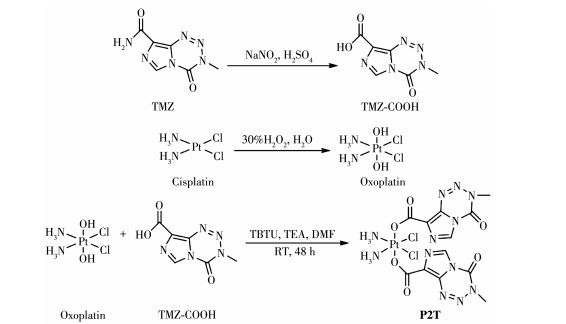
图 1.
配合物P1T的合成路线
Figure 1.
Synthetic route of complex P1T

胶质瘤亦称神经胶质瘤,是源于神经胶质细胞的肿瘤,约占所有颅内肿瘤的60%,具有发病率高、复发率高、死亡率高和治愈率低等“三高一低”的特点,是全身肿瘤中治疗难度最大和预后最差的肿瘤之一[1-6]。替莫唑胺(temozolomide,TMZ)是目前临床一线唯一的神经胶质瘤化疗药物,但存在着骨髓抑制、遗传毒性和生殖毒性等副作用,影响其长期使用[7-8]。TMZ本身没有活性,是一种前体药物,需要在生理pH条件下,经过非酶催化途径先转化为活性化合物5-(3-甲基三嗪-1-基)咪唑-4-酰胺(MTIC),随后进一步水解为活性代谢物5-氨基-咪唑-4-酰胺(AIC)和重氮甲烷[9]。重氮甲烷被认为是烷基化的活性片段,通过对DNA甲基化导致DNA复制的停滞,达到抗肿瘤作用[9]。相关研究也表明TMZ治疗过程中存在耐药性问题,O6-甲基鸟嘌呤-DNA甲基转移酶(MGMT)高表达增加肿瘤细胞对烷化剂类的耐药性,MGMT是一种DNA修复蛋白,能够移除DNA上鸟嘌呤O6位点的细胞毒性的烷基加合物,保护细胞对抗TMZ等烷化剂的损害,减弱TMZ的抗肿瘤效应[10-11]。尽管将TMZ添加到胶质瘤治疗方案中有效将胶质瘤患者中位总生存期延长到15个月,但患者的愈后5年总生存率仍低于10%[12-13]。因此研制新的抗胶质瘤药物迫在眉睫。
近年来以顺铂(cisplatin)为代表的铂类抗肿瘤药物已经成为无机药物领域中最为成功的金属药物之一[14-16]。顺铂主要是以细胞核内的DNA为作用靶标,Pt原子与DNA上的鸟嘌呤等碱基的N7原子进行配位,形成DNA链内或者链间交联产物,通过改变DNA的构象来抑制DNA的复制和转录等活动,最终引发细胞凋亡而达到抗癌的效果[17-20]。尽管金属药物具有巨大的发展潜力且顺铂目前已取得巨大成功,但是他们仍存在一些缺点,包括高发性的耐药性、较强的肾毒性和神经毒性及对靶标DNA的低选择性等[21-23]。在铂类新药开发的相关研究中,科学家逐渐将目光聚焦到了具有动力学惰性但被还原后又能发挥抗癌活性的Pt(Ⅳ)抗肿瘤化合物上。Pt(Ⅳ)配合物采用八面体构型[24],通常认为Pt(Ⅳ)配合物的配位取代缓慢,需被细胞内还原性物质还原成Pt(Ⅱ)配合物后才能与DNA反应,不仅可以改善传统Pt(Ⅱ)药物口服活性低的缺点,且不易与细胞内的水、氯离子、DNA碱基等亲核类物质发生取代反应,改善了传统Pt(Ⅱ)抗癌药物毒副作用和耐药性的缺点[25-26],因此其作为Pt(Ⅱ)配合物的前药被广泛开发。
在本研究中,我们将具有抗胶质瘤活性的TMZ分子引入到顺铂的轴向骨架中,得到2个多功能的Pt(Ⅱ)配合物P1T和P2T。当配合物进入肿瘤细胞后,被细胞内高浓度的含硫分子谷胱甘肽(GSH)等还原为Pt(Ⅱ)化合物和TMZ。Pt(Ⅱ)化合物进入细胞核与DNA结合,从而发挥抗肿瘤活性。TMZ通过对DNA进行甲基化,从而抑制DNA的复制,促进胶质瘤细胞的凋亡,协同增强Pt(Ⅱ)配合物对肿瘤细胞的杀伤作用。通过核磁共振氢谱和碳谱对其进行了基础表征。利用MTT法评估了配合物对多种肿瘤细胞株的抗增殖活性,并通过流式细胞术、免疫荧光、免疫印迹等实验探究了配合物诱导肿瘤细胞死亡作用机制,结果表明配合物协同加剧DNA损伤,最终诱导细胞凋亡。
实验所用溶剂均采用分析纯试剂,根据实验需要进行纯化。除特殊要求,所有反应均在氩气保护下进行。TMZ及顺铂购自安耐吉试剂公司。二甲基亚砜(DMSO)、N,N-二甲基甲酰胺(DMF)等试剂均购于阿拉丁试剂有限公司。磷酸缓冲盐溶液(PBS)、Dulbecco改良的细胞培养基(DMEM)、链霉素和青霉素、胰蛋白消化酶-EDTA、细胞凋亡检测试剂盒、细胞周期检测试剂盒等生物试剂均购于南京凯基生物科技有限公司。
1H NMR和13C NMR使用Bruker Advance Ⅱ400 MHz核磁共振仪检测。UV-Vis数据采用PerkinElmer Lambda 365紫外可见光谱仪进行收集。细胞毒活性通过多功能酶标仪LabServ K3测定。流式细胞仪型号为BD FACSverse。激光共聚焦显微镜为A1 Nikon。采用电感耦合等离子体质谱仪(ICP-MS,X Series 2,Thermo Fisher)测定金属元素Pt在细胞内的含量。在Mini-Protean Tetra System(BIO RAD)上进行蛋白免疫印迹(Western Blot)实验。
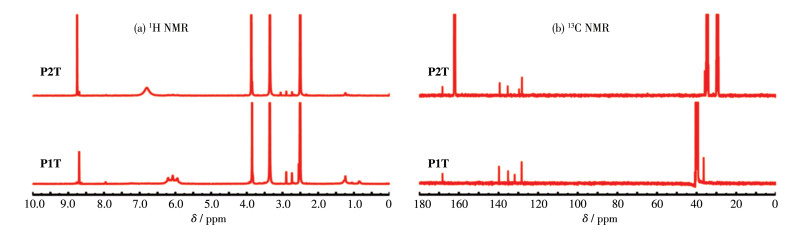
