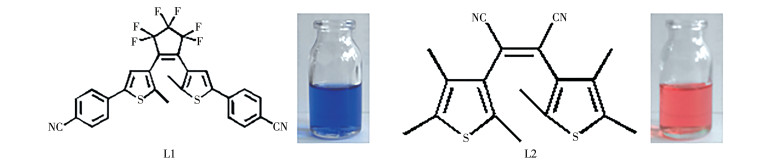
图 1.
L1和L2的结构和颜色
Figure 1.
Structures and colors of L1 and L2

日本科学家Masahiro Irie于1988年首次合成了具有光致变色性质的二芳基乙烯[1],其光致变色反应实质是光激发的异构化反应,由通常无色态的己三烯环化生成有色态的环己二烯。二芳基乙烯因具有灵敏的光响应性、特征的开/闭环双稳定性以及优异的光致变色和耐疲劳性,被公认为是最有可能实用化的高密度信息存储材料。
目前对二芳基乙烯的研究主要集中在单组分体系,如新分子设计和合成[2]、光信息的无损读取[3]、光致变色高聚物[4]以及功能配合物[5]等方面。单组分体系的读取记录位点上只含有一种光致变色组分,因此该点上只可表达2种状态,而多组分(多色)体系含有多种光致变色组分,就可在一个位点上记录更多的数据。例如,双组分体系一个记录位点可以表达4(即22)种状态,这使得多组分光致变色材料在超高密度的数据存储和无损读取方面具有很大优势,尤其适用于多频光学存储和多色显示器。目前研究多组分二芳基乙烯体系主要基于以下几种策略:
(1) 多组分混合体系,即将几种二芳基乙烯混合于一种溶液[6]或形成一种晶体[7-8]中。其突出特点是容易合成,可轻松实现多种组分的筛选,并且颜色的深浅可通过各组分的比例调节。但显而易见,溶液体系不具备实用性;晶相产物难以合成,或一种组分所占百分比很小,甚至X射线单晶衍射分析都无法进行[7],这毫无疑问制约了其多色性质的调控。
(2) 共价键结合的多组分单分子体系,即设计一种含有多种光致变色单元的新分子,各单元之间通过共价键连接。这种体系又可分为2类:一是多种光致变色单元都是二芳基乙烯[9-10],或通过桥连单元形成二聚体[11]、三聚体[12]和四聚体[13]等;二是多个组分中至少有一个是二芳基乙烯,其余的光致变色单元是啰吡喃[14]、萘并萘醌[15]、偶氮苯[16]和恶唑烷[17]等光致变色分子。这种体系比多组分混合体系具有更高的图像分辨率和更稳定的储存性,但相连的二芳基乙烯单元相互影响。此外,多色性质的调控必须重新设计新分子结构,调控周期长,不易实现。
(3) 多组分光致变色共/均聚物,即将二芳基乙烯接枝至聚合物侧链[18-19]。与前面的2个策略相比,多组分聚合物具有合成简便、易成膜、固相的光致变色性能稳定等突出优点,是一种可实用化的策略。但其不可避免的缺点是光致变色单元的百分含量低(最高也仅为53%[20])、多色性质差。通过将二芳基乙烯单元引入均聚物主链可以提高其含量,但聚合产率低,聚合物疲劳性差的问题仍然存在。
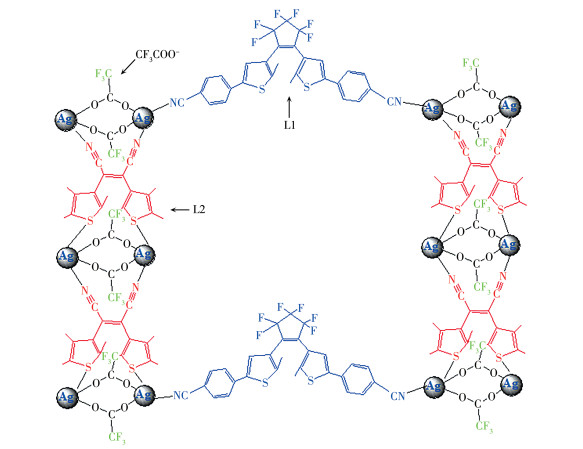
基于此研究背景,我们从晶体工程角度出发,提出设计一个配位键自组装形成的双组分金属配合物体系。为了成功地实现4个色态的转变,选择了2个二芳基乙烯化合物1,2-双(2′-甲基-5′-(4″-苯腈)-3′-噻吩)全氟环戊烯(L1)和顺-1,2-二氰基-1,2-双(2′,4′,5′-三甲基-3′-噻吩)乙烯(L2)为配体,其闭环体颜色分别为蓝色和红色(L1:λmax, closed=606 nm,L2:λmax, closed=512 nm,图 1),光环化反应的激发波长分别为254和405 nm[21],间隔较好;且2个配体具有相同的配位官能团氰基,可等同地与金属银离子组装形成结构稳定的配合物。合成的配合物1用红外光谱、核磁共振氢谱及质谱进行了结构表征。以直接混合的配体体系为对照,用UV-Vis光谱法研究了配合物1在四氢呋喃(THF)溶液和聚甲基丙烯酸甲酯(PMMA)薄膜中的多色态光致变色性质。
本实验过程所用试剂均为市售。红外测试以KBr为压片介质,在日本岛津公司的IR Prestige-21型傅里叶变换红外光谱仪上测定。UV-Vis光谱分析采用岛津UV-3600紫外分光光度计,溶液浓度约为1 μmol·L-1,光照采用100 W Xe灯,不同波长的光源通过滤光片实现(andover254 FS25、RANYAN BP 550-10 K)。核磁共振氢谱测试使用德国Bruker公司的AVANCE Ⅲ-400 MHz型超导核磁共振波谱仪(溶剂为CDCl3)。质谱测试采用德国Bruker公司的MicrOTOF-Q Ⅱ型电喷雾四级杆串联飞行质谱仪,采用电喷雾离子源(ESI),测定质量(m/z) 范围为100~3 000。
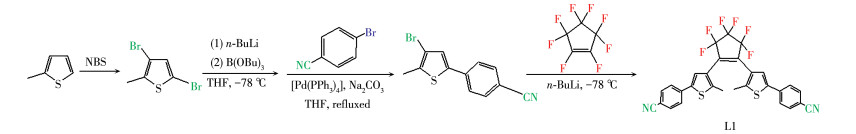
L1的合成:配体L1的合成采用改进的文献方法[22],如Scheme 1所示。以2-甲基噻吩为原料,经过溴化、锂化、硼酸化以及铃木偶联得到3-溴-2-甲基-5 -(4′-苯腈)噻吩,之后进行锂化反应以及全氟环戊烯的取代反应得到产物L1,然后用柱色谱提纯,经蒸发溶剂、重结晶后得到L1的固体粉末。IR(KBr压片,cm-1):2 360(m), 2 218(m), 1 595(m),1 261(m),1 101 (s),983(s),823(s),743(m)。1H NMR(CDCl3,400 MHz):δ 7.68(d,4H,J=8.5 Hz,cyanophenyl H),7.63(d,4H,J =8.6 Hz,cyanophenyl H),7.40(s,2H,thienyl H),2.00 (s,6H,methyl)。
配合物1的合成:称取L1(0.057 g,0.1 mmol)和L2(0.033 g,0.1 mmol)溶解于混合溶剂THF和CHCl2(1∶1,V/V)中,加入AgCF3COO(0.044 g,0.2 mmol),避光搅拌后缓慢蒸发溶液,一周后得到黄色晶状粉末,然后在THF中重结晶得到配合物1晶体。
PMMA膜样品制备:60 ℃下,将1.0 g PMMA加入10 mL的THF中至完全溶解。吸取约0.25 mL上述溶液旋涂于石英载玻片上,待溶剂挥发后得到无色透明的PMMA膜参比样品。配体混合体系和配合物1的PMMA膜的制备方法同上,L1和L2的物质的量相同,配体的质量分数约为2%。
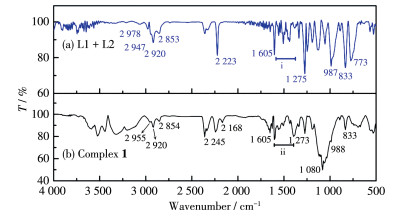
为了研究金属离子配位后的结构变化,以L1和L2直接混合的样品(1∶1,n/n,记为L1+L2)作为对照。如图 2b所示,红外光谱中2 976、2 951、2 920、2 858 cm-1的峰归属于配合物1中C—H的对称或非对称伸缩振动峰(配体的对应吸收在2 978、2 947、2 920和2 853 cm-1)。2 226 cm-1的峰归属于C≡N的伸缩振动峰(配体的对应吸收在2 223 cm-1)。1 603 cm-1的峰为C=C伸缩振动(配体的对应吸收在1 605 cm-1)。ⅱ区归属为苯环和噻吩环的骨架振动峰,分别位于1 558、1 543、1 506、1 437和1 339 cm-1处(配体ⅰ区在1 558、1 541、1 508、1 441和1 339 cm-1)。1 275 cm-1归属于C—F键的特征吸收峰(配体的对应吸收在1 275 cm-1)。
值得说明的是,配合物1的红外光谱中出现了2个L1+L2没有的强吸收峰,分别位于1 676和1 202 cm-1处,应归属为阴离子CF3COO-中C=O和C—O的伸缩振动峰(游离CF3COO-的对应吸收峰分别位于1 682和1 209 cm-1处)。而且与配体直接混合体系相比,C≡N的伸缩振动峰蓝移3 cm-1,表明配体中的氰基可能已经与银离子配位。由以上分析初步判定,配合物1已经形成。
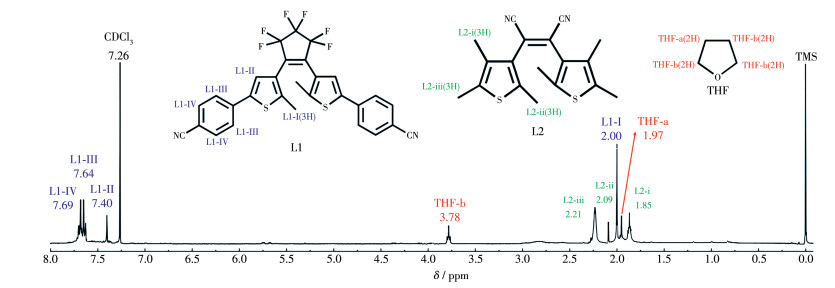
配合物1的核磁共振氢谱如图 3所示,溶剂为添加了标准物质四甲基硅烷(TMS)的CDCl3。图中δ=2.00(d,J=8.7 Hz,4H)和7.69(d,J=8.7 Hz,4H)的信号分别归属为配体L1的噻吩环甲基中的质子L1-Ⅰ和L1-Ⅱ,δ=7.64(s,6H)和7.40(s,2H)的信号分别归属于苯环上氰基间位的质子L1-Ⅲ和苯环上氰基邻位的质子L1-Ⅳ。δ=1.85(s,3H)、2.09(s,3H)和2.21(s,3H) 的信号分别归属为配体L2的噻吩环上甲基中的质子L2-ⅰ、L2-ⅱ和L2-ⅲ。这些信号峰进一步表明配合物1中包含配体L1和L2,与前面红外分析的结果一致。此外,δ=1.97(d,J=6.5 Hz,4H)和3.78(t,J=6.6 Hz,4H)的信号应归属为THF溶剂分子中的质子THF-a和THF-b,因此,配合物1的核磁测试样品中含有溶剂THF。通过对上述配体L1和L2的信号进行进一步的面积积分计算,可得配合物1中L1与L2之比为1∶2。
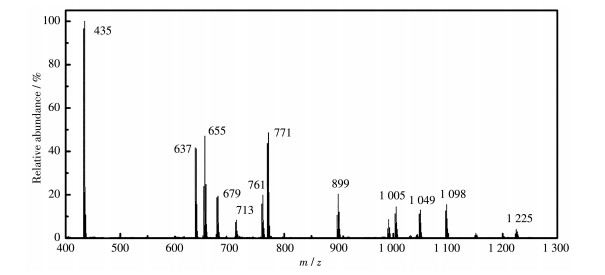
尽管红外光谱和核磁共振谱图的分析结果均初步证实形成了配合物1,但仍没有Ag(Ⅰ)离子直接配位的证据,因此继续对配合物1进行了质谱分析(图 4),表 1为质谱图的离子碎片归属,其中m/z为713、771、1 005、1 049、1 098和1 225的碎片离子全部都含有Ag(Ⅰ)、L1和L2,清楚地表明Ag(Ⅰ)与2个配体都发生了配位反应。m/z为655、899和1 225的碎片离子均含有CF3COO-,表明阴离子也参与了配位,进一步支持了前面红外分析得到的阴离子参与配位的结论。
 下载:
导出CSV
下载:
导出CSV
| m/z | Assignment | m/z | Assignment | |
| 435 | [Ag(L2)]+ | 771 | [Ag(L1)2(L2…CN)]2+* | |
| 637 | [Ag(CN)(L1)2]2+ | 899 | [Ag2(CF3COO)(L1)]+ | |
| 655 | [Ag2(CF3COO)(L2)]+ | 1 005 | [Ag(L1)(L2)]+ | |
| 679 | [Ag(L1)]+ | 1 049 | [Ag(COO)(L1)(L2)]+ | |
| 713 | [Ag2(L1)(L2)2]2+ | 1 098 | [Ag(L1)2(L2)2(L2…CN)]2+ | |
| 761 | [Ag(L2)2]+ | |||
 | ||||
综合以上红外光谱、核磁共振氢谱和质谱的分析结果,可以得出银离子与2个配体均配位,且2个配体的比例为1∶2,阴离子也参与配位,再结合我们前期得到的AgCF3COO分别和配体L1和L2合成的配合物单晶结构[23-24],初步推定合成的配合物1可能的结构式如Scheme 2所示。
在THF溶液中配体L1和L2光致闭环反应激发光波长分别为254和405 nm,两者相差151 nm;L1和L2的闭环体紫外最大吸收波长分别为606 nm[23, 25] (颜色为蓝色)和512 nm[1](颜色为红色),间隔较好,因此从理论上推断,通过不同波长的光激发L1+L2(1∶ 1,n/n)双组分混合体系应可以实现分步的环化-环转化反应,即实现光致多色态变化。为论述方便,以下用1o表示L1开环体,1c表示L1闭环体,2o表示L2开环体,2c表示L2闭环体。
未经紫外光照射前,双组分配体混合体系1o2o为淡黄色,最大吸收峰在301和324 nm处,这归因于其噻吩环结构产生的π-π*和n-π*的混合跃迁(图 5a)。在254 nm紫外光持续照射下,混合体系1o2o由淡黄色变为紫色,其UV-Vis光谱中的可见区出现2个新的吸收峰,分别位于537和605 nm(游离态2c最大吸收为512 nm,游离态1c最大吸收为606 nm),经过60 min光照后,可见区的这2个吸收峰强度不再增强,表明达到光静止状态(PSS)。这些颜色和光谱变化说明混合体系1o2o中2个配体在254 nm光照射下,各自发生了光致闭环反应,生成了闭环体混合体系1c2c。与游离配体相比较,混合体系中归属为配体L2的闭环体吸收峰位于537 nm,产生了25 nm红移,这可能归因于混合体系中2个配体间的弱相互作用及配体和溶剂间的弱相互作用。继续用大于550 nm的可见光照射上述溶液,颜色由紫色变回为黄色,且新的可见区吸收峰强度逐渐减弱直至恒定,表明光静止态的混合体系又变回为1o2o(图 5b)。因此,双组分配体混合体系在254 nm和大于550 nm的光的交替照射下可以实现溶液相的双色态可逆变化。
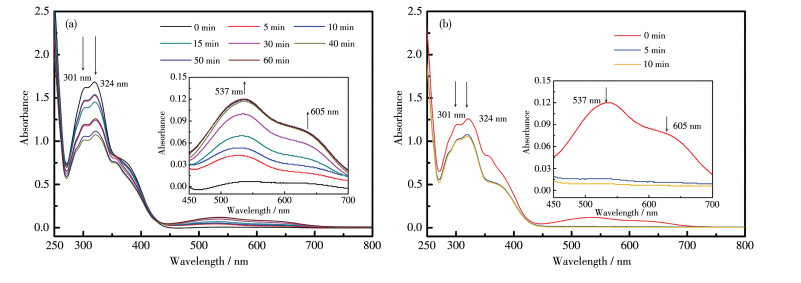
(a)λ=254 nm, (b) λ > 550 nm; Inset: corresponding enlarged view
前期研究表明,在405 nm的光激发下游离2o会变为2c[21, 26],而1o在此波长激发下不会发生光致闭环反应,因此选用405 nm波长作为激发光源可选择性地使L2发生闭环反应。
如图 6a,当采用405 nm波长照射上述1o2o的THF溶液,可见区在537和605 nm仍旧出现了新的归属为1c和2c的吸收峰,颜色由黄色变为红紫色,50 min后达到光静止态,这说明尽管1o在405 nm处吸收峰较弱,但配体L1在此波长光照射下也发生了闭环反应生成了1c,即采用405 nm波长导致2个配体同时闭环。继续用波长254 nm的光照射(图 6b),537 nm处的吸收不再增强,但605 nm处的吸收又逐渐增强,溶液变为紫色,20 min后605 nm处吸收峰达到光静止状态。这一现象表明之前用405 nm激发并未使1o最大限度地转化为1c,其红紫色光静止态应为1o1c2c。因此,采用波长为405和254 nm的光组合照射可以实现混合体系1o2o(黄色)→1o1c2c (红紫色)→1c2c(紫色)的颜色变化(图 7a)。
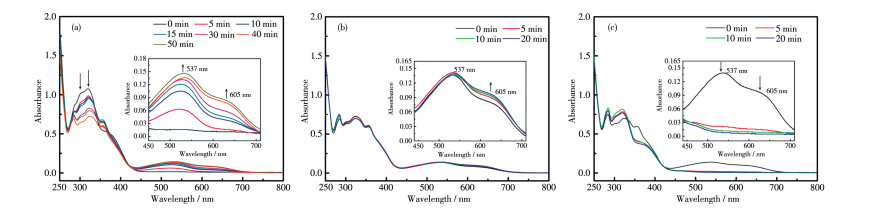
(a) λ=405 nm, (b) λ=254 nm, (c) λ > 550 nm; Inset: corresponding enlarged view
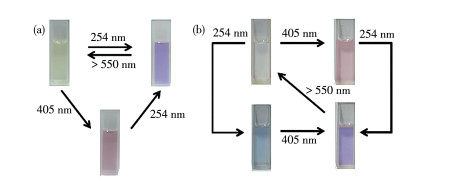
基于上述对混合配体体系的初步研究结果,以254和405 nm组合光作为激发光,继续研究配合物1在THF溶液中的光致多色态变化性质。
如图 8a,先用波长254 nm的紫外光照射配合物1的THF溶液,UV-Vis光谱在600 nm处出现归属为1c的吸收峰,溶液颜色由浅黄色变为蓝色,并随光照时间强度逐渐增加,80 min后不再变化,达到光静止状态,说明配合物1从1o2o变为1c2o。和配体混合体系(L1+L2)不同,配合物1在254 nm光照射下,配体L2并未转变为闭环体,很好地实现了选择性光致闭环反应。
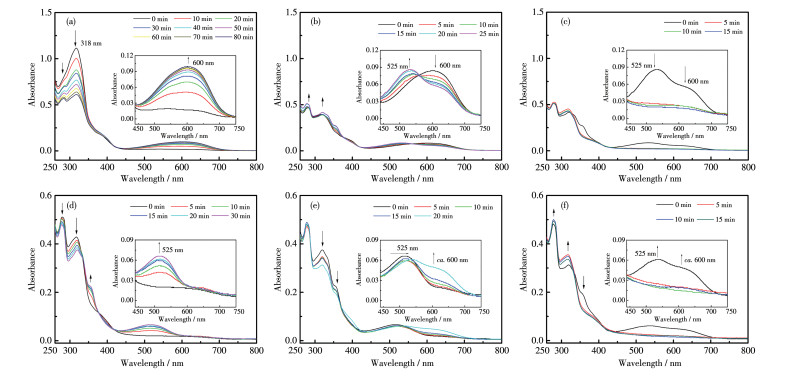
In the order of λ=254 nm (a), λ=405 nm (b) and λ > 550 nm (c); In the order of λ=405 nm (d), λ=254 nm (e) and λ > 550 nm (f); Inset: corresponding enlarged view
之后用波长405 nm光照射,UV-Vis光谱在525 nm处出现归属为2c的吸收峰,其强度随光照时间延长而不断增加,与此同时600 nm处的峰强度持续减弱(图 8b),表明配体L1在405 nm光照射下,部分闭环态又转变为开环态。光照25 min后,2处峰值均不再变化,溶液颜色变为紫色。这些颜色和光谱变化说明配合物1由1c2o转变为1c1o2c。值得指出的是,配合物1中归属为2c的吸收峰位于525 nm,与游离L2的最大吸收波长512 nm相比,红移13 nm,这归因于Ag(Ⅰ)与L2发生了配位反应,调节了其光致变色性质。
此后用λ>550 nm的光照射,525和600 nm峰的强度均不断地减弱,直至15 min后趋于平缓达到稳定(图 8c),溶液颜色恢复为黄色,说明体系回到1o2o状态。因此,通过254和405 nm组合光照射,配合物1可以实现黄色→蓝色→紫色→黄色的颜色变化(图 7b)。
在此基础上,改变组合光的辐照顺序,继续研究了配合物1在THF溶液中的光致多色态变化性质。先用波长为405 nm的光照射配合物1的THF溶液,UV-Vis光谱(图 8d)在525 nm处出现归属为2c的吸收峰,30 min后强度不再增强,溶液颜色由浅黄色变为微红色,表明体系由1o2o变为1o2c。即用405 nm光激发实现了配合物1中配体L2的选择性闭环反应。克服了配体混合体系中2个配体同时闭环的缺点。
接着用波长为254 nm的光继续照射,归属为1c的600 nm峰出现,并逐渐增强,525 nm峰强度基本不变(图 8e),溶液颜色由微红色变为紫色,20 min后到达光静止态。这表明配合物由1o2c变为1c2c。最后用λ>550 nm的光照射15 min后,配合物1回到初始的1o2o态(图 8f)。
综上,在THF溶液中通过引入Ag(Ⅰ)与2个配体配位形成配合物,采用254、405 nm和大于550 nm组合光作为激发光,配合物体系可以实现用254 nm光照选择性使L1闭环,用405 nm光照选择性使L2闭环,克服了混合体系2个配体相互干扰的缺点。通过调节组合光的辐照顺序,可以实现黄色、蓝色、红色、紫色4个色态的变化。
在实际中,光致变色分子通常需要附着在一定的载体上,如制成固相薄膜才能应用;且在固相薄膜中,配合物分子被限定在特定的构型,与溶液相的结构也可能不同。因此下面继续以双组分混合体系PMMA膜为对照,研究配合物1在PMMA膜中光致多色变化性质。
采用254、405 nm和大于550 nm的辐照顺序研究了L1+L2混合体系在PMMA膜中的光致变色性质,其UV-Vis光谱图见图 9。由光谱图变化可以看出,混合配体经历了1o2o(黄)→1c2o(蓝)→1c2c(紫) →1o2o(黄)的变化(图 10a)。和溶液相不同,采用254和405 nm的激发光在PMMA膜中可以独立激发2个配体的闭环反应。
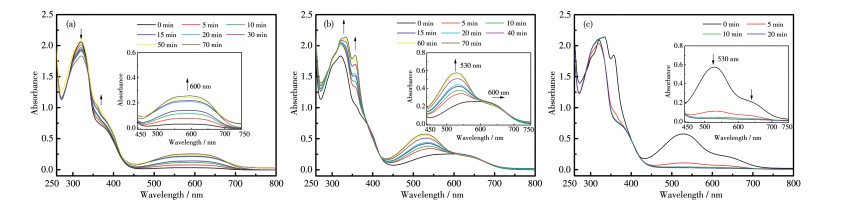
Inset: corresponding enlarged view

采用405、254 nm和大于550 nm的波长光的顺序辐照L1+L2混合体系的PMMA膜,得到的UV-Vis光谱图见图 11。可以发现,混合配体经历了1o2o (黄)→1o2c(红紫)→1c2o2c(紫)→1o2o(黄)的结构变化(图 10a)。即在PMMA膜中,254 nm光照射成功激发了L1的环化反应但同时也引发了L2的开环反应,这对于双组分体系是不利的。
Inset: corresponding enlarged view
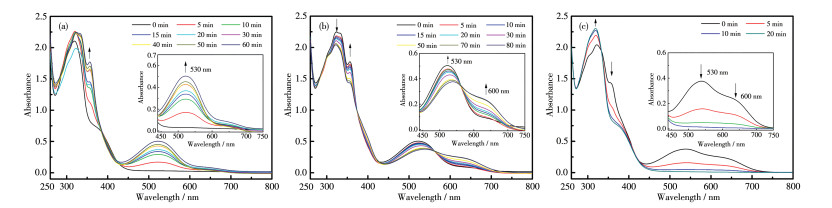
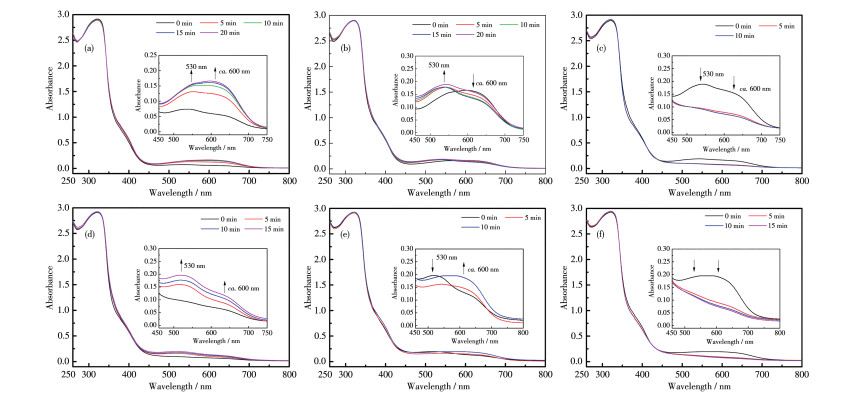
图 12a~12c为采用254、405 nm和大于550 nm组合光顺序照射配合物1的PMMA膜的UV-Vis光谱图,可以看出其经历了1o2o(黄)→1c2o2c(蓝)→1c2c(紫)→1o2o(黄)的结构变化(图 10b)。归属为2c的吸收峰位于530 nm,相比游离配体(512 nm)红移约18 nm,相比溶液相配合物归属为2c的吸收峰(525 nm)红移5 nm,表明配合物1在PMMA膜中的结构和溶液相不同。和溶液相配合物的光致变色性质相比,用254 nm光照射时PMMA膜中L1的光致环化时间由80 min缩短至20 min,但有部分L2也发生了环化反应;用405 nm光照射时,L1发生开环的比例减小;与配体混合体系在PMMA膜中的光致变色性质相比,3个波长照射到达光静止态的时间也大大缩短(表 2)。
In the order of λ=254 nm (a), λ=405 nm (b) and λ > 550 nm (c); In the order of λ=405 nm (d), λ=254 nm (e) and λ > 550 nm (f); Inset: corresponding enlarged view
下载:
导出CSV
| System | λmax(UV-Vis)/nm | Time/min | |
| 254, 405, > 550 nma | 405, 254, > 550 nmb | ||
| L1 | 606 | — | — |
| L2 | 512 | — | — |
| L1+L2 (THF) | 605, 537 | 60 (254, > 550 nm) | 70 |
| Complex 1 (THF) | 600, 525 | 80 | 30 |
| L1+L2 (PMMA) | 599, 530 | 70 | 60 |
| Complex 1 (PMMA) | 600, 530 | 20 | 15 |
| a Irradiation in the order of λ=254 nm, λ=405 nm and λ > 550 nm; b Irradiation in the order of λ=405 nm, λ=254 nm and λ > 550 nm | |||
图 12d~12f为采用405、254 nm和大于550 nm组合光顺序照射配合物1的PMMA膜的UV-Vis光谱图,可以发现,配合物1先后经历了1o2o(黄)→ 1o2c(红色)→1c2o2c→1c2c(紫)→1o2o(黄)的结构和颜色变化(图 10b),和混合体系在PMMA膜中的光致结构变化类似,但用254 nm光照射时,L2的开环反应比例显著减小。
综上,通过UV-Vis光谱研究发现,灵活采用254、405 nm和大于550 nm组合光的辐照顺序,配合物1在PMMA膜中可以实现红、黄、蓝、紫4个色态的变化(图 10b),实现了我们对于光致多色态转变的设想。与混合配体体系相比,金属Ag(Ⅰ)离子的引入在很大程度上抑制了2个配体间的相互影响。
由以上研究结果可以发现,引入Ag(Ⅰ)离子,并没有抑制L1和L2的光致变色性质。与游离配体相比,在溶液和薄膜相,配体L2的最大吸收峰都有不同程度的红移(表 2),如在溶液相红移13 nm,在PMMA膜中红移18 nm,表明配体L2与Ag(Ⅰ)配位后配体的结构发生了较大的变化,红移数值不同是因为L2在溶液相和薄膜相的结构不同。而配体L1的最大吸收峰配位后在溶液相和薄膜相均发生了6 nm蓝移,说明配合物中L1在溶液相和薄膜相的结构类似,且配位后配体L2的闭环体最大吸收波长变化大于L1,这是由于配体L2具有更为柔性的结构,而L1结构相对刚性较大,发生光致闭环反应后,柔性的L2的空间结构变化更大。
此外,与混合配体体系相比,配合物1在PMMA膜中达到光稳态时间大幅缩短,这些都反映出Ag(Ⅰ) 离子配位对光致变色性质的调控作用。
通过引入金属Ag(Ⅰ)离子与双光致变色配体发生配位反应形成配合物1,达到了调节和改善多色态光致变色性质的目的。在溶液相和PMMA薄膜中,通过设计和组合不同波长的光照射配合物,可选择性地使2个配体独立地发生光致闭环反应,成功实现黄色、蓝色、红色及紫色4个色态间的转变。在溶液相中配合物1的多色态性质克服了配体混合体系中2个配体同时闭环的缺点,且在PMMA薄膜中1的2个配体间光反应相互影响的比例降低。因此,通过金属离子与光致变色配体的配位反应可以改善配体混合体系的固有缺陷,成功实现4个色态间的转变。
在今后的研究中,可设计和采用具有更大闭环体最大吸收波长差值的2个配体,进一步克服配体间光致变色性质相互影响的弊病,或者通过在金属离子与配体间插入间隔基来减弱这种相互干扰。同时还可通过调节:(1) 光致变色单体的形状、大小和配位原子;(2) 金属离子的种类和配位数;(3) 阴离子的形状、半径和配位能力这3个途径对配合物的拓扑结构进行调控,从而得到具有不同维度和建筑结构特点的框架结构,最终实现灵活可调的多色光致变色性质。
Irie M, Mohri M. J. Org. Chem., 1988, 53(4): 803-808 doi: 10.1021/jo00239a022
Pu S Z, Ma L L, Liu G, Ding H C, Chen B. Dyes. Pigm., 2015, 113(1): 70-77
Wu T Q, Branda N R. Chem. Commun., 2016, 52(23): 8636-8644
(a) Chen H, Yao X Y, Ma X, Tian H. Adv. Opt. Mater., 2016, 4(9): 1397-1401
(b) Wesenhagen P, Areephong J, Landaluce T F, Heureux N, Katsonis N, Hjelm J, Rudolf P, Browne W R, Feringa B L. Langmuir, 2008, 24(12): 6334-6342
(a) Cao D K, Wei R H, Li X X, Chen F, Ward M D. Dalton Trans., 2015, 44: 4289-4296
(b) Li B, Wen H M, Wang J Y, Shi L X, Chen Z N. Inorg. Chem., 2013, 52(21): 12511-12520
(c) Nakai H, Matsuba K, Akimoto M, Nozaki T, Matsumoto T, Isobe K, Irie M, Ogo S. Chem. Commun., 2016, 52(23): 4349-4352
(a) Tsivgoulis G M, Lehn J M. Adv. Mater., 1997, 9(8): 627-630
(b) Acebes A F, Lehn J M. Adv. Mater., 1999, 11(11): 910-913
(a) Morimoto M, Kobatake S, Irie M. Adv. Mater., 2002, 14(15): 1027-1029
(b) Yamada T, Kobatake S, Masahiro I. Bull. Chem. Soc. Jpn., 2002, 75(1): 167-173
Morimoto M, Kobatake S, Irie M. J. Am. Chem. Soc., 2003, 125(36): 11080-11087 doi: 10.1021/ja035277o
(a) Higashiguchi K, Matsuda K, Irie M. Angew. Chem. Int. Ed., 2003, 42(30): 3537-3540
(b) Peters A, Branda N R. Adv. Mater. Opt. Electron., 2000, 10(6): 245-249
Higashiguchi K, Matsuda K, Tanifuji N, Irie M. J. Am. Chem. Soc., 2005, 127(25): 8922-8923 doi: 10.1021/ja051467i
(a) Kobatake S, Irie M. Tetrahedron, 2003, 59(42): 8359-8364
(b) Yagi K, Irie M. Chem. Lett., 2003, 32(9): 848-849
(c) Matsuda K, Irie M. J. Am. Chem. Soc., 2001, 123(40): 9896-9897
Zeng D X, Chen Y. Chin. J. Chem., 2006, 24(2): 264-268 doi: 10.1002/cjoc.200690050
Kaieda T, Kobatake S, Miyasaka H, Murakami M, Iwai N, Nagata Y, Irie M. J. Am. Chem. Soc., 2002, 124(9): 2015-2024 doi: 10.1021/ja0115722
(a) Frigoli M, Mehl G H. Angew. Chem. Int. Ed., 2005, 44(32): 5048-5052
(b) Delbaere S, Vermeersch G, Frigoli M, Mehl G H. Org. Lett., 2006, 8(21): 4931-4934
(c) Delbaere S, Micheau J C, Frigoli M, Mehl G H, Vermeersch G. J. Phys. Org. Chem., 2007, 20(11): 929-935
(d) Delbaere S, Vermeersch G, Frigoli M, Mehl G H. Org. Lett., 2010, 12(8): 4090-4093
Myles A J, Wigglesworth T J, Branda N R. Adv. Mater., 2003, 15(9): 745-748 doi: 10.1002/adma.200304917
He Y N, Zhu Y, Chen Z, He W, Wang X G. Chem. Commun., 2013, 49: 5556-5558 doi: 10.1039/c3cc41538d
Sevez V, Gan J, Delbaere S, Vermeersch G, Sanguinet L, Levillainc E, Pozzo J L. Photochem. Photobiol. Sci., 2010, 9(2): 131-135 doi: 10.1039/b9pp00127a
Wang S, Li X C, Chen B, Luo Q F, Tian H. Macromol. Chem. Phys., 2004, 205(11): 1497-1507 doi: 10.1002/macp.200400106
Wigglesworth T J, Branda N B. Chem. Mater., 2005, 17(22): 5473-5480 doi: 10.1021/cm051231d
Li X C, Tian H. Macromol. Chem. Phys., 2005, 206(17): 1769-1777 doi: 10.1002/macp.200500190
Han J, Nabei A, Suenaga Y, Maekawa M, Isihara H, Kuroda-Sowa T, Munakata M. Polyhedron, 2006, 25(13): 2483-2490 doi: 10.1016/j.poly.2006.02.013
Liu G, Tu Q, Zhang Q, Fan C, Yang T. Acta Cryst., 2006, 62(7): 3028-3030
Han J, Li Q, Yu Z, Quan C Y, Liu X, Han J C. Inorg. Chim. Acta, 2020, 509: 119666 doi: 10.1016/j.ica.2020.119666
Konaka H, Wu L P, Munakata M, Kuroda-Sowa T, Suenaga Y. Inorg. Chem., 2003, 42(6): 1928-1934 doi: 10.1021/ic020525n
Castagna R, Nardone V, Pariani G, Parisini E, Bianco A. J. Photochem. Photobiol. A, 2016, 325: 45-54 doi: 10.1016/j.jphotochem.2016.04.001
Han J, Li S, Yu Z, Chen H, Zang Y, Wang Q J. Inorg. Chim. Acta, 2017, 464: 11-17 doi: 10.1016/j.ica.2017.04.039

图 5 L1+L2的THF溶液经不同波长光照射的UV-Vis光谱图
Figure 5 UV-Vis spectra of L1+L2 in THF upon irradiation with different wavelengths
(a)λ=254 nm, (b) λ > 550 nm; Inset: corresponding enlarged view

图 6 L1+L2的THF溶液经不同波长光照射的UV-Vis光谱图
Figure 6 UV-Vis spectra of L1+L2 in THF upon irradiation with with different wavelengths
(a) λ=405 nm, (b) λ=254 nm, (c) λ > 550 nm; Inset: corresponding enlarged view

图 7 L1+L2 (a)和配合物1 (b)在THF中的光致变色图
Figure 7 Photochromic pictures of L1+L2 (a) and complex 1 (b) in THF

图 8 不同波长组合光照射配合物1的THF溶液后的UV-Vis光谱图
Figure 8 UV-Vis spectra of complex 1 in THF solution upon irradiation with combined light of different wavelengths
In the order of λ=254 nm (a), λ=405 nm (b) and λ > 550 nm (c); In the order of λ=405 nm (d), λ=254 nm (e) and λ > 550 nm (f); Inset: corresponding enlarged view

图 9 不同波长组合光照射L1+L2的PMMA膜后的UV-Vis光谱图: 按λ=254 nm (a), λ=405 nm (b)和λ > 550 nm (c)的顺序
Figure 9 UV-Vis spectra of L1+L2 in PMMA film upon irradiation with combined light of different wavelengths: in the order of λ=254 nm (a), λ=405 nm (b) and λ > 550 nm (c)
Inset: corresponding enlarged view

图 10 L1+L2 (a)和配合物1 (b)在PMMA膜中的光致变色图
Figure 10 Photochromic pictures of L1+L2 (a) and complex 1 (b) in PMMA films

图 11 不同波长组合光照射L1+L2的PMMA膜后的UV-Vis光谱图: 按λ=405 nm (a), λ=254 nm (b)和λ > 550 nm (c)的顺序
Figure 11 UV-Vis spectra of L1+L2 in PMMA film upon irradiation with combined light of different wavelengths: in the order of λ=405 nm (a), λ=254 nm (b) and λ > 550 nm (c)
Inset: corresponding enlarged view

图 12 不同波长组合光照射配合物1的PMMA膜后的UV-Vis光谱图
Figure 12 UV-Vis spectra of complex 1 in PMMA film upon irradiation with combined light of different wavelengths
In the order of λ=254 nm (a), λ=405 nm (b) and λ > 550 nm (c); In the order of λ=405 nm (d), λ=254 nm (e) and λ > 550 nm (f); Inset: corresponding enlarged view
表 1 配合物1质谱图的归属
Table 1. Assignments of MS spectrum of complex 1
| m/z | Assignment | m/z | Assignment | |
| 435 | [Ag(L2)]+ | 771 | [Ag(L1)2(L2…CN)]2+* | |
| 637 | [Ag(CN)(L1)2]2+ | 899 | [Ag2(CF3COO)(L1)]+ | |
| 655 | [Ag2(CF3COO)(L2)]+ | 1 005 | [Ag(L1)(L2)]+ | |
| 679 | [Ag(L1)]+ | 1 049 | [Ag(COO)(L1)(L2)]+ | |
| 713 | [Ag2(L1)(L2)2]2+ | 1 098 | [Ag(L1)2(L2)2(L2…CN)]2+ | |
| 761 | [Ag(L2)2]+ | |||
| | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 L1+L2和配合物1的光致变色性质
Table 2. Photochromism of L1+L2 and complex 1
| System | λmax(UV-Vis)/nm | Time/min | |
| 254, 405, > 550 nma | 405, 254, > 550 nmb | ||
| L1 | 606 | — | — |
| L2 | 512 | — | — |
| L1+L2 (THF) | 605, 537 | 60 (254, > 550 nm) | 70 |
| Complex 1 (THF) | 600, 525 | 80 | 30 |
| L1+L2 (PMMA) | 599, 530 | 70 | 60 |
| Complex 1 (PMMA) | 600, 530 | 20 | 15 |
| a Irradiation in the order of λ=254 nm, λ=405 nm and λ > 550 nm; b Irradiation in the order of λ=405 nm, λ=254 nm and λ > 550 nm | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们