Anhui Province Key Laboratory of Coal Clean Conversion and High Valued Utilization, School of Chemistry and Chemical Engineering, Anhui University of Technology, Maanshan, Anhui 243002, China
Received Date:
05 November 2020 Revised Date:
09 January 2021 Available Online:
10 May 2021
Abstract:
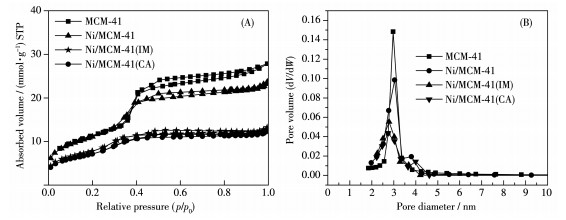
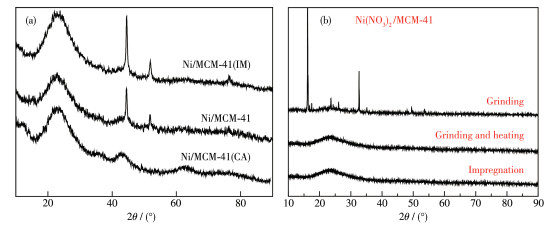
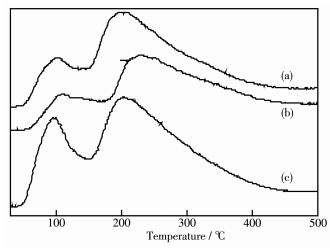
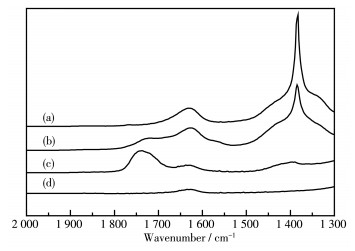
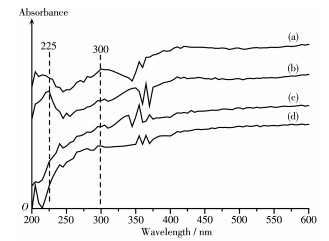
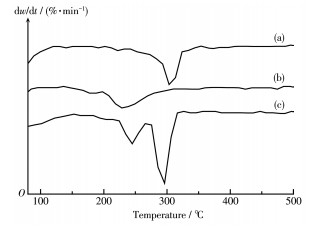
Ni/MCM-41 catalysts were prepared with a solvent-free method of mechanical grinding. Furtherly, Ni/MCM-41(CA) catalyst was also prepared by adding citric acid during preparation of Ni/MCM-41. Compared with Ni/MCM-41(IM) catalyst prepared with the conventional impregnation method, the physical properties of the catalyst with the solvent-free method were almost unchanged. And the dispersion of metallic nickel and the performance of naphthalene hydrogenation were slightly improved. After adding citric acid during preparation of Ni/MCM-41, the dispersion of metallic nickel in Ni/MCM-41(CA) catalyst was greatly increased from 6.9% to 67.9%, and the performance of naphthalene hydrogenation was nearly enhanced 100%. Through infrared spectroscopy, UV spectra and thermogravimetric analysis, the promotion mechanism for adding citric acid in the solvent-free method was attributed to forming a kind of nickel citrate by adding citric acid during preparation of Ni/MCM-41.
a Calculated from the volume of chemically adsorbed H2, loading mass fraction of Ni: 10.0%; b Estimated by the particle size using Scherer formula, d=0.89λ/(Bcos θB), where λ is the wavelength (0.154 18 nm), B is the full half-width (FWHM) of the peak, and θB is the Bragg angle (2θB=44.4°).
Dillen A J V, Robert J A M, Lensveld D J. J. Catal. , 2003, 216(1/2): 257-264
[2]
Ren S B, Zhang P, Shui H F, Lei Z P, Kang S G. Catal. Commun. , 2010, 12(2): 132-136 doi: 10.1016/j.catcom.2010.08.022
[3]
Ren S B, Zhao R, Zhang P, Lei Z P, Wang Z C, Kang S G, Pan C X, Shui H F. React. Kinet. Mech. Catal. , 2014, 111(1): 247-257 doi: 10.1007/s11144-013-0629-3
[4]
Ren S B, Shen Z, Zhang P, Lei Z P, Wang Z C, Kang S G, Pan C X, Shui H F. J. Fuel Chem. Technol. , 2014, 42(5): 591-596 doi: 10.1016/S1872-5813(14)60029-3
[5]
Li X, Quek X Y, Ligthart D A J M, Guo M, Zhang Y, Li C, Yang Q, Hensen E J M. Appl Catal. B, 2012, 123-124: 424-432 doi: 10.1016/j.apcatb.2012.05.009
[6]
Wang Y M, Wu Z Y, Shi L Y, Zhu J H. Adv. Mater. , 2005, 17(3): 323-327 doi: 10.1002/adma.200400860
[7]
Wang Y M, Wu Z Y, Zhu J H. J. Solid State Chem. , 2004, 177(10): 3815-3823 doi: 10.1016/j.jssc.2004.07.013
Table 2.
Hydrogen chemisorption data of catalysts prepared by different methods determined by H2-TPD
Catalyst
H2 Chemisorption uptake/(mL·g-1)
SNi/(m2·g-1)
D/%
d/nm
TPDa
XRDb
Ni/MCM-41
27.6
48
7.2
14.0
14.7
Ni/MCM-41(IM)
26.5
46
6.9
14.6
14.8
Ni/MCM-41(CA)
259.4
452
67.9
1.5
1.5
a Calculated from the volume of chemically adsorbed H2, loading mass fraction of Ni: 10.0%; b Estimated by the particle size using Scherer formula, d=0.89λ/(Bcos θB), where λ is the wavelength (0.154 18 nm), B is the full half-width (FWHM) of the peak, and θB is the Bragg angle (2θB=44.4°).

 下载:
下载:

 下载:
下载:







