Received Date:
11 May 2021 Revised Date:
06 August 2021 Available Online:
10 October 2021
Abstract:
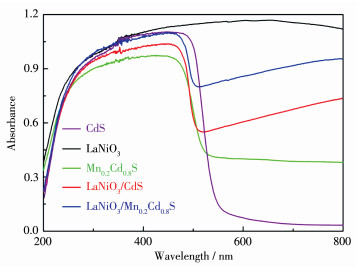
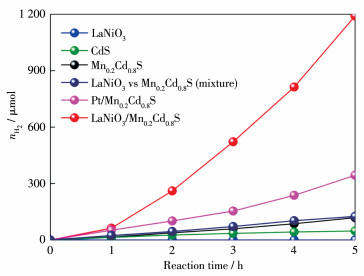
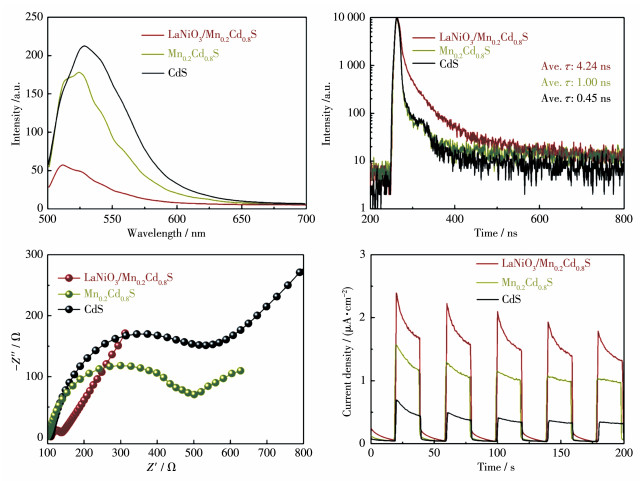
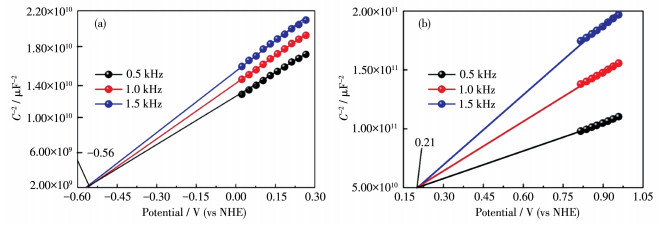
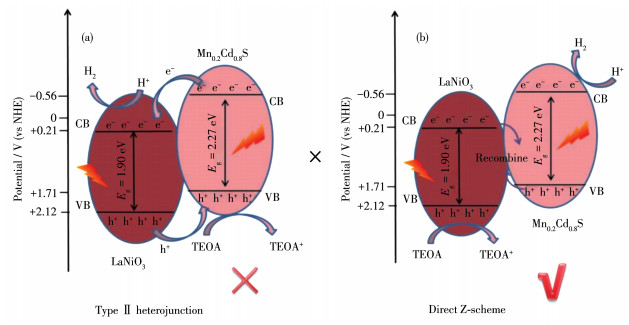
Direct Z-scheme LaNiO3/Mn0.2Cd0.8S heterojunction photocatalysts were synthesized in situ by the introduction of Mn2+ ions in the hydrothermal preparation of CdS using LaNiO3 nanoparticles matrix and fully characterized by field emission scanning electron microscope, powder X-ray diffraction, X-ray photoelectron spectroscopy, UV-Vis diffuse reflectance spectra, N2 adsorption-desorption test and electrochemistry test. LaNiO3/Mn0.2Cd0.8S heterojunction photocatalysts showed improved photocatalytic H2 evolution and stability. The photocatalytic H2 evolution over LaNiO3/Mn0.2Cd0.8S heterojunction photocatalysts reached to 1 190.3 μmol in 5 h, which was 25-fold and 10-fold enhanced compared with pure CdS and Mn0.2Cd0.8S. The enhanced performance of LaNiO3/Mn0.2Cd0.8S heterojunction photocatalysts was ascribed to the formation of heterojunction between LaNiO3 and Mn0.2Cd0.8S as well as the introduced Mn2+ ion, which both can inhibit the recombination of the photoinduced electron-hole. Based on the above results, the direct Z-scheme photocatalytic reaction mechanism was proposed to elucidate the improved performance of LaNiO3/Mn0.2Cd0.8S heterojunction photocatalysts.
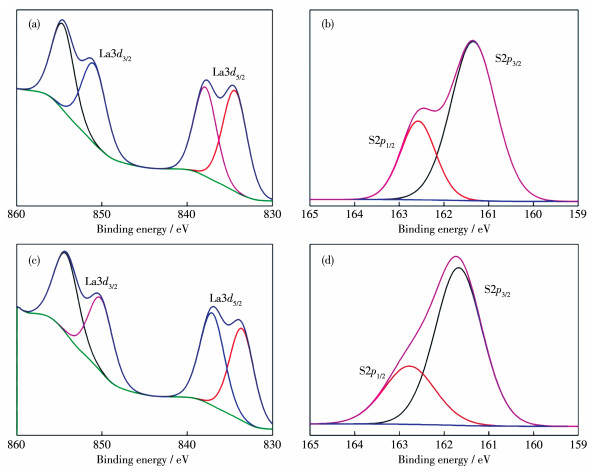
Figure 4.
La3d (a) and S2p (b) XPS spectra for LaNiO3/Mn0.2Cd0.8S heterojunction photocatalysts; La3d (c) and S2p (d) XPS spectra for LaNiO3 and Mn0.2Cd0.8S
Figure 6.