在氩气保护、冰浴条件下,将TMZ(971 mg,5 mmol)与10 mL浓硫酸H2SO4混合,用恒压滴液漏斗缓慢滴加NaNO2(1.062 6 g,15.4 mmol)的水溶液(240 mL),之后在室温下避光反应17 h。待反应结束后,在冰浴条件下将反应管温度降至0 ℃,将反应液转移至冰水混合物中,低温搅拌,有大量白色固体析出。过滤并用蒸馏水清洗几次后,得到较纯的产物TMZ-COOH。产率为67%。1H NMR(400 MHz,DMSO-d6):δ 13.36(s,1H),8.83(s,1H),3.88(s,3H)。
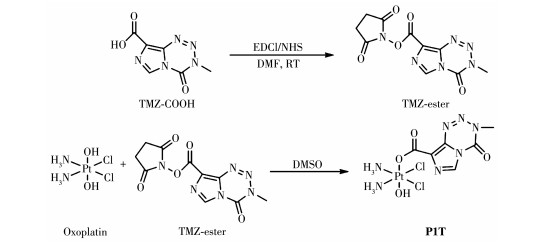
称取1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)(210.9 mg,1.10 mmol)、N-羟基丁二酰亚胺(NHS)(126.6 mg,1.10 mmol)和TMZ-COOH(195.2 mg,1.0 mmol)置于三颈烧瓶中,氮气保护下以无水DMF为溶剂(15 mL)在室温下反应12 h,得到无色溶液。旋转蒸馏溶液除去DMF,加入冰水后析出固体,过滤并用冰水洗涤2次,真空干燥得到配体TMZ-ester,产率约73.8%。1H NMR(400 MHz,DMSO-d6):δ 9.05 (s,1H),3.94(s,3H),2.91(d,J=5.6 Hz,4H)。
按照文献方法合成oxoplatin[27-28]。将过氧化氢(30%,20 mL)逐滴滴加到顺铂(400 mg,1.33 mmol)的水溶液(12 mL)中,60 ℃下回流搅拌4 h。待反应结束后将所得黄色溶液置于室温下冷却过夜,得到浅黄色晶体,过滤并用冰水洗涤,真空干燥得到黄色粉末状产物,产率约67.8%。
取TMZ-ester(105.2 mg,0.32 mmol)于无水DMSO (5 mL)中搅拌,向该溶液中加入oxoplatin(100 mg,0.30 mmol)的DMSO(5 mL)溶液,在氩气保护条件下,室温避光反应72 h。待反应结束后,向溶液中加入乙醚(100 mL)后析出固体,在-20 ℃下静置过夜。去除上层清液,将固体依次用乙醚和乙醇洗2遍后,真空干燥得到配合物P1T。产率约60.8%。1H NMR (400 MHz,DMSO-d6):δ 8.70(s,1H),6.22~5.93(m,6H),3.85(s,3H)。13C NMR(101 MHz,DMSO-d6):δ 168.29,139.79,135.20,131.84,128.33,36.47。元素分析按PtC6H11Cl2N7O4计算的理论值(%):C,14.09;H,2.15;N,19.17。实验值(%):C,14.61;H,2.23;N,19.42。
在氩气保护下,将oxoplatin(100 mg,0.3 mmol)、TMZ-COOH(175.6 mg,0.9 mmol)、三乙胺(TEA) (91 mg,0.9 mmol)和O-苯并三氮唑-N,N,N',N'-四甲基脲四氟硼酸(TBTU)(289 mg,0.9 mmol)溶解于10 mL的无水DMF中,室温下避光搅拌48 h。反应结束后旋蒸除去溶剂,加入乙醇/水(1∶1,V/V)后析出固体,离心后去除上层溶液,重新溶解于5 mL甲醇,再加入20 mL乙醚后析出固体,如此反复3次后,将析出的固体收集,真空干燥得到配合物P2T。产率约30%。1H NMR(400 MHz,DMSO-d6):δ 8.75(s,2H),6.81(s,6H),3.87(s,6H)。13C NMR (101 MHz,DMF-d7):δ 168.29,139.42,135.38,129.68,128.17,35.91。元素分析按PtC12H14Cl2N12O6计算的理论值(%):C,20.92;H,2.03;N,24.41。实验值(%):C,21.02;H,1.84;N,23.88。
通过摇瓶法测试化合物的油水分配系数。分别将P1T、P2T、TMZ溶于水相与有机相的混合溶液中,并采用紫外可见光谱进行定量分析。将水和正辛醇饱和2 d后分液,将化合物溶解于水相中,再向溶液中加入等体积正辛醇,置于摇床上匀速振动4 h,将混合相离心10 min(7 000 r·min-1)后分离有机相及水相,用紫外可见光谱分别检测两相中的吸光度,通过化合物在有机相和水相中的吸光度的比值求对数计算得出油水分配系数lg Po/w。
将配合物P1T与P2T及前体TMZ溶解在含有体积分数1% 的DMSO的PBS溶液中,浓度为50 μmol·L-1,置于1 cm光路路径的石英比色皿中,37 ℃下保持6 h,通过紫外可见光谱测定配合物的吸光度,间隔时间为30 min。将吸光度随时间变化的图拟合为适当的等式(A=C0+C1e-kt,其中C0和C1是计算机拟合的常数,A是配合物在时间t时的吸光度),用于拟合一级反应动力学,以计算半衰期t1/2。
原料TMZ、配合物P1T与P2T及其对照药物顺铂的细胞毒性实验采用MTT法测定,选择人脑星形胶质母细胞瘤细胞(U87MG)、小鼠胶质母细胞瘤细胞(A261)、人非小细胞肺癌细胞(A549)和正常小鼠海马神经元细胞株(HT-22)进行测试。细胞在含有10% FBS(胎牛血清)、100 μg·mL-1链霉素和100 μg· mL-1青霉素(Gibco BRL)的DMEM培养基(Gibco BRL) 中培养。在96孔细胞培养板中接种(每孔5 000个)对数生长期的细胞,将细胞在CO2(体积分数5%)、37 ℃条件下培养过夜以确保细胞贴壁正常生长,加入不同浓度的待测药物(培养基中含体积分数1% 的DMSO溶液)继续孵育48 h。然后每孔加入20 μL的浓度为5 mg·mL-1的MTT溶液,继续于培养箱中孵育4 h。吸出培养液,在每个孔中加入150 μL的DMSO溶液,振荡2 min后,使用LabServ K3酶标仪读取570 nm处吸光度值,使用SPSS软件计算半数抑制浓度IC50值。
将A261细胞接种到100 mm培养皿中,培养18 h后,加入含有20 μmol·L-1药物(顺铂、PIT和P2T) 的DMEM培养液继续培养12 h。结束之后,去除上层培养液,收集细胞并用PBS(4 ℃)洗涤2次。然后进行消化,每个样品中依次用浓硝酸(200 μL、95 ℃) 消化2 h、过氧化氢(100 μL、95 ℃)消化1 h和浓盐酸(200 μL、95 ℃)消化2 h。冷却后用超纯水将所得液体稀释至2 mL,通过ICP-MS测定样品中金属Pt的含量,再用各个样品中的细胞数对细胞内Pt含量进行归一化。
将处于对数生长期的A261细胞以每孔2×104个的初始密度接种到6孔细胞培养板中,培养24 h后,加入不同浓度的待测药物(含有体积分数1% 的DMSO的DMEM培养基溶液)。继续孵育24 h后,消化并收集细胞。用PBS洗涤2次后加入300 μL PBS重悬细胞,然后向其中缓慢滴加700 μL冰乙醇,过夜以固定细胞。固定结束后,5 000 r·min-1离心5 min,PBS洗涤2次,加入提前配好的碘化丙啶染色液(PI/RNase A,9∶1,V/V),室温下避光静置30 min,随后立即使用BD FACSverse流式细胞仪检测,激发波长为488 nm,发射波长为620 nm,使用FlowJo 7.6软件处理数据。
将处于对数生长期的A261细胞以每孔2×104个的初始密度接种到6孔细胞培养板中,培养24 h后,加入不同浓度的待测配合物(含有体积分数1% 的DMSO的DMEM培养基溶液)。继续孵育24 h后,收集细胞,用DMEM培养基洗涤2次,并向其中加入10 μmol·L-1活性氧探针DCFH-DA(2',7'-dichloroflu-orescein diacetate),避光继续孵育30 min。PBS洗涤2次后收集细胞并用PBS重悬细胞。随后立即使用BD FACSverse流式细胞仪检测,激发波长为488 nm,发射波长为510~540 nm,使用FlowJo 7.6软件处理数据。
将处于对数生长期的A261细胞以每孔2×104个的初始密度接种到6孔细胞培养板中,培养24 h后,加入不同浓度的待测配合物(含有体积分数1% 的DMSO的DMEM培养基溶液)。继续孵育48 h后,收集细胞,用PBS洗涤2次后用500 μL的Binding buf-fer重悬细胞。随后加入5 μL的Annexin V-FITC混匀,避光孵育15 min后,加入5 μL的PI,避光继续孵育15 min,使用BD FACSverse流式细胞仪对样品进行检测。FITC的激发波长为488 nm,发射波长为500~560 nm。PI的激发波长为488 nm,发射波长为620 nm。使用FlowJo 7.6软件处理数据。
在共聚焦小皿中种入A261细胞,培养箱中孵化24 h后去除培养液,代之以含有不同浓度的待测药物的DMEM培养基溶液(含有体积分数1% 的DMSO)。用培养液继续培养48 h。去除上层清液,用PBS洗2遍后立即用4% 的多聚甲醛固定15 min,PBS洗3遍,以0.3% Triton X-100(PBS配制)室温通透20 min。PBS洗3遍后吸干PBS,加入5% 的牛血清蛋白(BSA)溶液(其中含有0.3% Triton X-100),室温封闭30 min。吸干封闭液,加入一抗稀释液,4 ℃过夜。去除一抗稀释液,磷酸盐吐温缓冲液(PBST)洗3遍,加入稀释好的FITC荧光二抗,37 ℃避光孵育1 h,PBST洗3遍。滴加Hoechst 33342稀释液避光孵育5 min,PBST洗去残留液体,用共聚焦显微镜观察并采集图像。
细胞中全蛋白通过RIPA裂解液提取,于冰上裂解30 min后,高速离心(12 000 r·min-1,20 min),取上层清液通过BCA法测定蛋白浓度。通过加水和5×Loading buffer把各个样品的蛋白浓度稀释成4 μg·μL-1,95 ℃孵育10 min,获得蛋白样品。在电泳缓冲液中进行电泳实验,每孔中加入20 μL各蛋白样品。电泳结束后,把蛋白转于聚偏二氟乙烯(poly-vinylidene fluoride,PVDF)膜上,将载有蛋白的PVDF膜置于含5% 脱脂牛奶的1×TBST溶液中封闭1 h。然后用对应一抗稀释液孵育目标条带的PVDF膜,在4 ℃下过夜。第二天,用1×TBST缓冲液洗3次。继续在室温下用相应的二抗进行孵育1 h,1×TBST洗3次后,化学发光仪曝光成像。
将前体TMZ依据文献报道方法进行化学修饰得到TMZ-COOH(3-methyl-4-oxo-3,4-dihydroimidazo [5, 1-d][1, 2, 3, 5]tetrazine-8-carboxylic acid)和TMZ-ester (2,5-dioxopyrrolidin-1-yl-3-methyl-4-oxo-3,4-dihydro-imidazo[5,1-d][1, 2, 3, 5]tetrazine-8-carboxylate) [29]。Pt前体用过氧化氢氧化,随后在Pt中心的轴向位置引入TMZ-ester或TMZ-COOH配体得到最终产物P1T或P2T。合成路径如图 1、2所示。通过1H NMR和13C NMR对配体TMZ-COOH、TMZ-ester及配合物P1T和P2T进行了表征(图 3)。
化合物的亲脂性是衡量细胞摄取量的重要指标[30]。研究表明,化合物的亲脂性影响药物穿过细胞膜的能力,进而影响药物在细胞内的累积以及药效[31]。因此,评价药物的亲脂性是研究药物药理活性的基础。我们采用摇瓶法测定了配合物P1T和P2T、配体TMZ的脂溶性,即药物在正辛醇和水中的油水分配系数(lg Po/w),结果分别为-1.26±0.12、-1.30±0.08、-1.02±0.07,而顺铂的数值为-2.30[32],因此,通过对顺铂氧化修饰TMZ能够显著提高其脂溶性,进而可能提高其细胞摄取能力。
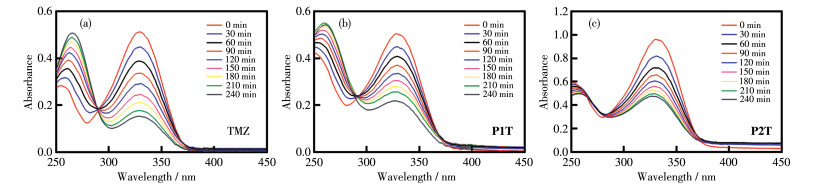
在生理条件下,TMZ可以很快转化为MITC,而MITC随后水解为甲基重氮盐,重氮甲烷被认为具有抗肿瘤作用[9]。因此我们利用紫外可见光谱研究配合物P1T和P2T及配体TMZ在PBS(含有1% DMSO) 中的水解速率。如图 4所示,TMZ的特征吸收峰为255和330 nm,配合物P1T和P2T也表现出类似的特征吸收峰。在240 min内,TMZ在255和330 nm处的吸收峰发生明显改变,在255 nm处的吸收峰减弱,在330 nm处的吸收峰增加,表明TMZ发生了水解。配合物P1T和P2T也表现出类似的现象,表明2个配合物也发生了水解。利用一级反应动力学方程计算出TMZ水解的t1/2为139 min,配合物P1T和P2T水解的t1/2分别为167和123 min,表明配合物进入细胞后能够在较快时间内水解,以发挥抗肿瘤作用。
通过MTT法研究了TMZ以及配合物P1T和P2T对U87MG、A261、A549和HT-22的48 h体外细胞毒性,并以顺铂作为阳性对照药物。从表 1可以看出,配体TMZ对胶质瘤细胞株和正常细胞毒性较小(IC50 > 1 000 μmol·L-1),远低于顺铂的毒性。与TMZ相比,配合物P1T和P2T对胶质瘤细胞株表现出与顺铂相当的细胞毒活性,其对A261细胞的IC50值分别为21.08和23.96 μmol·L-1,同时对正常神经细胞毒性较小,对HT-22细胞的IC50值分别为85.19和55.86 μmol·L-1,表明引入TMZ的四价铂配合物不仅对肿瘤细胞具有较好的细胞毒性,而且对正常细胞具有较低的毒副作用。为了探究配合物P1T和P2T对多种细胞株的作用机理,我们选用配合物细胞毒性最优的A261细胞深入研究配合物对细胞周 期的影响、诱导细胞死亡的方式以及相关分子机制。
 下载:
导出CSV
下载:
导出CSV
| Drug | A261 | U87MG | A549 | HT-22 |
| P1T | 21.08±0.85 | 49.19±2.04 | 45.57±1.30 | 85.19±2.76 |
| P2T | 23.96±0.01 | 35.61±0.61 | 34.76±0.24 | 55.86±0.91 |
| Cisplatin | 12.93±0.21 | 24.41±0.47 | 9.77±0.15 | 16.79±0.62 |
| TMZ | 1 372.09±185.72 | 1 465.16±65.47 | 1 573.48±98.29 | > 2 000 |
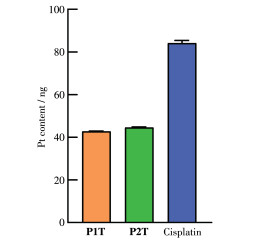
为了进一步探索配合物P1T和P2T进入细胞的能力,我们利用ICP-MS检测分析了经药物刺激后A261细胞内的金属Pt含量。如图 5所示,P1T、P2T和顺铂刺激后106个A261细胞内的Pt含量分别为42.79、44.61和84.10 ng。摄取数据的结果与上述检测的配合物的脂溶性的结果似乎不一致,说明药物的亲脂性并不是细胞摄取的唯一决定因素,其他因素如分子大小和电性结构等也会对细胞摄取产生影响。由于顺铂进入细胞内Pt摄取量最高,顺铂的细胞毒活性相比于P1T和P2T更优,但对正常细胞也表现出极高的细胞毒性,而P1T和P2T表现出较好的细胞选择性。
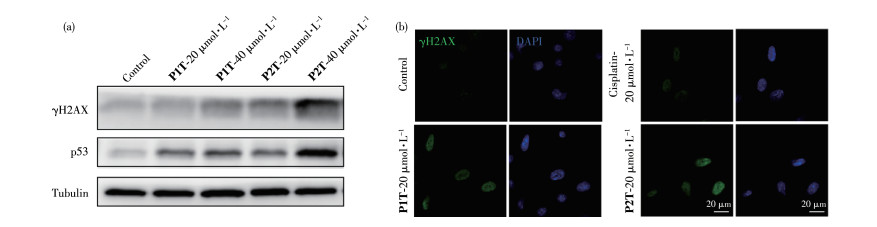
γH2AX是组蛋白H2AX的一种磷酸化蛋白,它是DNA双链断裂的标志蛋白[33]。文献报道顺铂能够诱导细胞中DNA损伤,进而活化γH2AX,所以γH2AX是研究铂类药物诱导细胞内DNA损伤的重要标志物[34]。我们采用免疫印迹的方法研究了药物刺激后细胞中γH2AX的表达,结果如图 6a所示。配合物P1T和P2T显著引起了细胞中γH2AX的表达,说明DNA损伤正在发生。同时我们也测试了DNA损伤下游蛋白p53的表达,它的表达也是DNA损伤通路中的一个重要事件,p53的活化能够启动细胞发生凋亡[35]。配合物P1T和P2T处理过的细胞中p53也显著激活,这意味着损伤的DNA没有被修复,p53开始引发细胞凋亡程序。我们利用免疫荧光进一步验证药物作用后细胞内γH2AX蛋白的表达变化。如图 6b所示,经配合物P1T和P2T作用后细胞核中γH2AX荧光信号明显增强,表明配合物诱导DNA损伤。
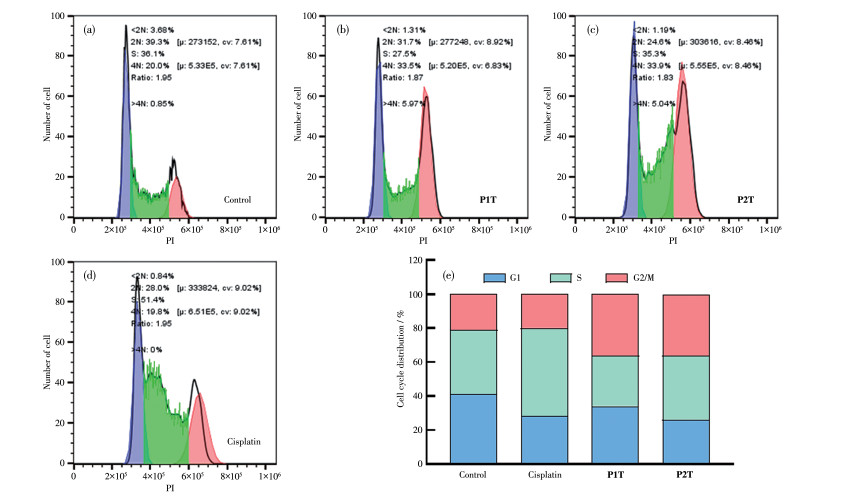
细胞周期是指连续分裂的细胞从一次有丝分裂结束到下一次有丝分裂完成所经历的全过程,一个细胞周期包括分裂间期与分裂期2个阶段,其中分裂间期又分为DNA合成前期(G1期)、DNA合成期(S期)以及DNA合成后期(G2/M期)。细胞内DNA损伤会引起细胞周期的阻滞[36]。我们用核酸染料PI标记DNA,通过流式细胞术研究药物对细胞周期的影响,实验结果如图 7所示,P1T和P2T以及顺铂均具有阻滞细胞周期的能力。P1T和P2T主要阻滞了细胞的G2/M期,而顺铂主要阻滞细胞的S期。推测配合物P1T和P2T主要通过干扰DNA合成复制,抑制肿瘤细胞增殖,进一步诱导细胞死亡。
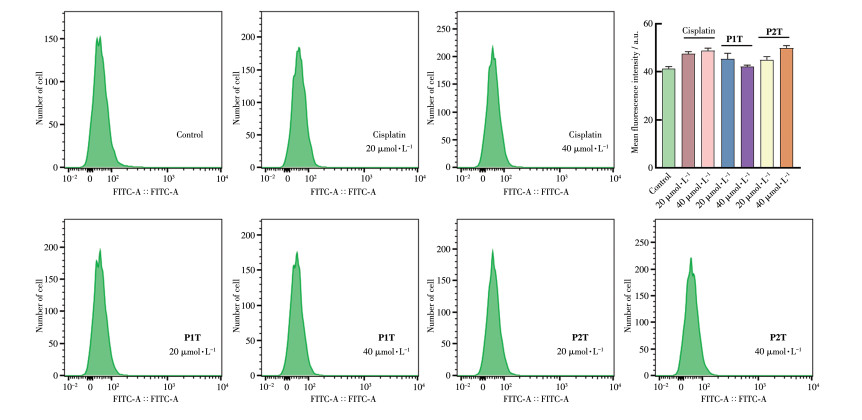
正常细胞通过控制细胞内活性氧(ROS)水平来调控细胞内信号传导、细胞增殖及细胞死亡等生理过程[37-38]。因此,为了进一步研究配合物P1T和P2T诱导细胞死亡的作用机理,我们选择商用ROS荧光探针DCFH-DA检测了肿瘤细胞中ROS水平。细胞中产生的ROS可以将无荧光的DCFH-DA氧化为有荧光的DCF(dichlorofluorescein),因此可通过DCF的荧光强度来指示细胞内ROS的水平[39]。我们先将配合物与A261细胞共孵育24 h,然后加入ROS荧光探针继续孵育30 min,利用流式细胞仪检测了细胞内ROS水平的变化。如图 8所示,与顺铂或对照组相比,配合物刺激后A261细胞中相对荧光强度无明显变化,这表明引入TMZ配体的配合物P1T和P2T无法诱导细胞产生过量的ROS,推测配合物主要是以其他方式诱导细胞死亡。
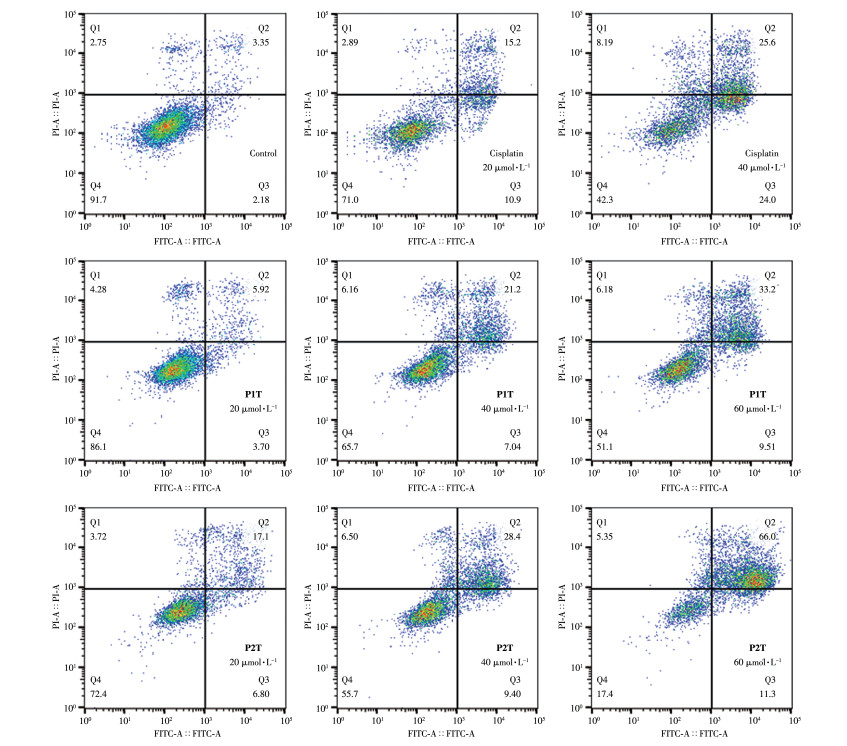
细胞凋亡是细胞的基本特征之一,普遍存在于生物界。对正常细胞而言,细胞膜内侧分布有大量磷脂酰丝氨酸(PS),而在细胞发生凋亡早期,PS会外翻到细胞膜表面。Annexin V能与PS特异性地结合,而Annexin V-FITC带有绿色荧光,因而可用其标识细胞凋亡[40]。核酸染料碘化丙啶(PI)不能透过完整的细胞膜,但是凋亡中晚期的细胞和死细胞的细胞膜通透性增加,PI能够透过细胞膜对细胞核进行染色,因此PI可以检测坏死细胞或凋亡晚期的细胞。我们采用流式细胞术研究了分别用P1T、P2T和顺铂刺激A261细胞48 h后的细胞死亡途径,利用Annexin V-FITC和PI对细胞进行双染,并用FlowJo软件对数据进行了分析。如图 9所示,与对照组相比,P1T和P2T主要诱导细胞发生晚期凋亡的能力。P1T和P2T以及顺铂均具有诱导A261细胞凋亡且表现出剂量依赖性,随着药物浓度的增加,晚期凋亡的细胞比例逐渐增加。
我们在修饰替莫唑胺的基础上合成并表征了2种新型四价铂配合物P1T和P2T,并研究了配合物的抗肿瘤活性及其诱导细胞死亡的作用机制。研究结果表明,与替莫唑胺相比,配合物的细胞毒活性明显提高,表现出与顺铂相当的细胞毒性,同时表现出较好的细胞选择性,其对正常细胞的细胞毒性较低。配合物进入细胞后,能在较短时间内发生水解,诱导DNA损伤,阻滞细胞周期在G2/M期,最终诱导细胞凋亡。总之,将替莫唑胺引入到四价铂配合物中,不仅提高了配合物的细胞毒活性和细胞选择性,而且拓展了铂类配合物在脑胶质瘤方面的应用,这些结果将为铂类金属抗肿瘤药物的研发提供一定的指导意义。
Louis D N, Perry A, Reifenberger G, Deimling A, Figarella-Branger D, Cavenee W K, Ohgaki H, Wiestler O D, Kleihues P, Ellison D W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary[J]. Acta Neuropathol., 2016, 131(6): 803-820. doi: 10.1007/s00401-016-1545-1
Giese A, Bjerkvig R, Berens M E, Westphal M. Cost of Migration: Invasion of Malignant Gliomas and Implications for Treatment[J]. J. Clin. Oncol., 2003, 21(8): 1624-1636. doi: 10.1200/JCO.2003.05.063
Affronti M L, Heery C R, Herndon J E, Rich J N, Reardon D A, Desjardins A, Vredenburgh J J, Friedman A H, Bigner D D, Friedman H S. Overall Survival of Newly Diagnosed Glioblastoma Patients Receiving Carmustine Wafers Followed by Radiation and Concurrent Temozolomide plus Rotational Multiagent Chemotherapy[J]. Cancer, 2009, 115(15): 3501-3511. doi: 10.1002/cncr.24398
de Souza R M, Shaweis H, Han C, Sivasubramaniam V, Brazil L, Beaney R, Sadler G, Al-Sarraj S, Hampton T, Logan J, Hurwitz V, Bhangoo R, Gullan R, Ashkan K. Erratum: Has the Survival of Patients with Glioblastoma Changed over the Years?[J]. Br. J. Cancer, 2016, 114(2): 146-150. doi: 10.1038/bjc.2015.421
Stupp R, Taillibert S, Kanner A, Read W, Steinberg D M, Lhermitte B, Toms S, Idbaih A, Ahluwalia M S, Fink K, Di Meco F, Lieberman F, Zhu J J, Stragliotto G, Tran D D, Brem S, Hottinger A F, Kirson E D, Lavy-Shahaf G, Weinberg U, Kim C Y, Paek S H, Nicholas G, Bruna J, Hirte H, Weller M, Palti Y, Hegi M E, Ram Z. Effect of Tumor-Treating Fields plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial[J]. JAMA - J. Am. Med. Assoc., 2017, 318(23): 2306-2316. doi: 10.1001/jama.2017.18718
Furnari F B, Fenton T, Bachoo R M, Mukasa A, Stommel J M, Stegh A, Hahn W C, Ligon K L, Louis D N, Brennan C, Chin L, DePinho R A, Cavenee W K. Malignant Astrocytic Glioma: Genetics, Biology, and Paths to Treatment[J]. Genes Dev., 2007, 21(21): 2683-2710. doi: 10.1101/gad.1596707
Fan C H, Liu W L, Cao H, Wen C, Chen L, Jiang G. O6-Methylgua-nine DNA Methyltransferase as a Promising Target for the Treatment of Temozolomide-Resistant Gliomas[J]. Cell Death Dis., 2013, 4(10): e876. doi: 10.1038/cddis.2013.388
Hirose Y, Berger M S, Pieper R O. p53 Effects both the Duration of G2 & M Arrest and the Fate of Temozolomide-Treated Human Glioblas- toma Cells[J]. Cancer Res., 2001, 61(5): 1957-1963.
Friedman H S, Kerby T, Calvert H. Temozolomide and Treatment of Malignant Glioma[J]. Clin. Cancer Res., 2000, 6(7): 2585-2597.
Gerson S L. MGMT: Its Role in Cancer Aetiology and Cancer Thera-peutics[J]. Nat. Rev. Cancer, 2004, 4(4): 296-307. doi: 10.1038/nrc1319
Lee S Y. Temozolomide Resistance in Glioblastoma Multiforme[J]. Genes Dis., 2016, 3(3): 198-210. doi: 10.1016/j.gendis.2016.04.007
Stupp R, Mason W P, van den Bent M J, Weller M, Fisher B, Taphoorn M J B, Belanger K, Brandes A A, Marosi C, Bogdahn U, Curschmann J, Janzer R C, Ludwin S K, Gorlia T, Allgeier A, Lacombe D, Cairncross J G, Eisenhauer E, Mirimanoff R O. Radio-therapy plus Concomitant and Adjuvant Temozolomide for Glioblas-toma[J]. N. Engl. J. Med., 2005, 352(10): 987-996. doi: 10.1056/NEJMoa043330
Stupp R, Hegi M E, Mason W P, Bent M J, Taphoorn M J B, Janzer R C, Ludwin S K, Allgeier A, Fisher B, Belanger K, Hau P, Brandes A A, Gijtenbeek J, Marosi C, Vecht C J, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross J G, Mirimanoff R O. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase Ⅲ Study: 5-Year Analysis of the EORTC-NCIC Trial[J]. Lancet Oncol., 2009, 10(5): 459-466. doi: 10.1016/S1470-2045(09)70025-7
Rosenberg B, Vancamp L, Trosko J E, Mansour V H. Platinum Com- pounds: A New Class of Potent Antitumour Agents[J]. Nature, 1969, 222(5191): 385-386. doi: 10.1038/222385a0
Wang X H, Wang X Y, Jin S X, Muhammad N, Guo Z J. Stimuli-Responsive Therapeutic Metallodrugs[J]. Chem. Rev., 2019, 119(2): 1138-1192. doi: 10.1021/acs.chemrev.8b00209
金素星, 郭子建, 王晓勇. 靶向能量代谢的抗肿瘤金属配合物[J]. 药学进展, 2020,44,(4): 280-293. JIN S X, GUO Z J, WANG X Y. Antitumor Metal Complexes Target-ing Energy Metabolism[J]. Progress in Pharmaceutical Sciences, 2020, 44(4): 280-293.
Klein A V, Hambley T W. Platinum Drug Distribution in Cancer Cells and Tumors[J]. Chem. Rev., 2009, 109(10): 4911-4920. doi: 10.1021/cr9001066
Dilruba S, Kalayda G V. Platinum-Based Drugs: Past, Present and Future[J]. Cancer Chemother. Pharmacol., 2016, 77(6): 1103-1124. doi: 10.1007/s00280-016-2976-z
Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular Mechanisms of Cisplatin Resis-tance[J]. Oncogene, 2012, 31(15): 1869-1883. doi: 10.1038/onc.2011.384
Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bel-lan A, Castedo M, Kroemer G. Systems Biology of Cisplatin Resis-tance: Past, Present and Future[J]. Cell Death Dis., 2014, 5(5): e1257. doi: 10.1038/cddis.2013.428
Wang X Y, Guo Z J. Targeting and Delivery of Platinum-Based Anti-cancer Drugs[J]. Chem. Soc. Rev., 2013, 42(1): 202-224. doi: 10.1039/C2CS35259A
Oun R, Moussa Y E, Wheate N J. The Side Effects of Platinum-Based Chemotherapy Drugs: A Review for Chemists[J]. Dalton Trans., 2018, 47(19): 6645-6653. doi: 10.1039/C8DT00838H
Karasawa T, Steyger P S. An Integrated View of Cisplatin-Induced Nephrotoxicity and Ototoxicity[J]. Toxicol. Lett., 2015, 237(3): 219-227. doi: 10.1016/j.toxlet.2015.06.012
Johnstone T C, Suntharalingam K, Lippard S J. The Next Generation of Platinum Drugs: Targeted Pt Agents, Nanoparticle Delivery, and Pt Prodrugs[J]. Chem. Rev., 2016, 116(5): 3436-3486. doi: 10.1021/acs.chemrev.5b00597
Wilson J J, Lippard S J. Synthetic Methods for the Preparation of Platinum Anticancer Complexes[J]. Chem. Rev., 2014, 114(8): 4470-4495. doi: 10.1021/cr4004314
Hall M D, Hambley T W. Platinum Antitumour Compounds: Their Bioinorganic Chemistry[J]. Coord. Chem. Rev., 2002, 232(1): 49-67.
Jin S X, Hao Y Y, Zhu Z Z, Muhammad N, Zhang Z Q, Wang K, Guo Y, Guo Z J, Wang X Y. Impact of Mitochondrion-Targeting Group on the Reactivity and Cytostatic Pathway of Platinum Complexes[J]. Inorg. Chem., 2018, 57(17): 11135-11145. doi: 10.1021/acs.inorgchem.8b01707
Shi Y, Liu S A, Kerwood D J, Goodisman J, Dabrowiak J C. Pt Complexes as Prodrugs for Cisplatin[J]. J. Inorg. Biochem., 2012, 107(1): 6-14. doi: 10.1016/j.jinorgbio.2011.10.012
Arrowsmith J, Jennings S A, Clark A S, Stevens M F G. Antitumor Imidazotetrazines. 41. Conjugation of the Antitumor Agents Mitozolo-mide and Temozolomide to Peptides and Lexitropsins Bearing DNA Major and Minor Groove-Binding Structural Motifs[J]. J. Med. Chem., 2002, 45(25): 5458-5470.
Huang H Y, Yu B L, Zhang P Y, Huang J J, Chen Y, Gasser G, Ji L N, Chao H. Highly Charged Ruthenium Polypyridyl Complexes as Lysosome-Localized Photosensitizers for Two-Photon Photodynamic Therapy[J]. Angew. Chem. Int. Ed., 2015, 54(47): 14049-14052. doi: 10.1002/anie.201507800
Huang H Y, Zhang P Y, Yu B L, Chen Y, Wang J Q, Ji L N, Chao H. Targeting Nucleus DNA with a Cyclometalated Dipyridophenazineru-thenium Complex[J]. J. Med. Chem., 2014, 57(21): 8971-8983. doi: 10.1021/jm501095r
Oldfield S P, Hall M D, Platts J A. Calculation of Lipophilicity of a Large, Diverse Dataset of Anticancer Platinum Complexes and the Relation to Cellular Uptake[J]. J. Med. Chem., 2007, 50(21): 5227-5237. doi: 10.1021/jm0708275
Bonner W M, Redon C E, Dickey J S, Nakamura A J, Sedelnikova O A, Solier S, Pommier Y. γH2AX and Cancer[J]. Nat. Rev. Cancer, 2008, 8(12): 957-967. doi: 10.1038/nrc2523
Revet I, Feeney L, Bruguera S, Wilson W, Dong T K, Oh D H, Dankort D, Cleaver J E. Functional Relevance of the Histone γH2Ax in the Response to DNA Damaging Agents[J]. Proc. Natl. Acad. Sci. U. S. A., 2011, 108(21): 8663-8667. doi: 10.1073/pnas.1105866108
Shieh S Y, Ikeda M, Taya Y, Prives C. DNA Damage-Induced Phos-phorylation of p53 Alleviates Inhibition by MDM2[J]. Cell, 1997, 91(3): 325-334. doi: 10.1016/S0092-8674(00)80416-X
Wang D, Lippard S J. Cellular Processing of Platinum Anticancer Drugs[J]. Nat. Rev. Drug Discovery, 2005, 4(4): 307-320. doi: 10.1038/nrd1691
Zhou Z J, Song J B, Nie L M, Chen X Y. Reactive Oxygen Species Generating Systems Meeting Challenges of Photodynamic Cancer Therapy[J]. Chem. Soc. Rev., 2016, 45(23): 6597-6626. doi: 10.1039/C6CS00271D
吴健, 陶钦, 葛超, 薛旭玲, 钱勇, 刘红科. 紫苏醇芳基钌配合物的合成及抗肿瘤性能[J]. 无机化学学报, 2020,36,(7): 1223-1232. WU J, TAO Q, GE C, XUE X L, QIAN Y, LIU H K. Synthesis and Antitumor Properties of Ruthenium-Arene Complexes Based on Nat-ural Product Perillol[J]. Chinese J. Inorg. Chem., 2020, 36(7): 1223-1232.
Wang F X, Liang J H, Zhang H, Wang Z H, Wan Q, Tan C P, Ji L N, Mao Z W. Mitochondria-Accumulating Rhenium Tricarbonyl Com-plexes Induce Cell Death via Irreversible Oxidative Stress and Gluta-thione Metabolism Disturbance[J]. ACS Appl. Mater. Interfaces, 2019, 11(14): 13123-13133. doi: 10.1021/acsami.9b01057
Carneiro B A, El-Deiry W S. Targeting Apoptosis in Cancer Therapy[J]. Nat. Rev. Clin. Oncol., 2020, 17(7): 395-417. doi: 10.1038/s41571-020-0341-y

图 4 P1T、P2T和配体TMZ在含1% DMSO的PBS中的稳定性
Figure 4 Stability of P1T, P2T, and ligand TMZ in PBS containing 1% DMSO

图 5 ICP-MS检测的P1T、P2T和顺铂在106个A261细胞内的铂含量
Figure 5 Intracellular Pt content of P1T, P2T and cisplatin in 106 A261 cells by ICP-MS

图 6 (a) 蛋白印迹显示经不同药物作用后A261细胞内γH2AX、p53蛋白的表达; (b) 共聚焦显微镜显示经不同药物作用后γH2AX在A261细胞内的表达
Figure 6 (a) Western blot showing the expression of γH2AX, p53 protein in A261 cells after treatment with different drugs; (b) Confocal microscope images of expression of γH2AX in A261 cells after treatment with different drugs

图 7 不同药物对A261细胞周期阻滞的影响: (a) control、(b) P1T、(c) P2T和(d) cisplatin的细胞周期分布; (e)细胞周期分布柱状图
Figure 7 Effect of different drugs on A261 cell cycle arrest: cell cycle distributions of (a) control, (b) P1T, (c) P2T and (d) cisplatin; (e) histogram of cell cycle distributions

图 8 不同药物作用后A261细胞内的ROS水平
Figure 8 ROS levels in A261 cell after treatment with different drugs

图 9 不同药物作用于A261细胞后导致的细胞凋亡
Figure 9 Apoptosis caused by different drugs acting on A261 cell
表 1 P1T、P2T、TMZ及顺铂对不同类型细胞株作用48 h后的IC50值
Table 1.
IC50 values towards different cell lines treated with P1T, P2T, TMZ and cisplatin for 48 h
| Drug | A261 | U87MG | A549 | HT-22 |
| P1T | 21.08±0.85 | 49.19±2.04 | 45.57±1.30 | 85.19±2.76 |
| P2T | 23.96±0.01 | 35.61±0.61 | 34.76±0.24 | 55.86±0.91 |
| Cisplatin | 12.93±0.21 | 24.41±0.47 | 9.77±0.15 | 16.79±0.62 |
| TMZ | 1 372.09±185.72 | 1 465.16±65.47 | 1 573.48±98.29 | > 2 000 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们