
引用本文:
陈金宝, 李开宁, 黎小芳, 范佳杰, 吕康乐. 结晶氮化碳的制备与改性策略[J]. 无机化学学报,
2021, 37(10): 1713-1726.
doi:
10.11862/CJIC.2021.206

Citation: Jin-Bao CHEN, Kai-Ning LI, Xiao-Fang LI, Jia-Jie FAN, Kang-Le LÜ. Preparation and Modification of Crystalline Carbon Nitride[J]. Chinese Journal of Inorganic Chemistry, 2021, 37(10): 1713-1726. doi: 10.11862/CJIC.2021.206
Citation: Jin-Bao CHEN, Kai-Ning LI, Xiao-Fang LI, Jia-Jie FAN, Kang-Le LÜ. Preparation and Modification of Crystalline Carbon Nitride[J]. Chinese Journal of Inorganic Chemistry, 2021, 37(10): 1713-1726. doi: 10.11862/CJIC.2021.206
结晶氮化碳的制备与改性策略
摘要:
石墨相氮化碳(g-C3N4)是最具代表性的二维有机聚合物半导体材料,其具有可见光响应性能、稳定化学结构和优良的生物相容性等优点,在环境和能源领域有非常广阔的应用前景。但是,普通g-C3N4材料的热聚合不完全,其体相和表面的缺陷多,因此光生载流子易复合,光催化活性不高。近年来,高活性结晶氮化碳(CCN)的研究得到了国内外学者的广泛关注。本文总结了目前CCN制备及其改性方法:5种代表性制备方法,包括传统熔盐法、预热熔盐法、固态盐法、溶剂法和质子化法;4种代表性CCN的改性方法,包括缺陷引入、形貌控制、单原子修饰和材料复合。文章重点介绍了CCN制备原理、结构特征与光催化性能。最后,对CCN的制备与改性方法进行了评价,并对其研究方向进行了展望。
English
Preparation and Modification of Crystalline Carbon Nitride
Abstract:
As a typical two-dimensional organic polymeric semiconductor photocatalyst, graphitic carbon nitride (g-C3N4) has attracted much attention due to its visible-light-responsive property, chemical structure stability and excellent biocompatibility, indicating its promising applications in fields of energy and environment. However, there are many defects both in the bulk and on the surface of g-C3N4 due to the incomplete polymerization, which become the charge recombination centers, resulting in a poor photoreactivity. Recently, the study on the high-photoreactive crystalline carbon nitride (CCN) has elicited considerable attention. In this mini review paper, typical methods in fabrication of CCN, together with the modification strategies, are summarized. The five methods in synthesizing CCN include traditional molten, precursor-preheated molten, solid salt, solvothermal reaction and protonation synthesis, and the modification strategies include defects engineering, morphology-controlling, single-atom modification and materials hybridization. We mainly focuse on the introduction of principles, structure properties and photocatalytic performances of CCN. Finally, the methods in preparation and modification of CCN are commented, and the study of CCN in the future is also outlooked.
-
Key words:
- crystalline carbon nitride
- / organic semiconductor
- / photocatalysis
- / visible light
- / defect
-
0. 引言
经济的快速发展和化石能源的大量使用,造成了日益严峻的能源短缺和环境污染问题。光催化作为一种环保型技术,可以利用太阳能来创造新能源和降解污染物,因而近年来引起了国内外科研人员的浓厚兴趣并得到了广泛研究[1-3]。作为一种非金属型光催化剂,氮化碳由于其良好的电子结构和光学性质、较低的成本以及较高的稳定性而成为光催化领域最有前途的催化剂之一[4-5]。
聚合物氮化碳(CxNy)因其独特的性质、丰富的原料来源、低廉的价格而受到越来越多的关注。CxNy的聚合特性和简单的合成过程使得其结构在分子水平上较容易被设计。人们不断设计和改进CxNy分子结构,如掺杂、控制聚合、官能团化以及层内氢键的调控等[6]。根据骨架中不同的C、N原子比例,利用不同的前驱体成功地合成了一系列CxNy材料[7]。这进一步引发了人们对CxNy结构、性能的研究,因此,CxNy在(光)催化、光电化学、生物传感器、生物成像和生物治疗等领域具有广阔的应用前景[8-9]。
在光催化领域研究最多的氮化碳是石墨相氮化碳(g-C3N4)。g-C3N4是一种聚合物半导体,禁带宽度为2.7 eV,是类似于石墨的层状结构,每一层都是由sp2轨道杂化的C原子和N原子组成的大π键共轭体系,层与层之间通过范德华力连接[10-11]。公认的组成g-C3N4的基本单元有2种,分别为三嗪(C3N3)和三-s-三嗪(C6N7)结构(图 1)。经过密度泛函理论(DFT)计算发现,三-s-三嗪结构的g-C3N4更加稳定,大部分常见的g-C3N4也是由三-s-三嗪单元所构成的。g-C3N4具有较好的化学稳定性,组成元素为自然界中最常见的C和N,二者成本低、安全无毒、对环境友好[12-13]。
图 1
传统的g-C3N4热聚合中,由于反应中间体的流动性不足,前驱体不完全脱氨导致形成以三-s-三嗪的melon结构(图 2a)为单位的链状聚合物,聚合物末端由氨基组成,链与链之间通过氢键作用连接在一起[14]。因此,普通热聚合得到的g-C3N4的结构不规则,结晶度较差[15-17]。体相C3N4(bulk carbon nitride,BCN)存在的大量缺陷、氨基、氢键以及折叠结构等,会阻碍电子在平面上的传导,从而导致低电导率,同时会形成电子-空穴的复合中心,大大限制了光催化中g-C3N4的电子-空穴分离、迁移和捕获的效率[18-20]。
图 2
为了促进光生载流子分离,增强g-C3N4的光催化活性,文献报道了许多改性策略,包括碳点修饰[21]、引入C[22-23]或者N[24-26]缺陷、利用表面等离子体效应[27]、制备具有纳米片形貌的g-C3N4[28-31]、元素掺杂[32-33]、建立层间载流子传输通道[34]以及材料复合[35-37]等手段。我们的研究还发现,酸处理可以显著促进g-C3N4的载流子分离效率,增强光活性[38]。关于普通g-C3N4的修饰改性策略,可以参考相应的综述论文[39-40]。
此外,相比于普通g-C3N4,体相缺陷少且晶体结构长程有序的结晶氮化碳(CCN)通常表现出优异的光催化性能[41]。由此可知结晶度是影响g-C3N4的光反应性的一个重要因素,想要获得较好的光催化活性,可以促进前驱体的完全聚合,从根本上改变组成BCN的melon结构单元。
到目前为止,文献已经报道了一些方法来增强g-C3N4的结晶度。比如,我们课题组曾经将双氰胺置于密闭的不锈钢高压釜中,通过高温高压环境下的热聚合反应增强g-C3N4的结晶性能[42]。研究发现,密闭反应产生的高压,不仅可以显著促进g-C3N4的晶化,而且可以有效消除g-C3N4分子的平面内氢键。高压环境虽然可以促进g-C3N4前体的缩聚,一定程度上提升了g-C3N4的晶化程度。但是,从相应的表征上来看,该结晶度的提升只是改善了melon结构,没有形成高结晶性的CCN。为此,本文简要介绍了具有高结晶度的CCN,并综述了CCN的制备方法及其改性手段。
1. CCN简介及其表征手段
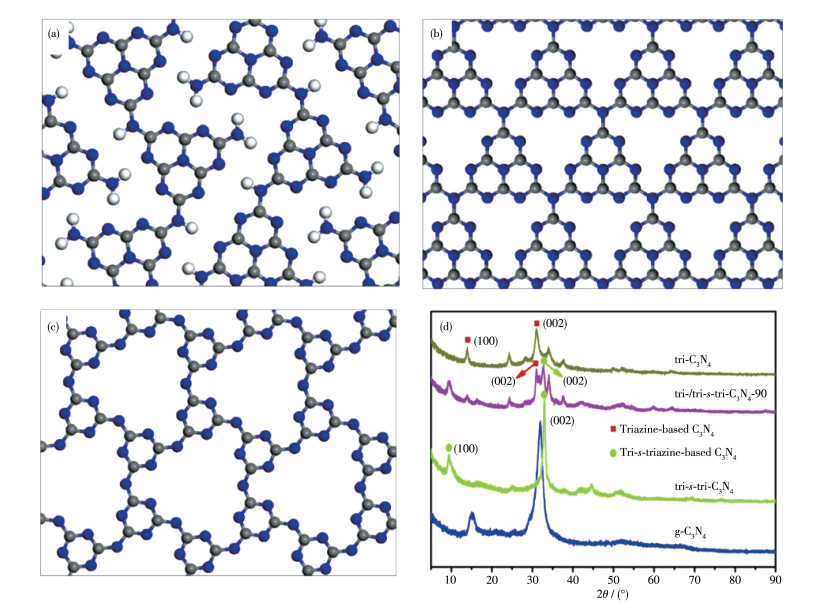
为了解决普通氮化碳BCN光催化活性不高的问题,科学家们开发了新方法来制备具有更好性能的CCN。目前文献报道的CCN主要有2种类型结构,分别是三嗪基(poly(triazine imide))的PTI型和三- s-三嗪基(poly(heptazine imide)) 的PHI型[43]。从图 2a~2c中可以看到,相比于BCN,CCN具有更有序的结构、更高的聚合度和更为扩展的共轭面。增强结构的有序性和减少缺陷的浓度,均有利于促进载流子的转移,进而提升氮化碳的光催化性能。
通常可以采用X射线衍射和透射电镜来确定CCN的分子结构[44]。图 2d为不同C3N4的X射线衍射图,对于三-s -三嗪单元的BCN(即图 2d中的g-C3N4),其X射线衍射图上有2个典型特征峰(13.1°和27.4°),分别代表g-C3N4的(100)面内重复的三-s-三嗪单元和(002)面的堆积。与BCN相比,三-s-三嗪单元的CCN(即图 2d中的tri-s-tri-C3N4)的(100)晶面衍射峰向小角度偏移,这很可能是共轭骨架充分缩合而形成的更扩展的共轭骨架所致。而(002)面衍射峰则偏移向更大的角度,表明层间距的减小,这更有利于光生载流子的传输和分离。此外,CCN的峰强度也略有增强,也表明其具有较高的结晶度。而三嗪基CCN(即图 2d中的tri-C3N4)具有不同于三-s-三嗪C3N4的X射线衍射图。图 3a、3b分别为BCN和CCN的高分辨率透射电镜图。从图中可以直观地看到熔融盐法合成的CCN具有明显的晶格,显示出其高结晶性。相反,从BCN的电镜图中看不到这种有序的晶格,意味着BCN的低结晶性。X射线衍射斑点的出现,也体现出CCN的高结晶度(图 3b内插图)。
图 3

较高的结晶度减少了缺陷造成的电荷复合中心,增加了光生电子和空穴的分离效率,从而促进更多的光生载流子到材料表面参与催化反应。此外,作为一种高分子类型的光催化剂,高结晶度会缩小带隙,增强对更多可见光的响应,并且共轭结构的扩展和结构有序度的增加减少了电荷移动过程中的阻碍[44-46]。
2. CCN的制备
2.1 传统熔融盐法
科学家们已经探究了多种制备CCN的方法。最近的研究发现,熔融盐的合理使用,可以促进前驱体的热聚合反应,提高结晶度进而提高光催化性能。加热使熔融盐(例如LiCl/KCl和LiBr/KBr)作为液体介质溶解缩聚前体,为前体的充分聚合提供条件[47]。Bojdys等[14]认为前体的不完全缩合是导致g-C3N4结晶度差的主要动力学原因,而这个问题可以通过使用合适的液体介质来解决,他们将双氰胺与LiCl和KCl的混合物混在一起彻底研磨,然后让反应混合物在惰性气氛下加热并持续保温一段时间,得到高聚合度的C3N4,并且通过傅里叶变换红外光谱等表明,该结构几乎没有缺陷和未反应的氨基,前体缩聚完全,因此是一个高度浓缩的框架(图 4a和4b)。这项工作为合成CCN开辟了新的思路。
图 4
 图 4. 傅里叶变换红外光谱图(a)和熔融盐处理的双氰胺的X射线衍射图(b)[14]; PTI/Li+Cl-的球差校正透射电镜图(c) 以及最大氢气和氧气释放速率与(1010)和(0001)面的平均表面积比的关系图(d)[49]Figure 4. Fourier transform infrared spectra (a) and X-ray diffraction patterns (b) of dicyandiamide prepared in molten salt[14]; Spherical aberration corrected transmission electron microscopy images (c) and plot of maximum hydrogen and oxygen evolution rate as a function of the mean surface area ratio of the (1010) and (0001) planes of PTI/Li+Cl-(d)[49]
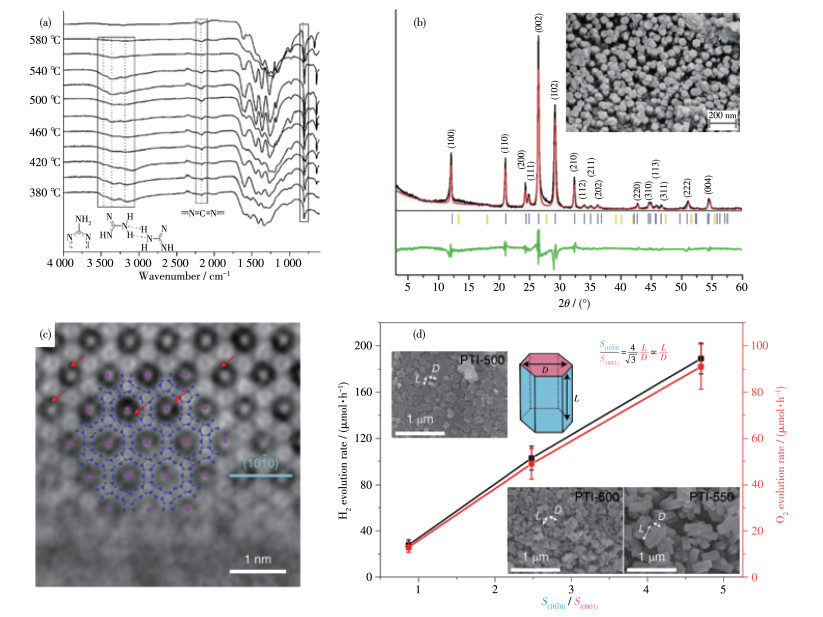
图 4. 傅里叶变换红外光谱图(a)和熔融盐处理的双氰胺的X射线衍射图(b)[14]; PTI/Li+Cl-的球差校正透射电镜图(c) 以及最大氢气和氧气释放速率与(1010)和(0001)面的平均表面积比的关系图(d)[49]Figure 4. Fourier transform infrared spectra (a) and X-ray diffraction patterns (b) of dicyandiamide prepared in molten salt[14]; Spherical aberration corrected transmission electron microscopy images (c) and plot of maximum hydrogen and oxygen evolution rate as a function of the mean surface area ratio of the (1010) and (0001) planes of PTI/Li+Cl-(d)[49]Chong等[48]采用类似的方法,运用其他熔融盐来合成CCN,将LiBr/KBr和双氰胺混合后600 ℃加热4 h,同样得到了三嗪基的CCN,其为一种卤素离子插层的二维层状网络,并发现材料的层间高度随插层材料的不同而不同,表面PTI型晶体的层间高度可通过控制盐熔体的组成和改性来调节。
近期Lin等[49]运用同样的方法,以LiCl/KCl熔融盐辅助双氰胺缩聚制成了结晶度非常高的C3N4(图 4c、4d),并深入研究了CCN单晶的反应面,打破传统观念,开创性地指出了棱侧面(1010)比基面(0001)光催化活性更强。随着棱侧面(1010)面面积比例的增加,CCN的产氢、产氧活性都显著提升,为反应面的控制和调整提供了方向。
2.2 预热的熔融盐法
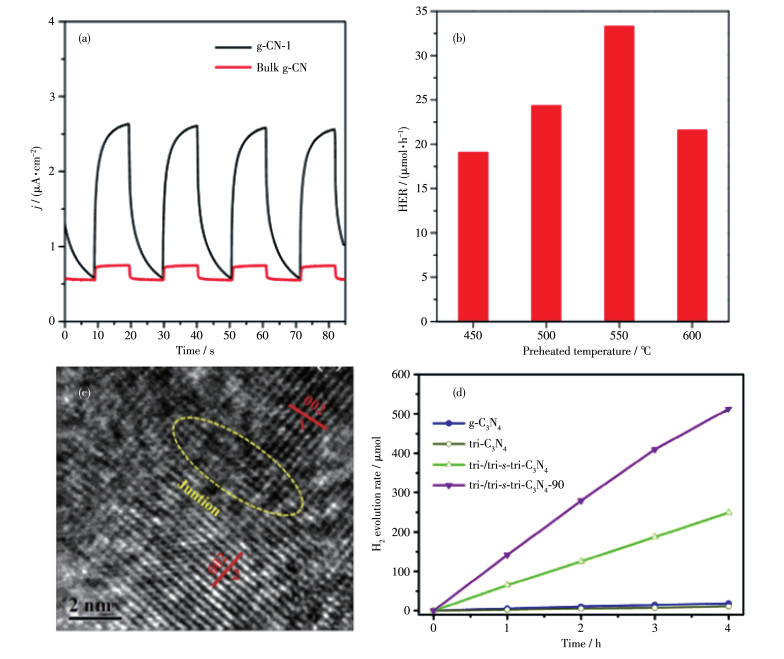
Lin等[43]指出传统的熔融盐法将C3N4的前驱体和混合盐研磨并加热反应,会将C3N4聚合物的结构改变为三嗪基的PTI型(图 2c),但是三嗪基序的PTI型的共轭面聚合度不如三-s-三嗪的PHI型,三-s-三嗪结构具有更快速的电子迁移和更窄的带隙,于是他们采取了预热的方法先将三聚氰胺加热到500 ℃预热4 h,再将其和混合盐LiCl/KCl加热反应,得到三-s-三嗪的PHI型CCN(即图 5a中的g-CN-1),其结晶度很高,核磁共振能谱图也证明得到的CCN基本单元为三-s-三嗪,其具有更好的光催化性能(图 5a)。在此基础上,Lin等[50]又探究了前驱体三聚氰胺预热的温度对CCN聚合物的性能和光催化活性的影响,发现550 ℃预热的产品具有最好的活性,可见光照射下也能表现出显著的光催化产氢能力(图 5b)。
图 5
Zeng等[44]将8 g三聚氰胺放入有盖的氧化铝坩埚中,500 ℃加热4 h,再将600 mg预处理过的聚合物和一定量的三聚氰胺与LiCl/KCl混合物研磨,然后在Ar气氛下加热到550 ℃,保温4 h,成功地在层状PHI型CCN上垂直生长了棒状PTI型的CCN(即图 5c中的tri-/tri-s-tri-C3N4-90),这种独特的形貌为光收集提供较长光路,增加了活性中心和减少了传质阻力。从X射线衍射图中可以看到高结晶的结构以及材料的成功复合,透射电镜图中2种CCN之间匹配良好的晶格条纹清楚地证明了三-s-三嗪基C3N4与三嗪基C3N4之间成功地构筑了紧密的晶体结构(图 5c、5d)。结果表明,这种三-s-三嗪/三嗪复合的材料具有优异的可见光催化析氢活性,是纯BCN的30倍。这种优异的光催化性能,可以归因于晶体的紧密连接,该结构可以极大地提高光生载流子的转移和分离效率。
2.3 固态盐法
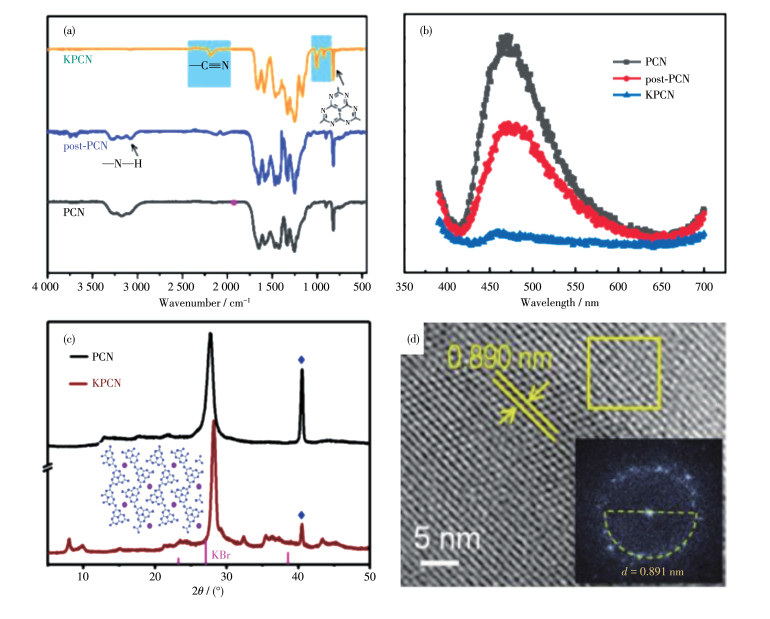
Xu等[51]认为与BCN相比,采用熔融盐辅助热缩聚制得的CCN具有较高结晶度和光催化活性,然而,这种方法需要大量熔融盐(m盐/m前体 > 10),产率低,而且不可缺少的水敏锂盐(LiCl、LiBr)使其广泛应用受到了限制,于是探究了单一KCl盐作为固体模板或结构导向剂来制备CCN材料。他们将三聚氰胺在管式炉中空气气氛下540 ℃加热,得到的产物用KCl和乙醇混合研磨,干燥后,在管式炉氮气气氛下550 ℃退火3 h即可得到CCN(图 6a,即KPCN)。固态KCl起到模板和黏结剂的作用,BCN(即图 6a和图 6b中的PCN)的非晶态结构在相邻KCl晶体提供的受限空间中重组,钾离子也融合在melon链之间,可以作为“黏合剂”重新排列BCN的结构,形成晶体,得到有序的电子传输通道,促进载流子的分离和迁移以及对可见光捕获的效率(图 6a、6b),在光催化产氢方面表现出近20倍的提升。Qiu等[52]将3.0 g三聚氰胺与2.0 g KBr在3 mL乙醇和1 mL乙二醇混合物中研磨,干燥后550 ℃加热3 h,也得到了高结晶度的钾离子插层的CCN(图 6c和图 6d的KPCN),高度结晶的结构使得其在光催化产氢方面较BCN提高了18倍以上。
图 6
Xu等[53]认为,通常的熔融盐方法制备CCN时得到的三嗪基是由于高温熔融盐破坏了三-s-三嗪的结构。他们探究了用CsCl和BCN混合后550 ℃加热的方法。由于改性温度低于盐的熔点,该方法较为温和。CsCl犹如温和的化学“剪刀”,提升了C3N4结晶度。得到的C3N4具有结晶性结构,其比表面积显著提高。这些优异的性能使产物表现出有效的电子-空穴对的捕获、迁移和转移性能,光催化产氢活性约为BCN的23倍。
2.4 溶剂热法
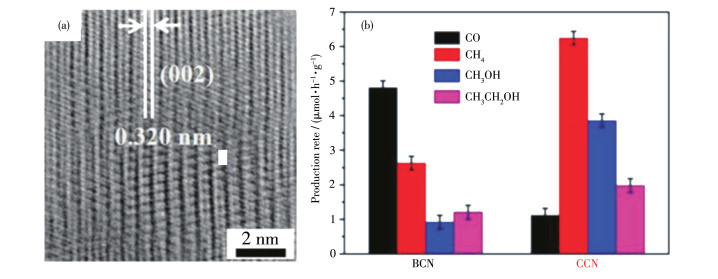
人们希望能以较低的温度来合成g-C3N4,而采用有机溶剂的方法可以利用有机溶剂的特性促进传质和缩聚动力学,从而实现该目的。Xia等[54]探究了低温溶剂热(180 ℃)合成法制备CCN,在乙腈促进合成的溶剂热条件下,通过盐酸胍和双氰胺的共缩合,合成了同时富含一定缺陷的CCN。得到的产物在光照下还原CO2的速率为12.07 μmol·h-1·g-1,选择性为91.5%,均显著高于BCN光催化剂(图 7)。在加热条件下,强极性溶剂乙腈作为亚临界流体,具有较高的溶解度和扩散系数,可以促进传质和化学反应动力学,与四氯化碳(CTC)、环己烷(CHX)和四氢呋喃(THF)等溶剂对比可知,所得产物结晶度均不如乙腈作溶剂时,说明只有强极性溶剂才能更好地促进前驱体生成CCN集成网络。
图 7

Cui等[55]在亚临界溶剂乙腈中由三嗪类化合物(如三聚氯氰和三聚氰胺)在相对较低的温度下缩合生成g-C3N4骨架。外观形貌控制比较简单,为纳米带组成的网络,g-C3N4层中三-s-三嗪类化合物有规则的平面内连接。
2.5 质子化法
Wang等[46]报道了在热缩合过程中,对传统热聚合方法进行轻微的修改,仅通过质子化合成g-C3N4的中间物种就可以极大地改善CCN的聚合动力学,得到具有高结晶度和择优取向的CCN。由于g-C3N4骨架中氮原子存在未配对电子,大块g-C3N4可以被HCl质子化,由此可以推测,g-C3N4边缘的质子化和断裂的界面氢键将更有利于g-C3N4前体的热缩聚。Wang等将双氰胺在N2中以一定的中间温度(350、400和450 ℃)加热2 h,然后将白色中间体置于HCl溶液(0、1、3、6、12 mol·L-1)中搅拌24 h,洗涤烘干后再在550 ℃下加热2 h得到最终产品。结果表明,所制备的CCN的光电化学性能明显提升,光催化降解染料的作用增强。与传统的BCN相比,其催化活性可提高7倍之多。该策略为制备高活性的CCN,提升其光催化活性开辟了新途径,作为一种通用、简便的方法有望在可持续能源生产中得到应用。
提升g-C3N4光催化性能的制备方法如表 1所示。
表 1
 表 1 CCN制备方法的比较Table 1. Comparison of preparation methods of CCN
表 1 CCN制备方法的比较Table 1. Comparison of preparation methods of CCN 下载:
导出CSV
下载:
导出CSV
Method Precursor Basic unit Photocatalytic condition Photocatalytic preformance Ref. Molten salt synthesis KCl/LiCl+dicyandiamide Triazine 300 W xenon lamp
(> 300 nm) 100 mg
catalyst; H2 and O2
evolution189 and 91 μmol·h-1 [14] Preheated and molten
salt synthesisKCl/LiCl+melamine Tri-s-triazine 300 W Xe lamp
(420 nm filter); 50 mg
catalyst; H2 evolution27.5 μmol·h-1 [43] Preheated and molten
salt synthesisCN+melamine+ KCl/LiCl Triazine+
tri-s-triazine300 W Xe lamp
(420 nm filter); 50 mg
catalyst; H2 evolution144 μmol·h-1
(30 times higher than BCN)[44] Solid salt synthesis CsCl+CN Tri-s-triazine 300 W Xe lamp
300 W Xe lamp(420 nm filter); 4 mg
catalyst; 4 h H2 evolution6.17 μmol
(23 times higher than BCN)[53] Solid salt synthesis KCl+CN Tri-s-triazine 300 W Xe lamp
(420 nm ≤ λ ≤ 780 nm);
50 mg catalyst;
H2 evolution59.4 μmol·h-1
(20 times higher than BCN)[51] Solid salt synthesis KBr+melamine Tri-s-triazine 500 W Xe lamp
(280 nm); H2 evolution1 600 μmol·h-1·g-1
(18 times higher than BCN)[52] Organic solvothermal
synthesisAcetonitrile+hydrochloride +
dicyandiamideTri-s-triazine 350 W Xe lamp
(AM1.5 filter);
CO2 photoreduction12.07 μmol·h-1·g-1 [54] Protonation synthesis HCl+dicyandiamide Tri-s-triazine 150 W Xe lamp
(λ > 420 nm); 300 mg
catalyst; RhB degradation0.012 96 min-1
(7 times higher than BCN)[46] 3. CCN的改性
为了进一步提升CCN的光催化性能,可以对其进行进一步改性;例如控制形貌可以增大材料的比表面积,增加传质[56];引入缺陷可以在保持高结晶度的条件下增加活性位点,同时缩短带隙,增强对可见光的响应[11, 57];复合其他材料能进一步促进载流子的分离[58]。下面介绍了一部分CCN的改性手段。
3.1 引入缺陷
结晶度的提高有利于光催化的进行,但是缺陷的减少,难免会减弱和反应分子的结合。缺陷的存在可以调整带隙,增加活性位点[59],但是过多的缺陷又会降低材料结晶度。所以可以在保持较高结晶度的条件下,适当地引入缺陷,从而使光催化活性最大化。
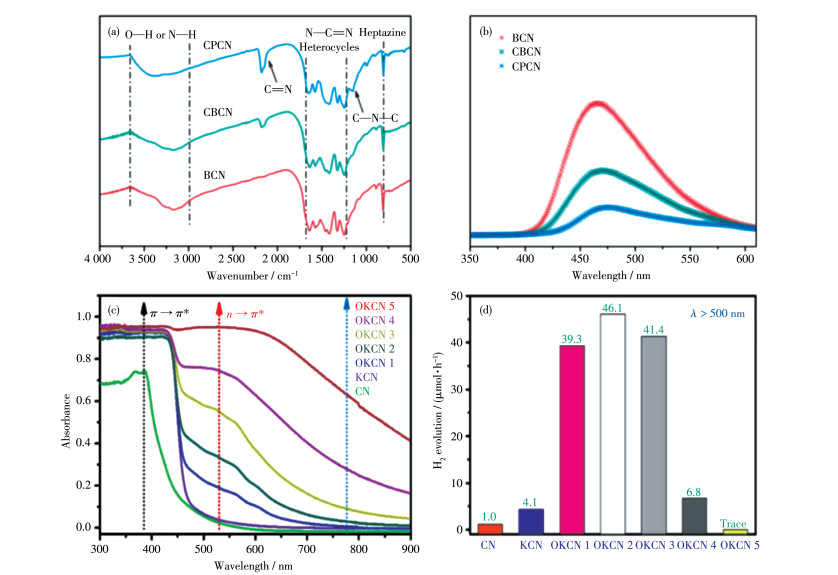
Guo等[60]将三聚氰胺的水热产物与KCl混匀,并加少量乙醇润湿研磨,然后于550 ℃下加热3 h进行热聚合反应,即得到了含氰基的CCN(图 8a的CPCN)。氰基的引入会导致CCN的分子结构发生部分扭曲,产生能量无序的界面。这种畸变导致电子可以被氰基储存,从而稳定K+离子,并促使光生电荷和空穴的空间分离(图 8b)。同时,较高的结晶度提升了载流子迁移率,显著提升了CCN的光催化活性。
图 8

能带工程对光催化具有重要意义,短的带隙可以增强对可见光的吸收范围。许多光催化剂由于带隙过长,只能吸收紫外光,这大大限制了光催化剂的应用[61]。g-C3N4材料的光吸收范围很难超过600 nm,为了进一步提升CCN的实用性,就需要采取一些手段来提升对可见光的响应范围。Zhang等[62]把尿素粉末与KCl和草酸机械混合后加热得到了O掺杂的CCN(即图 8c和8d中的OKCN 1~5)。O取代原来位置的N原子,就会产生孤电子对,并且可能导致非共面结构上C和N原子的对称性变化,从而激活更多的n→π*跃迁,使吸收边扩展到500 nm,极大地促进了从500到650 nm的光响应,并且在λ> 500 nm的情况下,其最高产氢活性分别是BCN(即图 8c、8d中的CN)和没有O掺杂的CCN(即图 8c和8d中的KCN)的45倍和10倍。
尽管g-C3N4结晶度的提高有助于促进电子的传输并使其到表面参加化学反应,但由于缺陷的减少,降低了对反应分子的结合和活化,为了增加光催化活性位点,Li等[63]提出了一种在保持良好结晶度的同时有效控制g-C3N4缺陷的方法。将三聚氰胺粉末加入KOH水溶液,在85 ℃油浴中蒸发至干燥,再将三聚氰胺、氢氧化钾和氯化钠的固体混合物均匀地混合在N2气氛中煅烧至600 ℃保温一段时间,最后清洗干燥即得到含N空位的CCN。在保持高结晶度的条件下,氮空位和末端氰基可以捕获光电子,抑制光生载流子的复合,同时作为光催化活性位点,使得光照还原CO2性能明显优于未改性的BCN和CCN。
此外,Liu等[64]也用类似的方法在普通的熔融盐法中引入KOH,将BCN和KCl/LiCl/KOH混合加热,得到了含有N空位的CCN,提升光催化活性和对可见光的响应范围、表面高结晶度和N空位对光催化的协同促进作用。
Ren等[65]在惰性气氛下用还原剂NaBH4破坏了所制备的CCN的局部有序结构,引入了缺陷,缺陷可以形成中间能带,从而显著提高了光吸收能力,产氢速率约为没有缺陷的CCN的8倍,即使光波长延长到610 nm,也能产生H2。
3.2 控制形貌
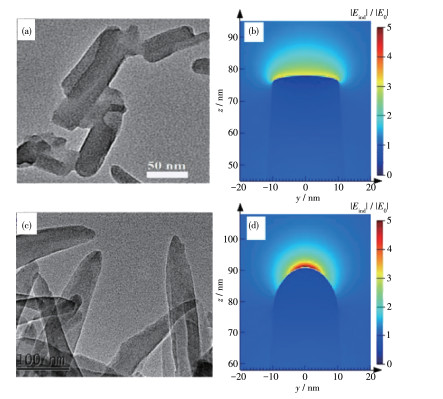
运用熔融盐法将g-C3N4与KCl/LiCl混合加热并冷却到室温可以得到三维分支的纳米棒组成的结构,在保持高结晶的同时,又增加了比表面积,极大促进了传质效率,并增加了吸收可见光的量[66]。在此基础上Zeng等[67]进一步对产物增加了淬火处理,用冰水对产物进行激冷,产生的热应力使纳米棒尖端曲率增加,得到了三维分支的纳米针。该形貌产生了微观的“避雷针效应”,由于尖端曲率的增加使纳米针尖端富集电荷,从而进一步促进了光生电荷和空穴的分离(图 9a~9d),实现了高效光催化。
图 9

二维材料是近些年研究的热点领域,特别是在光催化领域表现出了显著的优势。二维晶体相比其他晶体具有更大的比表面积和更有效的电荷传递,可以缩短光生电子从体相传递到表面的距离,增加对阳光的吸收,促进传质效率。Ou等[68]利用传统的超声和离心工艺,将三-s-三嗪基的CCN和异丙醇胺混合后超声处理后离心,即得到了剥离的超薄CCN纳米片,与BCN、块状CCN相比,具有显著增强的光催化活性。Li等[69]将三聚氰酸和三聚氰胺作为前驱体加热得到g-C3N4,然后用熔融盐法处理得到CCN,由于前驱体缩聚过程中放出气体,得到的产物形貌由实心结构转变为空心球状多孔结构,大大提升了比表面积,增加了活性位点并提升了对太阳光的利用。
3.3 单原子修饰
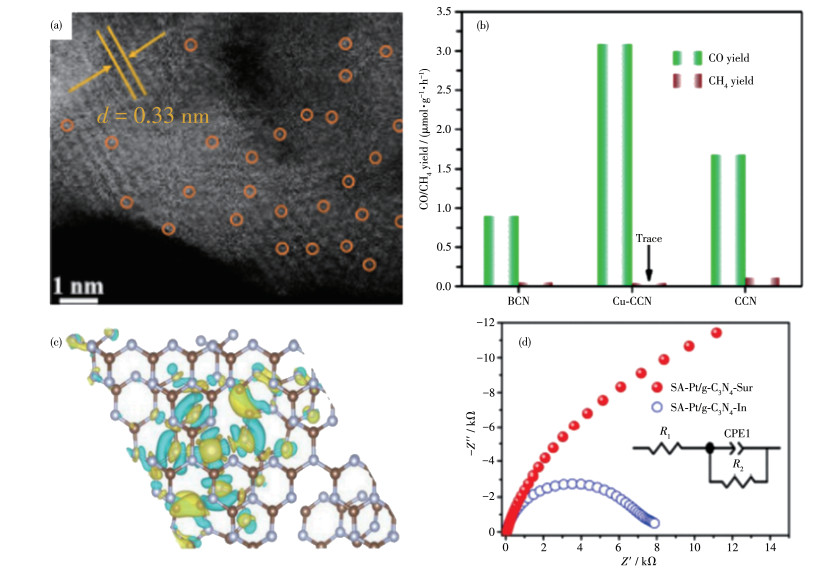
高度结晶的结构使光电子转移加快,但缺少了反应物的吸附中心。催化剂上负载金属助催化剂可以提供反应的活性位点。现在,单原子催化正在成为光催化的一个新的前沿,单原子助催化剂可使金属原子具有接近100%的利用率和不饱和的配位环境[70]。Li等[71]将6.8 g的CuCl2·2H2O溶解于300 mL的HCl溶液中,加入10 g三聚氰胺,收集沉淀物后在500 ℃煅烧4 h,得到BCN和铜化合物复合的材料,再将其与KCl(3.3 g)和LiCl(2.7 g)的混合物在550 ℃下加热,使g-C3N4结晶化,最后在2 mol·L-1 H2SO4溶液中浸取24 h后除去杂质即可得到单原子Cu修饰的CCN(Cu-CCN,图 10a)。单个Cu原子的引入可以作为CO2的吸附中心,从而提高Cu-CCN样品对CO2的吸附量,并且结果表明,将CO2还原为CO是一个熵减的过程,使得Cu-CCN样品在光催化CO2转化为CO方面表现出近100%的选择性(图 10b)。
图 10
Zeng等[72]采用简单的离子交换的方法,首先用熔融盐法将KCl/LiCl(质量比11∶9)和C3N4混合制备K插层的CCN,再将其加入到去离子水中搅拌,再加入Pt(NH3)4Cl2溶液搅拌1 d,洗涤烘干的粉末在Ar气氛下加热(用王水辅助洗涤,除去表面Pt原子),首次得到了Pt原子插入CCN层间的复合物。利用层状的CCN的层间亚纳米空间来限制Pt原子,可以显著改变Pt原子周围的电荷分布,促进质子吸附,降低反应能垒。所制备的单原子Pt插层的光催化剂具有较高的产氢性能(22 650 μmol·g-1·h-1),在420 nm处的表观量子产率(AQY)达到22.5%。和Pt负载在表面的CCN相比,位于层间的Pt电荷分布极化(图 10c),有利于质子吸附,电荷耗尽区减少,并且处于层间,载流子不需要转移到表面去反应,可以缩短电荷扩散距离,减小电荷复合(图 10d)。这项工作展示的创新设计可能会为高效催化的单原子催化剂的设计提供新的方向和思路。
3.4 碳材料复合
CCN的表面缺陷少,这不利于其复合材料的构建。因此,除了CCN的同质结(tri-s-tri-C3N4和tri-/tri-s-tri-C3N4)之外[41],关于CCN的复合材料制备相关的研究报道比较少[73]。
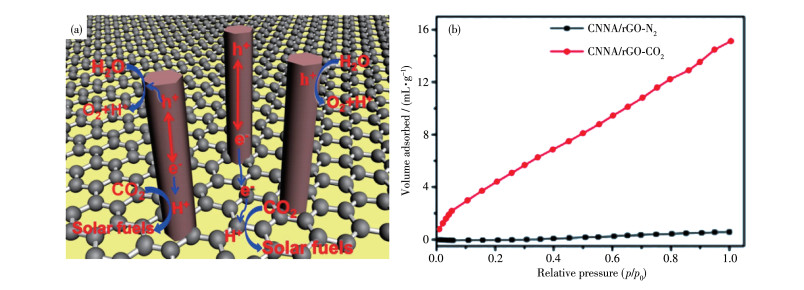
石墨烯表面有丰富的含氧官能团,如羟基(—OH)和羧基(—COOH)等,它们易于与CCN表面的氨基(—NH2)发生化学反应而键合在一起,从而形成紧密接触的界面,促进界面电荷转移,提高光催化活性。基于此原理,Xia等[73]通过熔融盐方法成功在石墨烯表面上原位生长一维阵列的CCN纳米棒(图 11a)。先将三聚氰胺粉末和石墨烯进行混合,然后将500 ℃下预热样品与混合熔融盐(KCl和LiCl)进行混合,加热到550 ℃反应,即可得到CCN和石墨烯复合的异质结(CNNA/rGO)。
图 11

CCN较低的缺陷可以减少电荷复合中心并增加电荷迁移率,和石墨烯复合后再形成异质结;石墨烯作为二维碳材料具有优异的导电性,并且有比g-C3N4更低的导带,这有助于电子转移到石墨烯相中积累而增强电荷空穴的分离,更多的光电子用于表面CO2还原反应,同时有序的一维晶体结构提供了良好的捕光通道和CO2的吸附。所得材料的光吸收、CO2的吸附和电荷转移都得到了改善。此外,石墨烯/CCN这种独特的二维/一维的异质结对吸附CO2有很高的选择性,使得即使在CO2浓度较低的情况下,也可以将湿CO2还原为化学燃料(图 11b)。
4. 结论与展望
g-C3N4因其可见光响应性能与良好的生物相容性,而成为最具代表性的非金属有机半导体光催化材料。但是,普通g-C3N4存在表面积小和体相缺陷多的问题,这导致其光催化活性不高。近年来发展起来的高活性的CCN相关研究为g-C3N4在环境与能源领域的应用提供了可能。本论文综述了制备CCN的5种代表性方法及改性CCN的4种策略,从中可以看出:用熔盐法制备CCN一般需要经历2次煅烧过程。熔盐法存在能耗高、制备程序复杂的问题,且煅烧过程伴随大量有毒气体的排放,不仅污染大气环境,而且导致催化剂的产率比较低。因此,需要大力发展CCN的绿色合成新方法。
在今后的CCN制备及其光催化性能研究过程中,下列问题值得我们注意:
(1) 无论是采用熔盐法还是固态盐法制备CCN,金属离子(如Li+和K+)等将会进入CCN的层内或层间,残余金属离子对CCN光吸收和光催化性能的影响,不能被忽略。
(2) 单原子修饰为CCN性能的提升提供了新的方案。但是,CCN良好的结晶性能同时也为其单原子修饰制造了障碍。在今后的发展中需要大力开展CCN的单原子修饰相关研究。
(3) CCN的表面缺陷少。因此,CCN复合材料的构建面临巨大挑战。需要大力开展CCN的材料复合新技术研究工作。
(4) 目前,关于CCN的光催化反应机理相关研究报道比较少,这不利于高效环境催化材料的开发。在今后的工作中,需要采用原位表征技术,结合DFT模拟计算,开展CCN的光催化机理研究,最终为高活性CCN材料的开发,奠定坚实的基础。
-
-
[1]
Tong H, Ouyang S X, Bi Y P, Umezawa N, Oshikiri M, Ye J H. Adv. Mater., 2012, 24(2): 229-251 doi: 10.1002/adma.201102752
-
[2]
Chen X B, Shen S H, Guo L J, Mao S S. Chem. Rev., 2010, 110(11): 6503-6570 doi: 10.1021/cr1001645
-
[3]
Chong M N, Jin B, Chbow C W, Saint C. Water Res., 2010, 44(10): 2997-3027 doi: 10.1016/j.watres.2010.02.039
-
[4]
Thomas A, Fischer A, Goettmann F, Antonietti M, Müller J O, Schlögl R, Carlsson J M. J. Mater. Chem., 2008, 18(41): 4893-4908 doi: 10.1039/b800274f
-
[5]
Zeng Z X, Quan X, Yu H T, Chen S, Zhang Y, Zhao H, Zhang S S. Appl. Catal. B, 2018, 236: 99-106 doi: 10.1016/j.apcatb.2018.05.003
-
[6]
Zhou Z X, Zhang Y Y, Shen Y F, Liu S Q, Zhang Y. Chem. Soc. Rev., 2018, 47(7): 2298-2321 doi: 10.1039/C7CS00840F
-
[7]
Yang H, Wang Z, Liu S Q, Shen Y F, Zhang Y J. Chin. Chem. Lett., 2020, 31(12): 3047-3054 doi: 10.1016/j.cclet.2020.07.048
-
[8]
Zhao T T, Zhou Q, Lv Y Q, Han D, Wu K Q, Zhao L F, Shen Y F, Liu S Q, Zhang Y J. Angew. Chem. Int. Ed., 2020, 59(3): 1139-1143 doi: 10.1002/anie.201911822
-
[9]
Huang C F, Wen Y P, Ma J, Dong D D, Shen Y F, Liu S Q, Ma H B, Zhang Y J. Nat. Commun., 2021, 12(1): 320 doi: 10.1038/s41467-020-20521-5
-
[10]
李开宁, 张梦曦, 欧小雨, 李睿娜, 李覃, 范佳杰, 吕康乐. 物理化学学报, 2020, 37(8): 2008010 https://www.cnki.com.cn/Article/CJFDTOTAL-DLXB201502001.htmLI K N, ZHANG M X, OU X Y, LI R N, LI Q, FAN J J, LV K L. Acta Phys. -Chim. Sin., 2020, 37(8): 2008010 https://www.cnki.com.cn/Article/CJFDTOTAL-DLXB201502001.htm
-
[11]
王薇, 黄宇, 王震宇. 物理化学学报, 2020, 37(8): 2011073 https://www.cnki.com.cn/Article/CJFDTOTAL-DLXB201502001.htmWANG W, HUANG Y, WANG Z Y. Acta Phys. -Chim. Sin., 2020, 37(8): 2011073 https://www.cnki.com.cn/Article/CJFDTOTAL-DLXB201502001.htm
-
[12]
Niu P, Zhang L L, Liu G, Cheng H M. Adv. Funct. Mater., 2012, 22(22): 4763-4770 doi: 10.1002/adfm.201200922
-
[13]
Zhao Z W, Sun Y J, Dong F. Nanoscale, 2015, 7(1): 15-37 doi: 10.1039/C4NR03008G
-
[14]
Bojdys M J, Müller J O, Antonietti M, Thomas A. Chem. Eur. J., 2008, 14(27): 8177-8182 doi: 10.1002/chem.200800190
-
[15]
Xu Y S, He X, Zhong H, Singh D J, Zhang L J, Wang R H. Appl. Catal. B, 2019, 246: 349-355 doi: 10.1016/j.apcatb.2019.01.069
-
[16]
Zhang G G, Liu M H, Heil T, Zafeiratos S, Savateev A, Antonietti M, Wang X C. Angew. Chem. Int. Ed., 2019, 58(42): 14950-14954 doi: 10.1002/anie.201908322
-
[17]
Yuan Y T, Wang T, Chen H, Mahurin S M, Luo H M, Veith G M, Yang Z Z, Dai S. Angew. Chem. Int. Ed., 2020, 59(49): 21935-21939 doi: 10.1002/anie.202009180
-
[18]
Li Y, Zhang D N, Feng X H, Xiang Q J. Chin. J. Catal., 2020, 41(1): 21-30 doi: 10.1016/S1872-2067(19)63427-3
-
[19]
Wang Y F, Jing B H, Wang F L, Wang S C, Liu X, Ao Z M, Li C H. Water Res., 2020, 180: 115925 doi: 10.1016/j.watres.2020.115925
-
[20]
Lin L H, Yu Z Y, Wang X C. Angew. Chem. Int. Ed., 2019, 58(19): 6164-6175 doi: 10.1002/anie.201809897
-
[21]
Fang S, Xia Y, Lv K L, Li Q, Sun J, Li M. Appl. Catal. B, 2016, 185: 225-232 doi: 10.1016/j.apcatb.2015.12.025
-
[22]
Li Y H, Ho W K, Lv K L, Zhu B C, Lee S C. Appl. Surf. Sci., 2018, 430: 380-389 doi: 10.1016/j.apsusc.2017.06.054
-
[23]
Li Y H, Gu M L, Shi T, Cui W, Zhang X M, Dong F, Cheng J S, Fan J J, Lv K L. Appl. Catal. B, 2020, 262: 118281 doi: 10.1016/j.apcatb.2019.118281
-
[24]
Li Y H, Gu M L, Zhang M, Zhang X M, Lv K L, Liu Y Q, Ho W K, Dong F. Chem. Eng. J., 2020, 389: 124421 doi: 10.1016/j.cej.2020.124421
-
[25]
Cao J, Nie W S, Huang L, Ding Y B, Lv K L, Tang H Q. Appl. Catal. B, 2019, 241: 18-27 doi: 10.1016/j.apcatb.2018.09.007
-
[26]
Niu P, Liu G, Cheng H M. J. Phys. Chem. C, 2012, 116(20): 11013-11018 doi: 10.1021/jp301026y
-
[27]
Li Y H, Lv K L, Ho W K, Zhao Z W, Huang Y. Chin. J. Catal., 2017, 38: 321-329 doi: 10.1016/S1872-2067(16)62573-1
-
[28]
Wu X F, Cheng J S, Li X F, Li Y H, Lv K L. Appl. Surf. Sci., 2019, 465: 1037-1046 doi: 10.1016/j.apsusc.2018.09.165
-
[29]
Duan Y Y, Li X F, Lv K, Zhao L, Liu Y. Appl. Surf. Sci., 2019, 492: 166-176 doi: 10.1016/j.apsusc.2019.06.125
-
[30]
Cheng J S, Hu Z, Li Q, Li X F, Fang S, Wu X F, Li M, Ding Y B, Liu B, Yang C J, Wen L L, Liu Y, Lv K L. Appl. Catal. B, 2019, 245: 197-206 doi: 10.1016/j.apcatb.2018.12.044
-
[31]
Zhao D M, Chen J, Dong C L, Zhou W, Huang Y C, Mao S S, Guo L J, Shen S H. J. Catal., 2017, 352: 491-497 doi: 10.1016/j.jcat.2017.06.020
-
[32]
Li X F, Hu Z, Li Q, Lei M, Fan J J, Carabineiro S A C, Liu Y, Lv K L. Chem. Commun., 2020, 56(91): 14195-14198 doi: 10.1039/D0CC05948J
-
[33]
Yang C, Zhang S S, Huang Y, Lv K L, Fang S, Wu X F, Li Q, Fan J J. Appl. Surf. Sci., 2020, 505: 144654 doi: 10.1016/j.apsusc.2019.144654
-
[34]
Xiong T, Cen W L, Zhang Y X, Dong F. ACS Catal., 2016, 6(4): 2462-2472 doi: 10.1021/acscatal.5b02922
-
[35]
Huang Z A, Sun Q, Lv K L, Zhang Z H, Li M, Li B. Appl. Catal. B, 2015, 164: 420-427 doi: 10.1016/j.apcatb.2014.09.043
-
[36]
Li Y H, Lv K L, Ho W, Dong F, Wu X F, Xia Y. Appl. Catal. B, 2017, 202: 611-619 doi: 10.1016/j.apcatb.2016.09.055
-
[37]
Yang C, Tan Q Y, Li Q, Zhou J, Fan J J, Li B, Sun J, Lv K L. Appl. Catal. B, 2020, 268: 118738 doi: 10.1016/j.apcatb.2020.118738
-
[38]
Fang S, Lv K L, Li Q, Ye H P, Du D Y, Li M. Appl. Surf. Sci., 2015, 358: 336-342 doi: 10.1016/j.apsusc.2015.07.179
-
[39]
Li Y H, Gu M L, Zhang X M, Fan J J, Lv K L, Carabineiro S A C, Dong F. Mater. Today, 2020, 41: 270-303 doi: 10.1016/j.mattod.2020.09.004
-
[40]
Ong W J, Tan L L, Ng Y H, Yong S T, Chai S P. Chem. Rev., 2016, 116(12): 7159-7329 doi: 10.1021/acs.chemrev.6b00075
-
[41]
Martin D J, Qiu K P, Shevlin S A, Handoko A D, Chen X W, Guo Z X, Tang J W. Angew. Chem. Int. Ed., 2014, 53(35): 9240-9245 doi: 10.1002/anie.201403375
-
[42]
Cheng J S, Hu Z, Lv K L, Wu X F, Li Q, Li Y H, Li X F, Sun J. Appl. Catal. B, 2018, 232: 330-339 doi: 10.1016/j.apcatb.2018.03.066
-
[43]
Lin L H, Ou H H, Zhang Y F, Wang X C. ACS Catal., 2016, 6(6): 3921-3931 doi: 10.1021/acscatal.6b00922
-
[44]
Zeng Z X, Yu H T, Quan X, Chen S, Zhang S X. Appl. Catal. B, 2018, 227: 153-160 doi: 10.1016/j.apcatb.2018.01.023
-
[45]
Li X H, Zhang J S, Chen X F, Fischer A, Thomas A, Antonietti M, Wang X C. Chem. Mater., 2011, 23(19): 4344-4348 doi: 10.1021/cm201688v
-
[46]
Wang J H, Shen Y F, Li Y, Liu S Q, Zhang Y J. Chemistry, 2016, 22(35): 12449-12454 doi: 10.1002/chem.201602095
-
[47]
Chen Z P, Savateev A, Pronkin S, Papaefthimiou V, Wolff C, Willinger M G, Willinger E, Neher D, Antonietti M, Dontsova D. Adv. Mater., 2017, 29(32): 1700555 doi: 10.1002/adma.201700555
-
[48]
Chong S Y, Jones J T A, Khimyak Y Z, Cooper A I, Thomas A, Antonietti M, Bojdys M J. J. Mater. Chem. A, 2013, 1(4): 1102-1107 doi: 10.1039/C2TA01068B
-
[49]
Lin L H, Lin Z Y, Zhang J, Cai X, Lin W, Yu Z Y, Wang X C. Nat. Catal., 2020, 3(8): 649-655 doi: 10.1038/s41929-020-0476-3
-
[50]
Lin L H, Ren W, Wang C, Asiri A M, Zhang J, Wang X C. Appl. Catal. B, 2018, 231: 234-241 doi: 10.1016/j.apcatb.2018.03.009
-
[51]
Xu Y S, Qiu C T, Fan X, Xiao Y H, Zhang G Q, Yu K Y, Ju H X, Ling X, Zhu Y F, Su C L. Appl. Catal. B, 2020, 268: 118457 doi: 10.1016/j.apcatb.2019.118457
-
[52]
Qiu C T, Xu Y S, Fan X, Xu D, Tandiana R, Ling X, Jiang Y N, Liu C B, Yu L, Chen W, Su C L. Adv. Sci., 2019, 6(1): 1801403 doi: 10.1002/advs.201801403
-
[53]
Xu W M, An X H, Zhang Q G, Li Z, Zhang Q H, Yao Z, Wang X K, Wang S, Zheng J T, Zhang J, Wu W T, Wu M B. ACS Sustainable Chem. Eng., 2019, 7: 12351-12357
-
[54]
Xia P F, Antonietti M, Zhu B C, Heil T, Yu J G, Cao S W. Adv. Funct. Mater., 2019, 29(15): 1900093 doi: 10.1002/adfm.201900093
-
[55]
Cui Y J, Ding Z X, Fu X Z, Wang X C. Angew. Chem. Int. Ed., 2012, 51(47): 11814-11818 doi: 10.1002/anie.201206534
-
[56]
王悦, 蒋权, 尚介坤, 许杰, 李永昕. 物理化学学报, 2016, 32(8): 1913-1928 https://www.cnki.com.cn/Article/CJFDTOTAL-YCJY201602001.htmWANG Y, JIANG Q, SHANG J K, XU J, LI Y X. Acta Phys. -Chim. Sin., 2016, 32(8): 1913-1928 https://www.cnki.com.cn/Article/CJFDTOTAL-YCJY201602001.htm
-
[57]
崔言娟, 杨传锋, 祝玉鑫, 滕伟, 唐盛. 无机化学学报, 2020, 36(2): 261-268 doi: 10.11862/CJIC.2020.019CUI Y J, YANG C F, ZHU Y X, TENG W, TANG S. Chinese J. Inorg. Chem., 2020, 36(2): 261-268 doi: 10.11862/CJIC.2020.019
-
[58]
Yang J H, Wang D, Han H X, Li C. Acc. Chem. Res., 2013, 46(8): 1900-1909 doi: 10.1021/ar300227e
-
[59]
Liao J Z, Cui W, Li J Y, Sheng J P, Wang H, Dong X A, Chen P, Jiang G M, Wang Z M, Dong F. Chem. Eng. J., 2020, 379: 122282 doi: 10.1016/j.cej.2019.122282
-
[60]
Guo H, Niu C G, Liang C, Niu H Y, Yang Y Y, Liu H Y, Tang N, Fang H X. Chem. Eng. J., 2021, 409: 128030 doi: 10.1016/j.cej.2020.128030
-
[61]
祝凯, 欧阳杰, 刘家满, 祝玉鑫, 曾黔, 崔言娟. 无机化学学报, 2019, 35(6): 1005-1012 doi: 10.11862/CJIC.2019.114ZHU K, OUYANG J, LIU J M, ZHU Y X, ZENG Q, CUI Y J. Chinese J. Inorg. Chem., 2019, 35(6): 1005-1012 doi: 10.11862/CJIC.2019.114
-
[62]
Zhang G Q, Xu Y S, He C X, Zhang P X, Mi H W. Appl. Catal. B, 2021, 283: 119636 doi: 10.1016/j.apcatb.2020.119636
-
[63]
Li H, Zhu B C, Cao S W, Yu J G. Chem. Commun., 2020, 56(42): 5641-5644 doi: 10.1039/D0CC01338B
-
[64]
Liu X, Jing B H, Lun G Q, Wang Y F, Wang X D, Fang C, Ao Z M, Li C H. Chem. Commun., 2020, 56(21): 3179-3182 doi: 10.1039/D0CC00280A
-
[65]
Ren W, Cheng J J, Ou H H, Huang C J, Titirici M M, Wang X C. ChemSusChem, 2019, 12(14): 3257-3262 doi: 10.1002/cssc.201901011
-
[66]
Du J Y, Fan Y F, Gan X R, Dang X M, Zhao H M. Electrochim. Acta, 2020, 330: 135336 doi: 10.1016/j.electacta.2019.135336
-
[67]
Zeng Z X, Quan X, Yu H T, Chen S, Zhang S S. J. Catal., 2019, 375: 361-370 doi: 10.1016/j.jcat.2019.06.019
-
[68]
Ou H H, Lin L H, Zheng Y, Yang P J, Fang Y X, Wang X C. Adv. Mater., 2017, 29(22): 1700008 doi: 10.1002/adma.201700008
-
[69]
Li Y, Zhang D N, Fan J J, Xiang Q J. Chin. J. Catal., 2021, 42(4): 627-636 doi: 10.1016/S1872-2067(20)63684-1
-
[70]
Hu Z, Yang C, Lv K L, Li X F, Li Q, Fan J J. Chem. Commun., 2020, 56(11): 1745-1748 doi: 10.1039/C9CC08578E
-
[71]
Li Y, Li B H, Zhang D N, Cheng L, Xiang Q J. ACS Nano, 2020, 14(8): 10552-10561 doi: 10.1021/acsnano.0c04544
-
[72]
Zeng Z X, Su Y, Quan X, Choi W Y, Zhang G H, Liu N, Kim B, Chen S, Yu H T, Zhang S S. Nano Energy, 2020, 69: 104409 doi: 10.1016/j.nanoen.2019.104409
-
[73]
Xia Y, Tian Z H, Heil T, Meng A Y, Cheng B, Cao S W, Yu J G, Antonietti M. Joule, 2019, 3(11): 2792-2805 doi: 10.1016/j.joule.2019.08.011
-
[1]
-


图 2 (a) Melon、(b) 三-s-三嗪基PHI型和(c) 三嗪基PTI型的结构模型[43]; (d) 不同结构C3N4的X射线衍射图[44]
Figure 2 Structure models of (a) melon, (b) tri-s-triazine-based PHI type, and (c) triazine-based PTI type[43]; (d) X-ray diffraction patterns of different structures of C3N4[44]
Gray, blue, and white spheres denote the C, N, and H atoms, respectively
图 4 傅里叶变换红外光谱图(a)和熔融盐处理的双氰胺的X射线衍射图(b)[14]; PTI/Li+Cl-的球差校正透射电镜图(c) 以及最大氢气和氧气释放速率与(1010)和(0001)面的平均表面积比的关系图(d)[49]
Figure 4 Fourier transform infrared spectra (a) and X-ray diffraction patterns (b) of dicyandiamide prepared in molten salt[14]; Spherical aberration corrected transmission electron microscopy images (c) and plot of maximum hydrogen and oxygen evolution rate as a function of the mean surface area ratio of the (1010) and (0001) planes of PTI/Li+Cl-(d)[49]

图 5 BCN和CCN的光电流响应图(a)[43]; 不同预热温度下CCN的产氢性能(b)[50]; tri-/tri-s-tri-C3N4-90的高分辨率透射电镜图(c); 产氢性能图(d)[44]
Figure 5 Photocurrent response of BCN and CCN (a)[43]; Hydrogen production performance of CCN at different preheating temperatures (b)[50]; High resolution transmission electron microscopy images of tri-/tri-s-tri-C3N4-90 (c); Hydrogen production performance (d)[44]

图 6 PCN、post-PCN和KCN的傅里叶红外变换光谱(a)和光致发光光谱(b)[51]; KPCN的X射线衍射图(c) 和高分辨透射电镜图像(d)[52]
Figure 6 Fourier transform infrared (a) and photoluminescence (b) spectra of PCN, post-PCN, and KCN[51]; X-ray diffraction patterns (c) and high resolution transmission electron microscopy images (d) of KPCN[52]
Inset in (d): fast Fourier transform pattern from the yellow square of KPCN

图 10 Cu-CCN的扫描透射电镜图(a); Cu-CCN、CCN、BCN还原CO2的光催化活性(b)[71]; Pt原子位于CCN内层内(SA-Pt/g-C3N4-In)的电荷分布(c)及电化学阻抗谱图(d)[72]
Figure 10 Scanning transmission electron microscopy image (a) of Cu-CCN; Photocatalytic activity of CO2 reduction of Cu-CCN, CCN, BCN (b)[71]; Charge distributions (c) of Pt atoms located within the inner layer of CCN (SA-Pt/g-C3N4-In), and electrochemical impedance spectroscopy (d)[72]
表 1 CCN制备方法的比较
Table 1. Comparison of preparation methods of CCN
Method Precursor Basic unit Photocatalytic condition Photocatalytic preformance Ref. Molten salt synthesis KCl/LiCl+dicyandiamide Triazine 300 W xenon lamp
(> 300 nm) 100 mg
catalyst; H2 and O2
evolution189 and 91 μmol·h-1 [14] Preheated and molten
salt synthesisKCl/LiCl+melamine Tri-s-triazine 300 W Xe lamp
(420 nm filter); 50 mg
catalyst; H2 evolution27.5 μmol·h-1 [43] Preheated and molten
salt synthesisCN+melamine+ KCl/LiCl Triazine+
tri-s-triazine300 W Xe lamp
(420 nm filter); 50 mg
catalyst; H2 evolution144 μmol·h-1
(30 times higher than BCN)[44] Solid salt synthesis CsCl+CN Tri-s-triazine 300 W Xe lamp
300 W Xe lamp(420 nm filter); 4 mg
catalyst; 4 h H2 evolution6.17 μmol
(23 times higher than BCN)[53] Solid salt synthesis KCl+CN Tri-s-triazine 300 W Xe lamp
(420 nm ≤ λ ≤ 780 nm);
50 mg catalyst;
H2 evolution59.4 μmol·h-1
(20 times higher than BCN)[51] Solid salt synthesis KBr+melamine Tri-s-triazine 500 W Xe lamp
(280 nm); H2 evolution1 600 μmol·h-1·g-1
(18 times higher than BCN)[52] Organic solvothermal
synthesisAcetonitrile+hydrochloride +
dicyandiamideTri-s-triazine 350 W Xe lamp
(AM1.5 filter);
CO2 photoreduction12.07 μmol·h-1·g-1 [54] Protonation synthesis HCl+dicyandiamide Tri-s-triazine 150 W Xe lamp
(λ > 420 nm); 300 mg
catalyst; RhB degradation0.012 96 min-1
(7 times higher than BCN)[46]  下载: 导出CSV
下载: 导出CSV
-













 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 56
- 文章访问数: 5545
- HTML全文浏览量: 1061