H2 evolution rate over CdS and Mn1-xCdxS catalysts (a); H2 evolution rate over LaNiO3/Mn0.2Cd0.8S heterojunction photocatalysts with different LaNiO3 contents
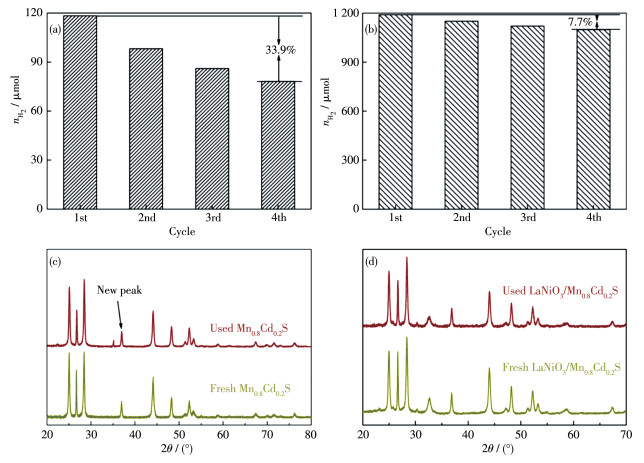
Figure 11.
Cycling runs of Mn0.2Cd0.8S (a) and LaNiO3/Mn0.2Cd0.8S heterojunction (b) photocatalysts for photocatalytic H2 evolution; XRD patterns of the used and fresh Mn0.2Cd0.8S (c) and LaNiO3/Mn0.2Cd0.8S heterojunction (d) photocatalysts
Fujishima A K, Honda K E. Nature, 1972, 238: 37-38 doi: 10.1038/238037a0
[2]
陈建军, 李永宇, 崔天露, 史妍, 王荣荣, 刘新民, 刘果, 陈可可. 无机化学学报, 2020, 36(5): 835-840 doi: 10.11862/CJIC.2020.095CHEN J J, LI Y Y, CUI T L, SHI Y, WANG R R, LIU X M, LIU G, CHEN K K. Chinese J. Inorg. Chem. , 2020, 36(5): 835-840 doi: 10.11862/CJIC.2020.095
[3]
Jiang X H, Zhang L S, Liu H Y, Wu D S, Wu F Y, Tian L, Liu L L, Zou J P, Luo S L, Chen B B. Angew. Chem. Int. Ed. , 2020, 59: 23112-23116 doi: 10.1002/anie.202011495
[4]
Zhu M, Zhang L S, Liu S S, Wang D K, Qin Y C, Chen Y, Dai W L, Wang Y H, Xing Q J, Zou J P. Chin. Chem. Lett. , 2020, 31: 1961-1965 doi: 10.1016/j.cclet.2020.01.017
[5]
Zheng L L, Wang D K, Wu S L, Jiang X H, Zhang J, Xing Q J, Zou J P, Luo S L. J. Mater. Chem. A, 2020, 8: 25425-25430 doi: 10.1039/D0TA10165F
[6]
Wang S, Wang Y, Zhang S L, Zang S Q, Lou X W. Adv Mater. , 2019, 31: 1903404 doi: 10.1002/adma.201903404
[7]
Yu F, Wang L C, Xing Q J, Wang D K, Jiang X H, Li G C, Zheng A M, Ai F R, Zou J P. Chin. Chem. Lett. , 2020, 31: 1648-1653 doi: 10.1016/j.cclet.2019.08.020
[8]
Ye L, Fu J L, Xu Z, Yuan R S, Li Z H. ACS Appl. Mater. Interfaces, 2014, 6: 3483-3490 doi: 10.1021/am5004415
[9]
Chen S S, Qi Y, Hisatomi T, Ding Q, Asai T, Li Z, Ma S K, Zhang F X, Domen K, Li C. Angew. Chem. Int. Ed. , 2015, 54: 8498-8501 doi: 10.1002/anie.201502686
Xiong M H, Qin Y, Chai B, Yan J R, Fan G Z, Xu F, Wang C L, Song G S. Chem. Eng. J. , 2022, 428: 131069-131078 doi: 10.1016/j.cej.2021.131069
[26]
Wan Y H, Du S W, Lu C R, Ren K K, Shi B Y, Liu S Y, Li C H, Dou W D, Fang P, Ye N. J. Alloys Compd. , 2021, 871: 159461-159470 doi: 10.1016/j.jallcom.2021.159461
[27]
Reckmeier C J, Wang Y, Zboril R, Rogach A L. J. Phys. Chem. C, 2016, 120: 10591-10604 doi: 10.1021/acs.jpcc.5b12294
[28]
Zhang H, Yu J L, Sun C X, Xu W H, Chen J, Sun H, Zong C, Liu Z, Tang Y, Zhao D Y. J. Am. Chem. Soc. , 2020, 142: 16177-16181 doi: 10.1021/jacs.0c07274
Figure 4
La3d (a) and S2p (b) XPS spectra for LaNiO3/Mn0.2Cd0.8S heterojunction photocatalysts; La3d (c) and S2p (d) XPS spectra for LaNiO3 and Mn0.2Cd0.8S
Figure 6
H2 evolution rate over CdS and Mn1-xCdxS catalysts (a); H2 evolution rate over LaNiO3/Mn0.2Cd0.8S heterojunction photocatalysts with different LaNiO3 contents
Figure 11
Cycling runs of Mn0.2Cd0.8S (a) and LaNiO3/Mn0.2Cd0.8S heterojunction (b) photocatalysts for photocatalytic H2 evolution; XRD patterns of the used and fresh Mn0.2Cd0.8S (c) and LaNiO3/Mn0.2Cd0.8S heterojunction (d) photocatalysts

 下载:
下载:


 下载:
下载:












