(15) O1—Sn1—Cx1 94.76(15) 94.38(13) 93.6(5) 97.50(17) O1—Sn1—Cx2 95.25(14) 94.77(13) 97.2(4) 98.05(18) Ny—Sn1—O3 85.03(11) 83.00(9) 82.29(18) 82.73(14) Ny—Sn1—Cx1 119.63(14) 130.95(12) 118.1(4) 110.30(18) Ny—Sn1—Cx2 120.58(14) 112.30(13) 107.1(5) 121.73(17) O3—Sn1—Cx1 95.98(15) 95.65(13) 91.5(5) 97.18(15) O3—Sn1—Cx2 95.29(15) 101.22(14) 97.4(4) 89.83(17) Cx1—Sn1—Cx2 119.39(15) 115.98(15) 134.7(6) 127.97(16) * x1=31, x2=37, y=1 for 1; x1=39, x2=45, y=1 for 2; x1=39a, x2=45a, y=2, Sn1=Sn1a for 3; x1=31, x2=37, y=2 for 4 Figure 1
 Figure 1. Molecular structures of complexes 1-4 with thermal ellipsoids drawn at the 30% probability level
Figure 1. Molecular structures of complexes 1-4 with thermal ellipsoids drawn at the 30% probability levelFor clarity, H atoms have been omitted, and the disorderly split C atoms (Cb) of 2 and 3 are deleted
Interestingly, for complexes 1-4, there are some interactions intermolecular in crystal stacking. For example, complex 1 has four σ…π weak action of adjacent intermolecular: C23—H23A…C32i, C27—H27B …C8ii, C35—H35…C49iii, and C40—H40…C3iv (Symmetry codes: i 2-x, -y, 1-z; ii 2-x, 2-y, -z; iii x, 1+y, -1+z; iv 1-x, 1-y, -1-z). The distances between the four H…C are 0.286 0, 0.289 5, 0.278 2, and 0.282 6 nm respectively, ∠C—H…C are 146.26°, 127.59°, 153.97°, and 152.96° respectively. A 3D supramolecular structure is formed by these weak interactions.
2.2 Spectral characteristics of the complexes
The infrared spectra of the ligands had four groups of characteristic peaks: (1) 3 400-3 500 cm-1 for phenolic hydroxyl group νO—H and ca. 3 300 cm-1 for amino νN—H stretching vibration peak; (2) C—H vibrational absorption peaks of the benzene ring (ca. 3 000 cm-1) and methyl and methylene (ca. 2 800 cm-1); (3) carbonyl (ca. 1 700 cm-1, νC=O) and imine (ca. 1 600 cm-1, νC=N) characteristic peak; (4) benzene C=C vibration absorption peak of the skeleton. There are three groups of chemical shift signals in the NMR spectrum: (1) phenolic hydroxyl hydrogen and amino hydrogen signals appearing at δ =10-13 in the low field region; (2) δ =6-9 for benzene hydrogen (Ar—H) and methylene hydrogen (HC=N); (3) δ =1-3 for methyl and methylene hydrogen proton signals in high field. After phenyltin or cyclohexyltin is coordinated with the ligands, the characteristic infrared absorption peak of the phenolic hydroxyl group νO—H and amino νN—H, and its NMR proton signal disappeared in the spectra of the complexes, and the weak peaks of O(N)→Sn and C—Sn[23-24] appeared in the low wavenumber region of the infrared spectrum of the complexes, indicating that the dehydrogenated ligand is coordinated with tin. The stretching peaks of the carbonyl group and the imine group of the ligands shifted towards the low wavenumber in the spectra of complexes, due to the carbonyl enol conversion and Schiff base imine coordination to tin weakening carbonyl and imine groups. After the ligands are coordinated to tin, the electron transfer occurs, and the characteristic lines of the 13C NMR spectra of the complexes shift to the low field region and divide into more lines. In the 119Sn NMR spectra, the chemical shifts of the complexes moved to ca. 330 (1, 2) and ca. 250 (3, 4) in the high field relative to SnMe4[25], which further indicates that the ligands coordinate with tin as an electron donor.
2.3 Fluorescence properties of ligands and their complexes
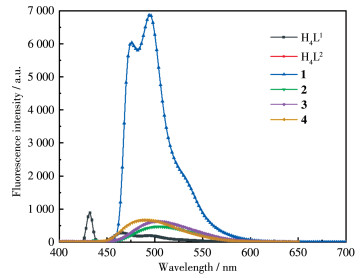
The ligands H4L1, H4L2, and the complexes (1-4)-chloroform solution with 50 µmol·L-1 were prepared. The solution was scanned in 3D on a fluorescence spectrometer and the excitation wavelength of the test solution was determined by referring to the UV spectrum. Then, the fluorescence spectrum of the solution was measured in a range of 400-800 nm at room temperature with 430 nm (H4L1), 290 nm (H4L2), 370 nm (1), 436 nm (2), 330 nm (3), and 330 nm (4) as excitation wavelengths respectively. The results are shown in Fig. 2. The H4L1-chloroform solution emitted weak fluorescence at 466 nm (I=282.2) and 494 nm (I=201.7). When complex 1 was formed by H4L1 and diphenyltin, the 466 nm peak red-shifted to 476 nm and emitted strong yellow fluorescence. The intensity was 6 037, which was 21.4 times higher than that of H4L1; The strong fluorescence of complex-chloroform solution was 34.1 times higher than that of H4L1 solution at 494 nm. No fluorescence was found in the H4L2-chloroform solution in a range of 400-800 nm. When coordinated with diphenyltin or dicyclohexyltin, complexes 2- and 3- chloroform solution produced strong fluorescence at 504 nm, and the fluorescence of solution 3 was 1.3 times that of solution 2. The luminescence property of complex 4 was similar to that of 3, with strong fluorescence at 490 nm. It can be seen that the ligand coordinates with diphenyl (dicyclohexyl)tin to form a planar conjugate rigid structure, which greatly increases the fluorescence intensity of the complex compared with the ligand[26]. Especially when the benzene ring of salicylaldehyde contains color enhancers such as the diethylamine group, the complex produces strong fluorescence and becomes a substance with luminescent properties, which can be further studied as luminescent materials.
Figure 2
 Figure 2. Fluorescence spectra of H4L1, H4L2, and their complexes 1-4 in CHCl3 solution
Figure 2. Fluorescence spectra of H4L1, H4L2, and their complexes 1-4 in CHCl3 solution3. Conclusions
The m-phthaloyl bis(substituted salicylaldehyde hydrazone) was prepared by reaction of substituted salicylaldehyde with m-phthaloyl hydrazine. m-phthaloyl bis(substituted salicylaldehyde hydrazone) tetraphenyl (tetracyclohexyl)ditin complexes were successfully synthesized by methanol solvothermal reaction of ligands with phenyl(cyclohexyl)tin hydroxide. In the preliminary test of the fluorescence properties of 50 µmol·L-1 ligands, the complexes- chloroform solution shows that when the ligands with weak fluorescence (H4L1) and non-fluorescence (H4L2) coordinated with phenyl(cyclohexyl)tin, the chloroform solution of the complexes emitted strong fluorescence, which can be further studied as fluorescent materials.
-
-
[1]
Gao H, Li J Q, Kang P W, Chigan J Z, Wang H, Liu L, Xu Y S, Zhai L, Yang K W. N-acylhydrazones Confer Inhibitory Efficacy against New Delhi Metallo-β-Lactamase-1[J]. Bioorg. Chem., 2021, 114: 105138-105138. doi: 10.1016/j.bioorg.2021.105138
-
[2]
Yao X D, Hu H M, Wang S B, Zhao W H, Song M X, Zhou Q G. Synthesis, Antimicrobial Activity, and Molecular Docking Studies of Aminoguanidine Derivatives Containing an Acylhydrazone Moiety[J]. Iran. J. Pharm. Res., 2021, 2021(2): 536-545.
-
[3]
Wu S T, Liang X, Luo F, Liu H, Shen L Y, Yang X J, Huang Y, Xu H, Wu N, Zhang Q L, Redshaw C. Synthesis, Crystal Structure and Bioactivity of Phenazine-1-Carboxylic Acylhydrazone Derivatives[J]. Molecules, 2021, 26(17): 5320-5320. doi: 10.3390/molecules26175320
-
[4]
Wanderson C S, Lucas D D, Jaqueline E Q, Hérika D A V, Vinícius B S, Andréia M L, Carlos H T P S, Giuliana M V V, Gilberto L B A. Synthesis and In Silico Studies of N-Acylhydrazone Derivatives as hnRNP K Ligands with Potential Anti-cancer Activity[J]. Curr. Bioact. Compd., 2020, 16(4): 432-441. doi: 10.2174/1573407215666190131121059
-
[5]
Liu Y Y, Peng Q C, Li Y Y, Hou H W, Li K. A Simple Strategy for Constructing Acylhydrazone Photochromic System with Visible Color/Emission Change and Its Application in Photo-Patterning[J]. Chin. Chem. Lett., 2020, 31(12): 3271-3275. doi: 10.1016/j.cclet.2020.05.007
-
[6]
Huang M L, Qiu R X, Pan Z H, Tian D, Tao Y W, Lin J Q, Luo G G. Thermally Triggered Isomerization in a Naphthalene-Based Acylhydrazone with Solid-State Optical Nonlinearity Response[J]. Eur. J. Inorg. Chem., 2020, 2020(45): 4313-4317. doi: 10.1002/ejic.202000767
-
[7]
Yang Z Y, Wu D Q, Dai K, Cao S Q, Li Z C, Huang H Y, Shang Z B, Liang H, Yan M H, Xie S Y. A Facile Accessible Acylhydrazone as Al3+ Sensor with Excellent Sensitivity and Selectivity[J]. J. Mol. Struct., 2020, 1201: 1-8.
-
[8]
Feng X S, Li X Z, Hu S J, Yan D N, Zhou L P, Sun Q F. Base- and Metal-Dependent Self-Assembly of Lathanide-Organic Coordination Polymers or Macrocycles with Tetradentate Acylhydrazone-Based Ditopic Ligands[J]. Chem. Asian J., 2021, 16(11): 1392-1397. doi: 10.1002/asia.202100256
-
[9]
Popov L D, Levchenkov S I, Lukov V V, Gishko K B, Borodkin S A, Tupolova Y P, Askalepova O I, Vlasenko V G, Spiridonova D V, Lazarenko D A, Burlov A S, Shcherbakov I N. Acylhydrazone Based on 2-N-Tosylaminobenzaldehyde and Girard T Reagent: Synthesis, Structure, and Coordination Ability[J]. Russ. J. Gen. Chem., 2021, 91(1): 90-97. doi: 10.1134/S1070363221010102
-
[10]
蒲小华. 2-[(E)-(3-氯苯胺基)甲基]-6-溴4-氯苯酚Schiff碱的铜及锌的配合物的合成及晶体结构[J]. 无机化学学报, 2012,28,(10): 2211-2216. PU X H. Synthesis and Crystal Structure of Copper(Ⅱ) and Zinc(Ⅲ) Complexes with Schiff Base 2-((E)-(3-Chlorophenylimino)methyl)-6-bromo-4-chlorophenol[J]. Chinese J. Inorg. Chem., 2012, 28(10): 2211-2216.
-
[11]
Luo W, Meng X G, Cheng G Z, Ji Z P. Synthesis, Characterization and Bioactivity of Two Novel Trinuclear Copper(Ⅱ)/Ni(Ⅱ) Complexes with Pentantate Ligand N-2-Methyl-acryl-salicylhydrazide[J]. Inorg. Chim. Acta, 2009, 362: 551-555. doi: 10.1016/j.ica.2008.05.007
-
[12]
Gupta P, Panda T, Allu S, Borah S, Baishya A, Gunnam A, Nangia A, Naumov P, Nath N K. Crystalline Acylhydrazone Photoswitches with Multiple Mechanical Responses[J]. Cryst. Growth. Des., 2019, 19(5): 3039-3044. doi: 10.1021/acs.cgd.8b01860
-
[13]
Chakraborty S, Bhattacharjee C R, Mondal P, Prasad S K, Rao D S S. Synthesis and Aggregation Behaviour of Luminescent Mesomorphic Zinc(Ⅱ) Complexes with'Salen'Type Asymmetric Schiff Base Ligands[J]. Dalton Trans., 2015, 44(16): 7477-7488. doi: 10.1039/C4DT03989K
-
[14]
Maria P, Georgia K M, Maria V. Photo- and Acid-Degradable Polyacylhydrazone-Doxorubicin Conjugates[J]. Polymers, 2021, 13(15): 2461. doi: 10.3390/polym13152461
-
[15]
Lin Z H, Emge T J, Warmuth R. Multicomponent Assembly of Cavitand-Based Polyacylhydrazone Nanocapsules[J]. Chem. Eur. J., 2011, 17(34): 9395-9405. doi: 10.1002/chem.201100527
-
[16]
Terrence J C, Brian G F, Zheng G H, Kimberly L K, Eckard M, Clifford E F R, Wright L J. High Valent Transition Metal Chemistry.Synthesis and Characterization of an Intermediate-Spin Iron(Ⅳ) Complex of a Strong π-Acid Ligand[J]. J. Am. Chem. Soc., 1992, 114(22): 8724-8725. doi: 10.1021/ja00048a069
-
[17]
Liu J J, Liu X R, Zhao S S, Yang Z W, Yang Z. Syntheses, Crystal Structures, Thermal Stabilities, CT-DNA, and BSA Binding Characteristics of a New Acylhydrazone and Its Co(Ⅱ), Cu(Ⅱ), and Zn(Ⅱ) Complexes[J]. J. Coord. Chem., 2020, 73(7): 1159-1176. doi: 10.1080/00958972.2020.1758316
-
[18]
Dolomanov O V, Bourhis L J, Gildea R J, Howard J A K, Puschmann H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program[J]. J. Appl. Cryst., 2009, 42: 339-341. doi: 10.1107/S0021889808042726
-
[19]
Sheldrick G M. SHELXT-Integrated Space-Group and Crystal-Structure Determination[J]. Acta Crystallogr. Sect. A, 2015, A71: 3-8.
-
[20]
Wu L M, Li Q, Jin L F, Zhou Z Q.. Synthesis and Structure of the Acylhydrazone Schiff Base[J]. Chin. J. Struct. Chem., 2010, 29(9): 1399-1403.
-
[21]
Lin H W. Synthesis and Crystal Structure of Isonicotinic Acid[1-(3, 5- Dibromo-2-hydroxyphenyl)methylidene]hydrazide Methanol[J]. Chin. J. Struct. Chem., 2007, 26(7): 773-776.
-
[22]
Feng Y L, Zhang F X, Kuang D Z, Yang C L. Two Novel Dibutyltin Complexes with Trimers and Hexanuclear Based on the Bis(5-Cl/Mesalicylaldehyde) Carbohydrazide: Syntheses, Structures, Fluorescent Properties and Herbicidal Activity[J]. Chin. J. Struct. Chem., 2020, 39(4): 682-692.
-
[23]
Nath M, Saini P K. Chemistry and Applications of Organotin(Ⅳ) Complexes of Schiff Bases[J]. Dalton Trans., 2011, 40: 7077-7121. doi: 10.1039/c0dt01426e
-
[24]
Yan F F, Ma C L, Li Q L, Zhang S L, Ru J, Cheng S, Zhang R F. Syntheses, Structures and Anti-tumor Activity of Four Organotin(Ⅳ) Dicarboxylates Based on (1, 3, 4-Thiadiazole-2, 5-diyldithio) Diacetic Acid[J]. New J. Chem., 2018, 42: 11601-11609. doi: 10.1039/C8NJ00431E
-
[25]
Jiang W J, Mo T Z, Zhang F X, Kuang D Z, Tan Y X. Syntheses, Crystal Structures and In Vitro Anticancer Activities of Dibenzyltin Compounds Based on the N-(2-Phenylacetic acid)-aroyl Hydrazone[J]. Chin. J. Struct. Chem., 2020, 39(4): 673-681.
-
[26]
冯泳兰, 杨春林, 张复兴, 邝代治. 双(取代水杨醛)缩卡巴肼及其苄基锡配合物的合成、表征、荧光性质和除草活性[J]. 无机化学学报, 2019,35,(10): 1737-1745. doi: 10.11862/CJIC.2019.205FENG Y L, YANG C L, ZHANG F X, KUANG D Z. Synthesis, Characterization, Fluorescence Properties and Herbicidal Activity of Bis (substituted salicylaldehyde) Carbohydrazide and Its Benzyltin Complexes[J]. Chinese J. Inorg. Chem., 2019, 35(10): 1737-1745. doi: 10.11862/CJIC.2019.205
-
[1]
-
Figure 1 Molecular structures of complexes 1-4 with thermal ellipsoids drawn at the 30% probability level
For clarity, H atoms have been omitted, and the disorderly split C atoms (Cb) of 2 and 3 are deleted
Figure 2 Fluorescence spectra of H4L1, H4L2, and their complexes 1-4 in CHCl3 solution
Table 1. Crystal data and structure refinement for complexes 1⁃4
Parameter 1 2 3 4 Empirical formula C54H52N6O4Sn2 C62H66N4O4Sn2 C62H92N4O4Sn2 C54H74N4O4Sn2 Formula weight 1 086.39 1 168.70 1 194.77 1 080.55 Temperature/K 296.15 296.15 296.15 296.15 Crystal system Triclinic Triclinic Monoclinic Triclinic Space group P1 P1 P21/c P1 a/nm 1.018 44(7) 0.928 60(5) 2.121 75(13) 0.830 4(14) b/nm 1.384 59(9) 1.445 83(8) 1.019 40(6) 1.749(3) c/nm 1.854 86(12) 2.245 08(13) 2.885 28(17) 1.769(3) α/(°) 74.457 0(10) 99.703 0(10) 86.64(3) β/(°) 84.644 0(10) 90.020 0(10) 92.824 0(10) 79.44(2) γ/(°) 89.875 0(10) 91.683 0(10) 78.39(2) V/nm3 2.508 2(3) 2.969 8(3) 6.233 0(6) 2.473(7) Z 2 2 4 2 Dc/(Mg·m-3) 1.438 1.308 1.273 1.451 μ/mm-1 1.046 0.888 0.847 1.059 F(000) 1 100.0 1 198.0 2 496.0 1 116.0 Crystal size/mm 0.23×0.2×0.18 0.14×0.14×0.12 0.22×0.2×0.15 0.15×0.12×0.12 θ range/(°) 2.141-25.749 2.375-25.478 2.217-25.719 2.343-25.098 Limiting indices (h, k, l) -12-12,
-16-16,
-22-22-11-11,
-17-16,
-27-27-25-25,
-12-12,
-35-35-9-9,
-20-20,
-21-21Reflection collected 27 617 32 238 66 746 25 880 Unique reflection (Rint) 9 531 (0.016 3) 10 970 (0.019 0) 11 869 (0.031 6) 8 752 (0.020 8) Completeness to θ/% 99.7 99.6 99.8 99.5 Data, restraint, parameter 9 531, 156, 595 10 970, 1 671, 720 11 869, 2 865, 896 8 752, 24, 559 Goodness-of-fit on F2 1.024 1.049 1.020 1.090 Final R indices [I>2σ(I)] R1=0.039 3,
wR2=0.103 8R1=0.036 3,
wR2=0.089 9R1=0.074 1,
wR2=0.214 4R1=0.037 0,
wR2=0.098 1R indices (all data) R1=0.045 7,
wR2=0.110 6R1=0.045 5,
wR2=0.097 3R1=0.094 4,
wR2=0.238 5R1=0.046 1,
wR2=0.104 8Largest diff. peak and hole/(e·nm-3) 1 220 and -1 560 760 and -970 1 220 and -1 010 1 340 and -740  下载: 导出CSV
下载: 导出CSV
Table 2. Selected bond lengths (nm) and bond angles (°) of complexes 1⁃4
Length or angle* 1 2 3 4 Sn1—O1 0.214 2(3) 0.214 4(2) 0.215 6(5) 0.210 4(3) Sn1—O3 0.208 0(3) 0.208 1(2) 0.210 4(5) 0.204 3(4) Sn1—Ny 0.212 4(3) 0.215 4(3) 0.214 7(5) 0.214 6(4) Sn1—Cx1 0.211 7(4) 0.211 8(4) 0.209 0(13) 0.211 7(5) Sn1—Cx2 0.212 4(4) 0.212 7(4) 0.220 6(16) 0.213 7(5) O1—Sn1—O3 158.83(11) 155.03(10) 153.72(19) 153.76(11) O1—Sn1—Ny 73.80(10) 73.18(9) 72.58(17) 71.81(15) O1—Sn1—Cx1 94.76(15) 94.38(13) 93.6(5) 97.50(17) O1—Sn1—Cx2 95.25(14) 94.77(13) 97.2(4) 98.05(18) Ny—Sn1—O3 85.03(11) 83.00(9) 82.29(18) 82.73(14) Ny—Sn1—Cx1 119.63(14) 130.95(12) 118.1(4) 110.30(18) Ny—Sn1—Cx2 120.58(14) 112.30(13) 107.1(5) 121.73(17) O3—Sn1—Cx1 95.98(15) 95.65(13) 91.5(5) 97.18(15) O3—Sn1—Cx2 95.29(15) 101.22(14) 97.4(4) 89.83(17) Cx1—Sn1—Cx2 119.39(15) 115.98(15) 134.7(6) 127.97(16) * x1=31, x2=37, y=1 for 1; x1=39, x2=45, y=1 for 2; x1=39a, x2=45a, y=2, Sn1=Sn1a for 3; x1=31, x2=37, y=2 for 4
下载: 导出CSV
-



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 4337
- HTML全文浏览量: 145
