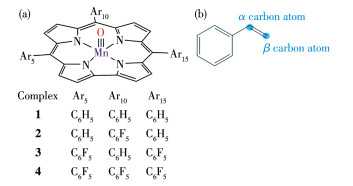
图 1.
(a) 配合物1~4和(b) 苯乙烯的结构
Figure 1.
Structures of (a) complexes 1-4 and (b) styrene

氧原子转移(oxygen atom transfer,OAT)反应是使氧原子从过渡金属配合物转移到底物以实现氧化底物的反应[1]。这个过程通常会产生具有M—O键的作为反应活性中间体的高价过渡金属氧配合物[2-3]。高价锰氧配合物作为光合系统Ⅱ(Photosys- tem Ⅱ)产氧关键中间体具有优异的氧化活性[2, 4]。近些年人们对模拟产氧活性中心锰配合物的研究取得了长足的进步[5-8],但由于高活性瞬态物种的分离困难[9],以高价锰氧配合物作为中间体的氧转移反应过程机理成为研究的热点。
咔咯(corrole)具有独特的三价阴离子和较小的空腔结构,能稳定结合较高氧化态金属离子形成配合物[1]。咔咯锰配合物的OAT反应广泛应用于催化领域[10-11]以及硫醚磺化[12-13]、烷烃羟基化[14]和烯烃环氧化[15-17]等化学反应中。研究发现咔咯锰(Ⅴ)氧配合物的OAT反应主要存在歧化反应和直接转移2种机理[18-21]。我们研究咔咯锰(Ⅴ)氧配合物与苯乙烯环氧化反应的实验发现,其OAT反应是采用直接转移的机理[18],但是氧原子是先进攻苯乙烯双键,还是双键上的碳原子,无法从实验上确认,因此该反应的具体途径尚待研究。另外,取代基效应将显著影响OAT反应的产率,例如吸电子的取代基能明显提升咔咯锰氧配合物氧化硫醚的反应速率[22]。
我们对咔咯锰(Ⅴ)氧配合物与苯乙烯的OAT反应路径进行理论计算,进一步确认氧原子转移的具体位置,结合内禀反应坐标法(IRC)和最低能量交叉点(MECP)计算,推导出氧原子转移的途径。选取具有吸电子诱导效应的五氟苯基(—C6F5)对咔咯大环进行中位取代,探究吸电子效应对咔咯锰(Ⅴ)氧配合物的OAT反应的影响,从理论上推进对咔咯锰(Ⅴ)氧配合物氧化性能研究的发展。
我们一共设计4种咔咯锰(Ⅴ)氧配合物(图 1a),即MnⅤO corrole、10-五氟苯基- MnⅤO corrole、5,15-二(五氟苯基)- MnⅤO corrole、5,10,15-三(五氟苯基)- MnVO corrole配合物,依次编号为配合物1、2、3 和4。根据本课题组以往的研究[23],采用B3LYP方法计算咔咯锰所得的结果与实验值相近,故所有计算均采用DFT/B3LYP方法[24-25],C、N、F、H、O等非金属元素采用6-31G**基组,金属元素Mn采用LanL2DZ基组[26]。N,N-二甲基甲酰胺(DMF)是咔咯锰(Ⅴ)氧配合物反应实验中最常用的有机溶剂[11],动力学实验表明轴向配位作用可显著提升咔咯锰(Ⅴ)氧配合物与苯乙烯的OAT反应速率[21],故以DMF作为溶剂本体,采用可极化连续介质模型(PCM)考虑溶剂效应。研究表明[23, 27] MnⅤO corrole的基态通常是单重态,在自旋轨道耦合作用下易形成三重态,因此分别对单重态和三重态下的4种配合物与苯乙烯(图 1b)进行OAT反应的过渡态进行计算,并利用IRC验证,找出相应的反应途径。所有计算均采用Gaussian 03程序完成,另外使用Harvey的MECP程序[28]的修改版sobMECP[29]进一步获取反应路径中的MECP。
配合物1~4 在单重态、三重态下的优化结构大致相似,所得几何结构参数见表 1。由于锰原子与氧原子的成键作用,原本几乎位于咔咯环平面的锰原子[23]向氧原子方向偏离。单重态下,随中位—C6F5的增多,dMn—4N和∠MnNNN数值逐渐减小,锰原子偏离咔咯环平面程度减小,锰、氧原子之间相互作用减弱。与此同时,dMn≡O从0.154 9 nm稍缩短至0.154 3 nm,Mn≡O的Wiberg键级随取代数增加而逐渐增强(表 2),这均说明了锰氧键强度随吸电子效应的增强而轻微增大。考察各配合物在单重态和三重态下的NPA(natural population analysis,自然布居分析)电荷发现,吸电子取代基的诱导效应导致QMn(锰原子所带NPA电荷)、QO(氧原子所带NPA电荷)越来越大(表 2),而反应活性与锰原子的电荷密度密切相关,锰原子缺电子性的增大有利于亲核试剂对其进攻发生反应;另一方面,随着—C6F5取代数的增加,中位取代基所带负电荷QSub数值趋向变大,导致其与氧原子所带电荷产生的负-负静电排斥作用显著增强,该作用已被理论计算证明能明显加大氧原子脱离体系束缚实现氧转移的趋势[30],导致咔咯锰(Ⅴ)氧配合物的氧化性增加。
 下载:
导出CSV
下载:
导出CSV
| Complex | dMn—Na/nm | dMn≡Ob/nm | dMn—4Nc/nm | ∠MnNNNd/(°) | ∠NMnNe/(°) | |||||||||
| Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | |||||
| 1 | 0.191 6 | 0.194 5 | 0.154 9 | 0.158 9 | 0.054 7 | 0.047 2 | 22.90 | 19.51 | 85.31 | 86.67 | ||||
| 2 | 0.191 5 | 0.194 4 | 0.154 6 | 0.158 8 | 0.054 3 | 0.046 9 | 22.73 | 19.40 | 85.38 | 86.73 | ||||
| 3 | 0.191 5 | 0.194 3 | 0.154 5 | 0.158 7 | 0.054 2 | 0.046 7 | 22.69 | 19.34 | 85.40 | 86.77 | ||||
| 4 | 0.191 4 | 0.194 2 | 0.154 3 | 0.158 6 | 0.053 8 | 0.046 5 | 22.54 | 19.29 | 85.46 | 86.77 | ||||
| aAverage distance between the manganese atom and the four nitrogen atoms; bLength of Mn≡O bond; cDistance from the manganese atom to the corrole plane; dDihedral angle between the manganese atom and the corrole plane; eAverage value of the angles between nitrogen atoms, manganese atoms and nitrogen atoms on the corrole plane. | ||||||||||||||
下载:
导出CSV
| Complex | QMn | QO | Qcora | QSubb | WIMn≡O | |||||||||
| Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | |||||
| 1 | 0.345 | 0.436 | -0.132 | -0.314 | -0.303 | -0.266 | -0.042 | -0.170 | 2.078 | 1.940 | ||||
| 2 | 0.348 | 0.436 | -0.120 | -0.308 | -0.274 | -0.225 | -0.075 | -0.211 | 2.089 | 1.943 | ||||
| 3 | 0.349 | 0.437 | -0.112 | -0.302 | -0.231 | -0.169 | -0.119 | -0.268 | 2.096 | 1.947 | ||||
| 4 | 0.359 | 0.437 | -0.101 | -0.296 | -0.199 | -0.118 | -0.155 | -0.320 | 2.107 | 1.952 | ||||
| a NPA charge of the skeleton of corrole; b Total NPA charge of the three substituents. | ||||||||||||||
三重态计算结果显示,配合物结构参数和电荷分布变化规律与单重态的情况相似,吸电子效应能增强MnⅤO corrole的稳定性与氧转移趋势。虽然同一多重态下变化幅度较小,但当MnⅤO corrole从单重态跃迁到三重态时,各配合物的分子结构发生明显变化:dMn—4N和∠MnNNN数值减小,且dMn≡O增大了约0.004 2 nm,锰在三重态下偏离咔咯环平面的程度更小,锰氧键的强度减弱,更容易断裂;dMn—N和∠NMnN增大,环空腔位阻更小。以上几何结构变化均表明了三重态下的MnⅤO corrole更容易发生OAT反应。
我们分别计算配合物1、2、3 和4 与苯乙烯的OAT反应过程,结果发现,在单重态和三重态反应过程中,均仅存在一个过渡态。对过渡态的频率计算发现只有一个虚频,确认了相应结构为反应的过渡态。通过IRC验证,计算结果确认了反应路径的反应物、过渡态和产物的构型,其中三重态的配合物1 的各构型见图 2。
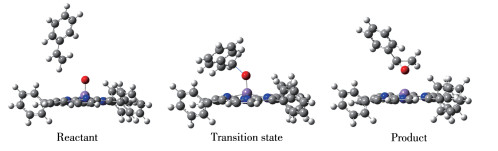
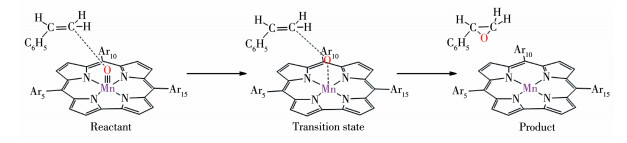
观察OAT过程中配合物体系的几何优化结构变化(图 2),可见随着反应的进行,咔咯锰(Ⅴ)氧配合物逐渐接近苯乙烯,其Mn≡O上的氧原子不是进攻苯乙烯中的双键或者连接苯环的α 碳原子,而是进攻远离苯环的β 碳原子。例如三重态下配合物1 作为反应物,进攻苯乙烯上的β 碳原子形成具有Mn… O…C键的过渡态结构,其Mn≡O键上氧原子与β碳原子距离dC—O从反应物时的0.403 3 nm缩短到0.221 1 nm(表 3),Mn≡O键长dMn≡O从0.158 9 nm增长至0.171 0 nm。反应结束,dC—O进一步缩短到0.144 9 nm,dM≡O则增长至0.226 4 nm,氧原子直接转移到苯乙烯上,发生直接氧原子转移反应,形成环氧苯乙烷结构。反应中β 碳原子作为反应位点,显然是因为β碳原子位阻比α 碳原子小得多。计算结果也表明单重态和三重态的配合物1~4 与苯乙烯的OAT反应过程都是相同的,由此得到了咔咯锰(Ⅴ) 氧配合物与苯乙烯氧转移反应的路径示意图,如图 3所示。
下载:
导出CSV
| Complex | Multiplicity | Substance | dMn≡O/nm | dC—O/nm | ∠MnNNN / (°) |
Qβa |
| 1 | Singlet | Reactant | 0.155 1 | 0.383 5 | 22.99 | -0.423 |
| Transition state | 0.164 4 | 0.170 0 | 20.50 | -0.262 | ||
| Product | 0.179 8 | 0.138 7 | 18.60 | -0.158 | ||
| Triplet | Reactant | 0.158 9 | 0.403 3 | 19.69 | -0.423 | |
| Transition state | 0.171 0 | 0.221 1 | 17.55 | -0.354 | ||
| Product | 0.226 4 | 0.144 9 | 12.96 | -0.109 | ||
| 2 | Singlet | Reactant | 0.154 6 | 0.381 2 | 22.77 | -0.423 |
| Transition state | 0.164 1 | 0.170 9 | 29.21 | -0.265 | ||
| Product | 0.180 2 | 0.138 4 | 18.72 | -0.158 | ||
| Triplet | Reactant | 0.158 8 | 0.399 6 | 19.40 | -0.423 | |
| Transition state | 0.168 7 | 0.205 7 | 16.75 | -0.296 | ||
| Product | 0.221 9 | 0.145 0 | 13.60 | -0.107 | ||
| 3 | Singlet | Reactant | 0.154 5 | 0.617 8 | 22.42 | -0.423 |
| Transition state | 0.163 9 | 0.171 3 | 20.36 | -0.268 | ||
| Product | 0.173 8 | 0.146 1 | 18.98 | -0.189 | ||
| Triplet | Reactant | 0.158 6 | 0.459 7 | 19.27 | -0.423 | |
| Transition state | 0.167 9 | 0.248 8 | 17.75 | -0.378 | ||
| Product | 0.215 6 | 0.145 3 | 14.37 | -0.100 | ||
| 4 | Singlet | Reactant | 0.154 2 | 0.392 0 | 22.52 | -0.424 |
| Transition state | 0.163 6 | 0.172 1 | 20.20 | -0.271 | ||
| Product | 0.180 1 | 0.138 4 | 18.30 | -0.162 | ||
| Triplet | Reactant | 0.158 5 | 0.398 1 | 19.19 | -0.424 | |
| Transition state | 0.166 6 | 0.258 3 | 17.66 | -0.373 | ||
| Product | 0.202 2 | 0.146 0 | 14.88 | -0.094 |
取代基效应对咔咯配合物的氧化性能有显著的影响。Kuma等[12]在研究咔咯配合物的OAT反应时发现当中位连有吸电子基团的三芳基时,尤其是β 位全溴代的咔咯锰(Ⅴ)氧配合物氧化活性很强。何婧[22]通过理论计算得到在β 位连有吸电子取代基的MnⅤO corrole对二甲基硫醚(DMS)氧转移的效果更加明显,李皎[31]模拟计算咔咯锰(Ⅲ)配合物催化RNA磷酸二酯类似物断裂时也发现了吸电子取代基效应能显著降低反应能垒。本课题组研究发现以PhIO为氧源时,全氟取代的咔咯锰配合物在氧化烯烃时比非全氟取代的配合物表现出更高的反应速度[32]。
OAT反应的难易程度可以由理论计算的反应能垒的大小所反映。以单重态下配合物1、2、3 和4的反应物体系能量分别作零参考势能面,对直接转移机理的OAT反应过程中各状态体系经ZPE校正后的分子电子总能量进行考察,作单重态、三重态下各配合物与苯乙烯反应的势能图(图 4)。在单重态下,配合物1~4 的反应能垒分别为91.59、86.28、88.54和81.06 kJ·mol-1。而在三重态下各体系对应能垒分别为38.63、39.25、30.92和25.84 kJ·mol-1,显著低于单重态的数值,表明该OAT反应在三重态下更容易反应。该反应的能垒数值与配合物4 氧化二甲基硫醚的能垒大小接近,其单重态和三重态的能垒分别为92.46和21.33 kJ·mol-1 [22]。
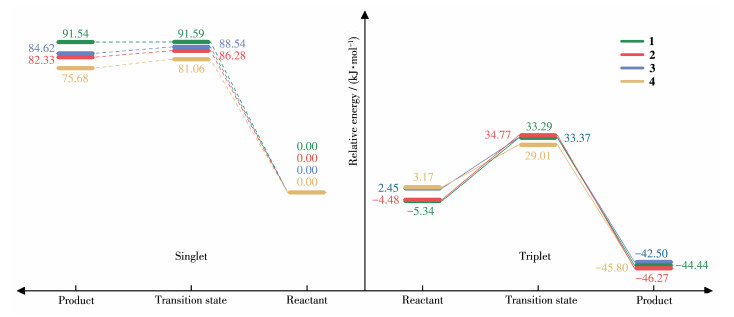
各体系的单重态和三重态能垒曲线揭示了吸电子效应对MnⅤO corrole氧化苯乙烯反应能垒的显著影响。体系反应能垒大致随—C6F5取代数的增加而逐渐降低,其中吸电子效应最强的配合物4 反应能垒最低,其规律与相关文献[12]咔咯锰氧配合物氧化实验结果相符合。观察1~4 反应物的能量情况(相对能量见图 4,具体能量数值见表S1,Supporting information)可知,反应物1 和2 三重态的能量低于单重态能量值,因此体系可直接遵循三重态过渡态的路径进行反应。但是反应物3 和4 在单重态时能量低于三重态,其基态为单重态,而产物则是能量更低的三重态体系更稳定。所以在体系3 和4 在OAT历程中,单重态的反应物易发生自旋翻转,使得反应最终向能量更低的三重态过渡态的路径进行。
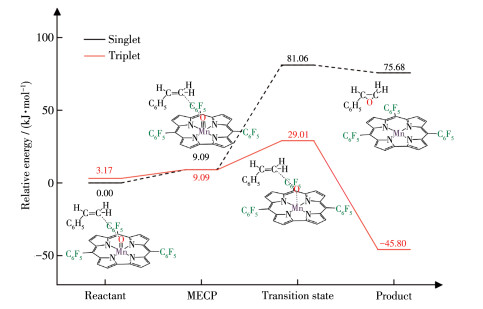
配合物3 和4 的反应物单重态的能量低于对应三重态能量值,所以MECP成为反应物从单重态经过自旋翻转成为三重态进行OAT反应路线的关键步骤。经过MECP搜索,发现配合物3 和4 的反应过程中均存在一个MECP。配合物与苯乙烯OAT反应中交叉点的构型变化主要体现在dMn≡O中。配合物4 的交叉点(MECP位置)dMn≡O为0.157 0 nm,该键长稍短于配合物处于三重态时的0.158 5 nm,稍长于单重态的0.154 2 nm(表 4),其相对分子能量约为9 kJ· mol-1(图 5),高于单重态和三重态的能量值。在氧转移过程中,随体系4 单重态的总能量上升,dMn≡O不断增大,到达MECP对应的0.157 0 nm时,体系4 可以发生自旋翻转成为三重态,并沿着低能垒(25.84 kJ· mol-1)三重态反应路线进行。另外,我们注意到无论是反应物还是交叉点构型的dC—O均大于0.3 nm,如此长的距离表明苯乙烯对反应物和交叉点的构型变化几乎没有影响。因此,咔咯配合物的OAT反应MECP和自旋翻转现象还可以发生在其他氧化底物[22, 27] (例如硫醚)中。另一方面,配合物3 和4 的MECP相对能量约为8.32和9.09 kJ·mol-1,可见五氟苯基的不断加入能逐渐升高体系的MECP相对能量,减少与过渡态的能量差。
 下载:
导出CSV
下载:
导出CSV
| Reaction state | dMn≡O/nm | dC—O/nm | |||
| Singlet | Triplet | Singlet | Triplet | ||
| Reactant | 0.154 2 | 0.158 5 | 0.392 0 | 0.398 1 | |
| MECP | 0.157 0 | 0.157 0 | 0.333 9 | 0.333 9 | |
| Transition state | 0.163 6 | 0.166 6 | 0.172 1 | 0.258 3 | |
| Product | 0.180 1 | 0.202 2 | 0.138 4 | 0.146 0 | |
采用DFT/B3LYP方法对中位连有吸电子基团的—C6F5的MnⅤO corrole和苯乙烯之间的OAT反应路径进行理论计算。结果显示MnⅤO corrole进攻苯乙烯中位阻较小的β 碳原子,以直接氧转移方式形成环氧化合物。三重态时的体系的反应能垒明显低于单重态的反应能垒。在氧转移过程中,随着反应体系单重态的总能量上升,经过最小能量交叉点构型时,体系可发生自旋翻转成为三重态,并沿着低能垒三重态反应路线进行。五氟苯基取代数的增加能提高MnVO corrole的氧转移能力并显著降低OAT反应能垒。
Supporting information is available at http://www.wjhxxb.cn
Baglia R A, Zaragoza J P T, Goldberg D P. Biomimetic Reactivity of Oxygen-Derived Manganese and Iron Porphyrinoid Complexes[J]. Chem. Rev., 2017, 117(21): 13320-13352. doi: 10.1021/acs.chemrev.7b00180
Larson V A, Battistella B, Ray K, Lehnert N, Nam W. Iron and Manganese Oxo Complexes, Oxo Wall and Beyond[J]. Nat. Rev. Chem., 2020, 4(8): 404-419. doi: 10.1038/s41570-020-0197-9
Zhang R, Klaine S, Alcantar C, Bratcher F. Visible Light Generation of High - Valent Metal - Oxo Intermediates and Mechanistic Insights into Catalytic Oxidations[J]. J. Inorg. Biochem., 2020, 212: 111246-11259. doi: 10.1016/j.jinorgbio.2020.111246
Mcevoy J P, Brudvig G W. Water-Splitting Chemistry of Photosystem Ⅱ[J]. Chem. Rev., 2006, 106(11): 4455-4483. doi: 10.1021/cr0204294
Neu H M, Quesne M G, Yang T, Prokop-Prigge K A, Lancaster K M, Donohoe J, Debeer S, de Visser S P, Goldberg D P. Dramatic Influence of an Anionic Donor on the Oxygen-Atom Transfer Reactivity of a MnⅤ-Oxo Complex[J]. Chem. Eur. J., 2014, 20(45): 14584-14588. doi: 10.1002/chem.201404349
Zou H B, Yang H, Liu Z Y, Mahmood M H R, Mei G Q, Liu H Y, Chang C K. Iron (Ⅳ) - Corrole Catalyzed Stereoselective Olefination of Aldehydes with Ethyl Diazoacetate[J]. Organometallics, 2015, 34(12): 2791-2795. doi: 10.1021/acs.organomet.5b00069
Kim S H, Park H, Seo M S, Kubo M, Ogura T, Klajn J, Gryko D T, Valentine J S, Nam W. Reversible O—O Bond Cleavage and Formation Between Mn (Ⅳ) - Peroxo and Mn (Ⅴ) - Oxo Corroles[J]. J. Am. Chem. Soc., 2010, 132(40): 14030-14032. doi: 10.1021/ja1066465
Zaragoza J P, Baglia R A, Siegler M A, Goldberg D P. Strong Inhibition of O-Atom Transfer Reactivity for Mn (Ⅳ) (O)(π-Radical-Cation) (Lewis Acid) versus Mn(Ⅴ)(O) Porphyrinoid Complexes[J]. J. Am. Chem. Soc., 2015, 137(20): 6531-6540. doi: 10.1021/jacs.5b00875
Ka W K, Ngo F L, Ranburger D, Malone J, Zhang R. Visible Light-Induced Formation of Corrole-Manganese(Ⅴ)-Oxo Complexes: Observation of Multiple Oxidation Pathways[J]. J. Inorg. Biochem., 2016, 163: 39-44. doi: 10.1016/j.jinorgbio.2016.08.004
Liu H Y, Mahmood M H, Qiu S X, Chang C K. Recent Developments in Manganese Corrole Chemistry[J]. Coord. Coord. Chem. Rev., 2013, 257(7/8): 1306-1333.
Wang Q, Zhang Y, Yu L, Yang H, Mahmood M H R, Liu H Y. Solvent Effects on the Catalytic Activity of Manganese (Ⅲ) Corroles[J]. J. Porphyrins Phthalocyanines, 2014, 18(4): 316-325. doi: 10.1142/S1088424614500059
Kumar A, Goldberg I, Botoshansky M, Buchman Y, Gross Z. Oxygen Atom Transfer Reactions from Isolated (Oxo)manganese (Ⅴ) Corroles to Sulfides[J]. J. Am. Chem. Soc., 2010, 132(43): 15233-15245. doi: 10.1021/ja1050296
Xu Y, Xu Z G, Zhang X H, Chen H B, Xu Xuan, Liu H Y. Oxygen Atom Transfer Reaction of Manganese-Oxo Corrole toward Dimethyl Sulfide: A Density Functional Study[J]. Chin. J. Struct. Chem., 2019, 38(11): 1857-1866.
Bose S, Pariyar A, Biswas A N, Das P, Bandyopadhyay P. Electron Deficient Manganese(Ⅲ) Corrole Catalyzed Oxidation of Alkanes and Alkylbenzenes at Room Temperature[J]. Catal. Commun., 2011, 12(13): 1193-1197. doi: 10.1016/j.catcom.2011.04.026
Gross Z, Golubkov G, Simkhovich L. Epoxidation Catalysis by a Manganese Corrole and Isolation of an Oxo - Manganese (Ⅴ) Corrole[J]. Angew. Chem. Int. Ed., 2000, 39(22): 4045-4047. doi: 10.1002/1521-3773(20001117)39:22<4045::AID-ANIE4045>3.0.CO;2-P
Collman J P, Zeng L, Decréau R A. Multiple Active Oxidants in Competitive Epoxidations Catalyzed by Porphyrins and Corroles[J]. Chem. Commun., 2003, (24): 2974-2975. doi: 10.1039/B310763A
Gross Z, Simkhovich L, Galili N. First Catalysis by Corrole Metal Complexes: Epoxidation, Hydroxylation, and Cyclopropanation[J]. Chem. Commun., 1999, (7): 599-600. doi: 10.1039/a900571d
Liu H Y, Yam F, Xie Y T, Li X Y, Chang C K. A Bulky Bis-Pocket Manganese (Ⅴ) - Oxo Corrole Complex: Observation of Oxygen Atom Transfer Between Triply Bonded MnⅤ ≡O and Alkene[J]. J. Am. Chem. Soc., 2009, 131(36): 12890-12891. doi: 10.1021/ja905153r
Zhang R, Harischandra D N, Newcomb M. Laser Flash Photolysis Generation and Kinetic Studies of Corrole-Manganese(Ⅴ)-Oxo Intermediates[J]. Chem. Eur. J., 2005, 11(19): 5713-5720. doi: 10.1002/chem.200500134
Zhang R, Newcomb M. Laser Flash Photolysis Generation of High-Valent Transition Metal-Oxo Species: Insights from Kinetic Studies in Real Time[J]. Acc. Chem. Res., 2008, 41(3): 468-477. doi: 10.1021/ar700175k
Liu H Y, Zhou H, Liu L L, Ying X, Jiang H F, Chang C K. The Effect of Axial Ligand on the Reactivity of Oxo - Manganese (Ⅴ) Corrole[J]. Chem. Lett., 2007, 36(2): 274-275. doi: 10.1246/cl.2007.274
He J, Xu Z G, Xu X, Gong L Z, Mahmood M H R, Liu H Y. Reactivity of (Oxo)manganese (Ⅴ) Corroles in One - Electron Redox State: Insights from Conceptual DFT and Transition Sate Calculations[J]. J. Porphyrins Phthalocyanines, 2013, 17(12): 1196-1203. doi: 10.1142/S1088424613500971
Gong L Z, Xu Z G, Xu X, He J, Wang Q, Liu H Y. Axial Coordination Behavior of Corrole Mn-Ⅲ and (MnO)-O-Ⅴ Complexes with NBased Ligands[J]. Acta Phys. Chim. Sin., 2014, 30(2): 265-272. doi: 10.3866/PKU.WHXB201312181
Lee C, Yang W, Parr R G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density[J]. Phys. Rev. B: Condens. Matter, 1988, 37(2): 785-789. doi: 10.1103/PhysRevB.37.785
Becke A D. Density - Functional Exchange - Energy Approximation with Correct Asymptotic Behavior[J]. Phys. Rev. A: At. Mol. Opt. Phys., 1988, 38(6): 3098-3100. doi: 10.1103/PhysRevA.38.3098
Couty M, Hall M B. Basis Sets for Transition Metals: Optimized Outer p Functions[J]. J. Comput. Chem., 1996, 17(11): 1359-1370. doi: 10.1002/(SICI)1096-987X(199608)17:11<1359::AID-JCC9>3.0.CO;2-L
Zhu C, Liang J X, Wang B J, Zhu J, Cao Z X. Significant Effect of Spin Flip on the Oxygen Atom Transfer Reaction from (Oxo)manganese (Ⅴ) Corroles to Thioanisole: Insights from Density Functional Calculations[J]. Phys. Chem. Chem. Phys., 2012, 14(37): 12800-12806. doi: 10.1039/c2cp41647f
Harvey J N, Aschi M, Schwarz H, Koch W. The Singlet and Triplet States of Phenyl Cation[J]. A Hybrid Approach for Locating Minimum Energy Crossing Points Between Non - Interacting Potential Energy Surfaces. Theor. Chem. Acc., 1998, 99: 95-99.
Lu T. SobMECP Program, http://sobereva.com/286.
何婧, 徐志广, 曾允秀, 许旋, 喻兰, 王琦, 刘海洋. 取代基对咔咯锰(Ⅴ)-氧配合物Mn—O的成键影响[J]. 物理化学学报, 2012,7,(28): 1658-1664. HE J, XU Z G, ZENG Y X, XU X, YU L, WANG Q, LIU H Y. Effect of Substituents on Mn—O Bond in Oxo-Manganese(Ⅴ) Corrole Complexes[J]. Acta Phys.-Chim. Sin., 2012, 7(28): 1658-1664.
李皎, 徐艳, 许旋, 徐志广, 刘海洋. 咔咯锰(Ⅲ)配合物催化RNA磷酸二酯类似物HpPNP的水解断裂反应机理[J]. 无机化学学报, 2020,36,(3): 435-442. LI J, XU Y, XU X, XU Z G, LIU H Y. Mechanism of Catalytic Hydrolysis Cleavage of DNA Phosphodiester Analogue HpPNP by Corrole Manganese(Ⅲ) Complex[J]. Chinese J. Inorg. Chem., 2020, 36(3): 435-442.
Liu H Y, Lai T S, Yeung L L, Chang C K. First Synthesis of Perfluorinated Corrole and Its MnO Complex[J]. Org. Lett., 2003, 5(5): 617-620. doi: 10.1021/ol027111i

图 2 三重态时配合物1在OAT过程中各反应状态的几何优化结构
Figure 2 Optimized geometrical structures of each reaction state of complex 1 in the OAT process in the triplet state

图 3 咔咯锰(Ⅴ)氧配合物与苯乙烯氧的转移机理
Figure 3 Oxygen transfer mechanism of manganese(Ⅴ)-oxo corrole complexes with styrene

图 4 配合物1~4与苯乙烯在单重态和三重态下的OAT反应势能图
Figure 4 Potential energy diagram of OAT reaction between complexes 1-4 and styrene in the singlet and triplet states

图 5 配合物4与苯乙烯OAT反应的MECP过程图
Figure 5 MECP process diagram of OAT reaction between complex 4 with styrene
表 1 配合物1~4在单重态和三重态下的几何结构参数
Table 1. Geometric structure parameters of complexes 1-4 in the singlet and triplet states
| Complex | dMn—Na/nm | dMn≡Ob/nm | dMn—4Nc/nm | ∠MnNNNd/(°) | ∠NMnNe/(°) | |||||||||
| Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | |||||
| 1 | 0.191 6 | 0.194 5 | 0.154 9 | 0.158 9 | 0.054 7 | 0.047 2 | 22.90 | 19.51 | 85.31 | 86.67 | ||||
| 2 | 0.191 5 | 0.194 4 | 0.154 6 | 0.158 8 | 0.054 3 | 0.046 9 | 22.73 | 19.40 | 85.38 | 86.73 | ||||
| 3 | 0.191 5 | 0.194 3 | 0.154 5 | 0.158 7 | 0.054 2 | 0.046 7 | 22.69 | 19.34 | 85.40 | 86.77 | ||||
| 4 | 0.191 4 | 0.194 2 | 0.154 3 | 0.158 6 | 0.053 8 | 0.046 5 | 22.54 | 19.29 | 85.46 | 86.77 | ||||
| aAverage distance between the manganese atom and the four nitrogen atoms; bLength of Mn≡O bond; cDistance from the manganese atom to the corrole plane; dDihedral angle between the manganese atom and the corrole plane; eAverage value of the angles between nitrogen atoms, manganese atoms and nitrogen atoms on the corrole plane. | ||||||||||||||
 下载: 导出CSV
下载: 导出CSV
表 2 配合物1~4在单重态和三重态下的NPA电荷和Wiberg键级(WI)
Table 2. NPA charges and Wiberg bond orders (WI) of complexes 1-4 in the singlet and triplet states
| Complex | QMn | QO | Qcora | QSubb | WIMn≡O | |||||||||
| Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | |||||
| 1 | 0.345 | 0.436 | -0.132 | -0.314 | -0.303 | -0.266 | -0.042 | -0.170 | 2.078 | 1.940 | ||||
| 2 | 0.348 | 0.436 | -0.120 | -0.308 | -0.274 | -0.225 | -0.075 | -0.211 | 2.089 | 1.943 | ||||
| 3 | 0.349 | 0.437 | -0.112 | -0.302 | -0.231 | -0.169 | -0.119 | -0.268 | 2.096 | 1.947 | ||||
| 4 | 0.359 | 0.437 | -0.101 | -0.296 | -0.199 | -0.118 | -0.155 | -0.320 | 2.107 | 1.952 | ||||
| a NPA charge of the skeleton of corrole; b Total NPA charge of the three substituents. | ||||||||||||||
下载: 导出CSV
表 3 反应物、过渡态和产物在单重态和三重态下的部分几何结构参数和NPA电荷
Table 3. Selected geometric structure parameters and NPA charges of reactants, transition states and products in the singlet and triplet states
| Complex | Multiplicity | Substance | dMn≡O/nm | dC—O/nm | ∠MnNNN / (°) |
Qβa |
| 1 | Singlet | Reactant | 0.155 1 | 0.383 5 | 22.99 | -0.423 |
| Transition state | 0.164 4 | 0.170 0 | 20.50 | -0.262 | ||
| Product | 0.179 8 | 0.138 7 | 18.60 | -0.158 | ||
| Triplet | Reactant | 0.158 9 | 0.403 3 | 19.69 | -0.423 | |
| Transition state | 0.171 0 | 0.221 1 | 17.55 | -0.354 | ||
| Product | 0.226 4 | 0.144 9 | 12.96 | -0.109 | ||
| 2 | Singlet | Reactant | 0.154 6 | 0.381 2 | 22.77 | -0.423 |
| Transition state | 0.164 1 | 0.170 9 | 29.21 | -0.265 | ||
| Product | 0.180 2 | 0.138 4 | 18.72 | -0.158 | ||
| Triplet | Reactant | 0.158 8 | 0.399 6 | 19.40 | -0.423 | |
| Transition state | 0.168 7 | 0.205 7 | 16.75 | -0.296 | ||
| Product | 0.221 9 | 0.145 0 | 13.60 | -0.107 | ||
| 3 | Singlet | Reactant | 0.154 5 | 0.617 8 | 22.42 | -0.423 |
| Transition state | 0.163 9 | 0.171 3 | 20.36 | -0.268 | ||
| Product | 0.173 8 | 0.146 1 | 18.98 | -0.189 | ||
| Triplet | Reactant | 0.158 6 | 0.459 7 | 19.27 | -0.423 | |
| Transition state | 0.167 9 | 0.248 8 | 17.75 | -0.378 | ||
| Product | 0.215 6 | 0.145 3 | 14.37 | -0.100 | ||
| 4 | Singlet | Reactant | 0.154 2 | 0.392 0 | 22.52 | -0.424 |
| Transition state | 0.163 6 | 0.172 1 | 20.20 | -0.271 | ||
| Product | 0.180 1 | 0.138 4 | 18.30 | -0.162 | ||
| Triplet | Reactant | 0.158 5 | 0.398 1 | 19.19 | -0.424 | |
| Transition state | 0.166 6 | 0.258 3 | 17.66 | -0.373 | ||
| Product | 0.202 2 | 0.146 0 | 14.88 | -0.094 |
下载: 导出CSV
表 4 配合物4在OAT反应中MECP的部分几何构型
Table 4. Selected geometric structure parameters of MECP of complex 4 in OAT process
| Reaction state | dMn≡O/nm | dC—O/nm | |||
| Singlet | Triplet | Singlet | Triplet | ||
| Reactant | 0.154 2 | 0.158 5 | 0.392 0 | 0.398 1 | |
| MECP | 0.157 0 | 0.157 0 | 0.333 9 | 0.333 9 | |
| Transition state | 0.163 6 | 0.166 6 | 0.172 1 | 0.258 3 | |
| Product | 0.180 1 | 0.202 2 | 0.138 4 | 0.146 0 | |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
