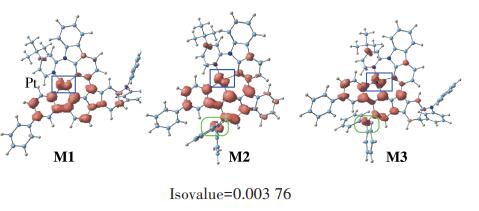
Figure 1.
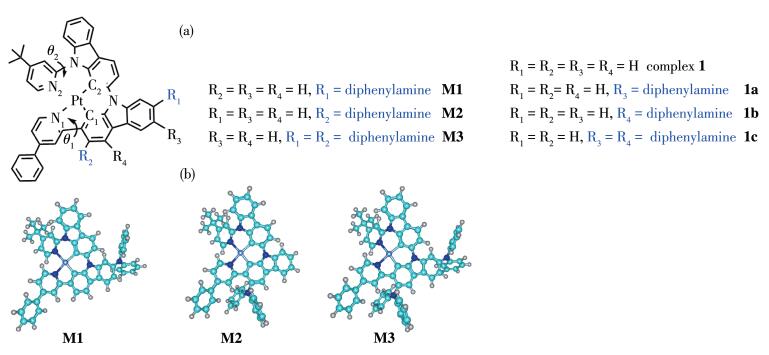
(a) Schematic structures with atomic labeling for metal-ligand bond of studied complexes M1~M3 and referenced complexes 1, 1a~1c[14]; (b) Optimized structures of M1~M3 in ground states

Organometallic complexes of Pt(Ⅱ) and/or Ir(Ⅲ) play important roles on the phosphorescent organic light-emitting diodes (PhOLEDs). Due to the strong spin-orbit coupling (SOC) induced by the metal atoms, the singlet and triplet excitions contribute to the emission[1-4]. For many years, many efforts have been made to synthesize highly efficient and stable materials[5-8]. However, it is still a crucial task to work out detailed understanding of the deactivation mechanism because of the complicated intersystem crossing (ISC). Nowadays, density functional theory (DFT) and time -dependent DFT (TD-DFT) calculations are the most widely used techniques to provide the relationship between structure and optical properties[9-13]. Especially, a type of efficient Pt(Ⅱ) emitters is obtained by introducing the diphenylamine functional substitutions (1a~1c) as shown in Fig. 1a[14]. These primary results show the functionalized diphenylamine moiety can tune the non-radiative process. However, the most important position (R2 position of carbazole ring, as shown in Fig. 1a), which is closed to biphenyl substitutions, may show greatly influence on reaching triplet metal centered (3MC) state. In particular, once populating in 3MC state, it can return to the local minimum structure on T1 surface (T1) or non-radiative decay process to S0 state through the minimal energy crossing point (MECP) between T1 and S0 state. Therefore, these interesting prospects lead us to investigate this type pf Pt(Ⅱ) emitters (M1~M3) in order to set a firmer basis and obtain a deeper understanding of its properties.
In this work, the hybrid density functional, PBE0, was employed for all calculations using the program package in Gaussian 09 program package[15]. The geom-etry optimizations of the ground-state (S0) and lowest-lying triplet excited state (T1) were calculated using restricted and unrestricted DFT with hybrid-type Perdew-Burke-Ernzerh of exchange correlation functional (PBE0) [16-17]. The effective core potential (ECP) basis sets of the Stuttgart group (SDD)[18-19] and 6-31G (d) basis set was used for Pt(Ⅱ) and non-metal atoms, respectively. Frequencies calculations were calculated at the same level to ensure that they were minimums on the potential energy surface. The stability calculations were performed for all these optimized geometries to confirm these wavefunctions were stable. Based on these optimized S0 and T1 geometries, the absorption and emission spectra of the investigated complexes were calculated by TDDFT/PBE0 and TDDFT/PBE38 in a CH2Cl2 solution, respectively. The 3MC states were computed by UPBE0 method. The transition state (TS) geometries between T1 and 3MC state were searched. The connection of T1 and 3MC state through the TS was confirmed by the intrinsic reaction coordinate (IRC) calculation. The minimal energy crossing points (MECP) for T1/S0 surface crossing were calculated by sobMECP program[20-21], which is modified version of Harvey's MECP program and is performed with the Gaussian 09 program. Frequency calculations were run at the same level of theory and the absence of imaginary frequencies ascertained the nature of these points as minima.
The molecular structures of complexes M1~M3 are depicted in Fig. 1. The optimized geometries of these complexes in S0 and T1 state are collected in Table 1. As shown in Table 1, the N1—Pt—N2 and N1—Pt—C2 bond angles of M1 in S0 state were 100.35° and 166.28° respectively, while its N1—N2—C2—C1 angle was 14.95° The N1—Pt—N2, N1—Pt—C2, as well as N1—N2—C2—C1 bond angles of M2 and M3 are similar with those of M1. Clearly, all the complexes adopt distorted square-planar coordination geometries around the metal center, which are similar with the referenced complexes 1 and 1a~1c[14]. Compared with the synthesized molecule 1, the introduction of diphenylamine moiety can effectively strengthen the π-conjugation interaction between the metal and ligand, and thus the metal bond lengths are shortened. Especially, the Pt—N1 and Pt—C1 bond of M2 and M3 were decreased by ca. 0.002 nm and ca. 0.004 nm, respectively, compared with complex 1[14]. On going from S0 state to T1 state, the large structural modifications of these complexes happened on the same ligand. The Pt—N1 and Pt—C1 bonds of M2 and M3 were shortened by ca. 0.004 nm and ca. 0.006 nm, while the Pt—N1 and Pt—C1 of M1 were decreased by ca. 0.005 nm and ca. 0.002 nm, respectively. The strengthened metal-ligand interaction may raise the probability of charge transfer from the metal to the ligand, which will be further discussed in Section 2.2.
 下载:
导出CSV
下载:
导出CSV
| M1 | M2 | M3 | ||||||
| S0 | T1 | S0 | T1 | S0 | T1 | |||
| Pt-N1 | 0.214 | 0.209 | 0.212 | 0.208 | 0.212 | 0.208 | ||
| Pt-C1 | 0.195 | 0.193 | 0.199 | 0.193 | 0.199 | 0.193 | ||
| Pt-N2 | 0.214 | 0.215 | 0.214 | 0.215 | 0.214 | 0.214 | ||
| Pt-C2 | 0.199 | 0.199 | 0.195 | 0.199 | 0.195 | 0.199 | ||
| N1-Pt-N2 | 100.35 | 98.93 | 100.18 | 99.33 | 100.27 | 98.89 | ||
| N1-Pt-C2 | 166.28 | 167.39 | 165.63 | 168.09 | 165.71 | 166.50 | ||
| N1-N2-C2-C1 | 14.95 | 13.55 | 14.28 | 12.82 | 14.22 | 14.74 | ||
The spin density of T1 state of studied complexes is shown in Fig. 2. For M1~M3, the electron density was mainly located at the metal center and ligands, thus T1 state of these complexes is MLCT characteristic. T1 state with MLCT character is useful to tune spinorbit coupling and therefore facilitates the ISC. Besides, a distinguish contribution from the diphenylamine moiety have been detected on M2 and M3, indicating that the photophysical properties can be well tuned by diphenylamine moiety.
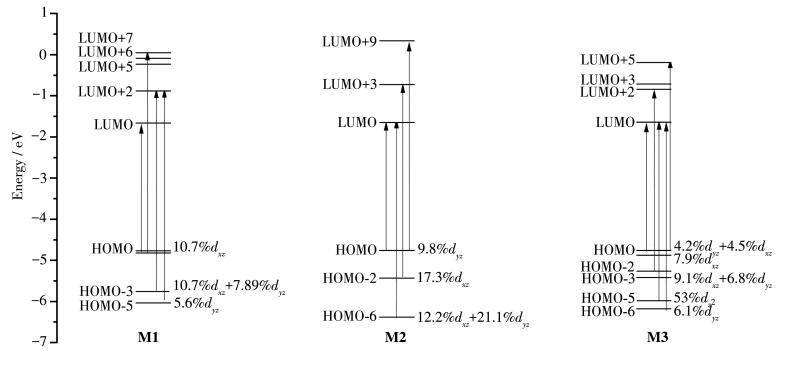
The calculated absorption wavelength (λ, nm), transition energy (E, eV), oscillator strengths (f), dominant configurations, and transition assignments of main absorption spectra are collected in Table 2. The simulated absorption spectra of these complexes are depicted in Fig. 3, together with the absorption spectrum of synthesized compound 1[8]. To analyze nature of these excitations, the molecular orbital composition and the characteristics of Ptd orbitals are shown in Fig. 4.
下载:
导出CSV
| λ/nm | E/eV | f | Main configuration | Assignment | Contribution of MLCT /% | |
| M1 | ||||||
| S1 | 483.8 | 2.56 | 0.036 1 | H-1→L(0.68) | LLCT | 0 |
| S2 | 442.7 | 2.80 | 0.487 5 | H→L(0.68) | MLCT/LLCT | 8.6 |
| S19 | 303.4 | 4.09 | 0.252 4 | H→L+6(0.46) | MLCT/LLCT | 4.32 |
| H-1→L+5(0.37) | ||||||
| S28 | 281.4 | 4.41 | 0.299 5 | H-3→L+2(0.45) | MLCT/LLCT | 7.84 |
| H-1→L+7(-0.29) | ||||||
| H-5→L+2(-0.26) | ||||||
| M2 | ||||||
| S1 | 496.9 | 2.50 | 0.090 4 | H→L(0.69) | MLCT/LLCT | 5.86 |
| S17 | 305.3 | 4.06 | 0.323 2 | H-6→L(0.39) | MLCT/LLCT | 15.1 |
| H-2→L+3(0.36) | ||||||
| S29 | 277.8 | 4.46 | 0.341 4 | H→L+9(0.47) | MLCT/LLCT | 7.49 |
| M3 | ||||||
| S1 | 494.5 | 2.51 | 0.088 8 | H→L(0.64) | MLCT/LLCT | 6.28 |
| S2 | 449.8 | 2.76 | 0.311 2 | H-1→L(0.58) | LLCT | 0 |
| S18 | 312.5 | 3.97 | 0.203 9 | H-5→L(0.35) | MLCT/LLCT | 19.75 |
| H-3→L+2(0.25) | ||||||
| H-2→L+2(0.29) | ||||||
| H→L+5(0.24) | ||||||
| S22 | 305.4 | 4.06 | 0.219 2 | H-6→L(0.42) | MLCT/LLCT | 6.99 |
| H-3→L+3(-0.38) |
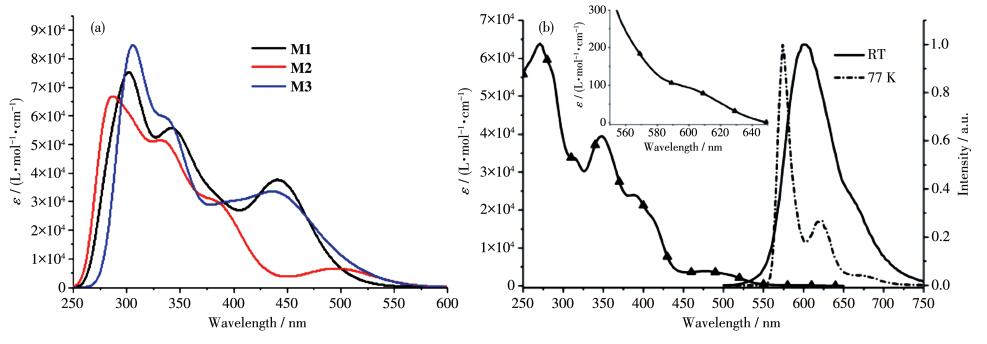
Inset: T1 absorption transition, reproduced with permission from Ref.[8]
As depicted in Fig. 3, complex M2 displayed intense high-energy absorption band (230~300 nm) and weaker band in the region of 450~550 nm, which can display similar curves to the absorption spectrum of experimental molecule 1[14]. Especially, the absorption spectra of M1 and M3 were slightly blue-shifted and the absorption intensity were greatly enhanced. Therefore, the possibility of the intersystem crossing (ISC) from the singlet and triplet state may be well tuned by the introduction of diphenylamine groups. As listed in Table 2, the first strong absorption peak of M1 was at 442.7 nm. The dominant orbital transition is HOMO → LUMO. Because of the large contribution of Ptd (10.7%dxz), the absorption peak of M1 at 442.7 nm was 8.6%MLCT character. The largest absorption peak of M1 at 281.4 nm (S28 state, 7.84%MLCT) shared a similar transition character with that of synthesized 1 at 294 nm (S18 state) with 9.6% MLCT. For M2, the largest absorption peaks were located at 305.3 and 277.8 nm. According to Table 2 and Fig. 4, the peaks at 305.3 nm (15.1%) and 277.8 nm (7.49%) possessed large amounts of MLCT character. Clearly, introducing the diphenylamine at the R2 position is more suitable for enhancing the light energy collection and singlet-triplet transitions than at R1 position. Besides, for M3, the distinguishable absorption peaks around 312.5 and 305.4 nm possessed 19.75% and 6.99%MLCT character, respectively. Obviously, the participation of MLCT in the absorption spectra can be increased by gradually increasing the amount of diphenylamine moiety. The increased amount of MLCT might be efficient to collect light energy participation of metals and increase the spin-orbital coupling effect. Meanwhile, the intensities of the absorption bands of M3 were much larger than those of M1, which may result in high intensity for the triplet excited states through the ISC procedure, and hence increasing the phosphorescence efficiency, which is consistent with the analyses of molecular geometry.
The emission energies and wavelength, transition characters are listed in Table 3. The calculated phosphorescent spectrum of M1 was 631.6 nm, demonstrating that it is potential candidates for red emitting materials. Different from the referenced molecules (1a~1c) and M1, the calculated phosphorescent spectrum of complexes M2 (615.6 nm) and M3 (602.7 nm) were blue-shifted, which is caused by that the internal occupied orbitals are dominant transition orbitals, and the energy gaps of transition orbitals increased. In addition, due to the significant contribution of Ptd orbital in the transition orbital, the phosphorescence spectra of these compounds can be described as 3MLCT/3LLCT character. As well accepted, the contribution of 3MLCT in T1 state will be beneficial to enhance the SOC and transition probability. The calculation results show that the introduction of the diphenylamine moiety at different position could influence the emission spectra, which can provide useful information for further designing highly efficient emitters.
下载:
导出CSV
| λ/nm | Main configuration | Orbital contribution | Assignment | ||
| M1 | 631.6 | 0.50L → H | 12.2%dxz+84.2%π(ligand) → 95.6%π*(ligand) | 3MLCT/3LLCT | |
| -0.30L → H-2 | 16.5%dyz+76.2%π(ligand) → 95.6%π*(ligand) | ||||
| M2 | 615.6 | 0.54L → H | 8.9%dyz+89.3%π(ligand) → 95.8%π*(ligand) | 3MLCT/3LLCT | |
| 0.24L → H-1 | 7.4%dyz+88.2%π(ligand) → 95.8%π*(ligand) | ||||
| -0.23L → H-2 | 15.3%dyz+82.9%π(ligand) → 95.8%π*(ligand) | ||||
| M3 | 602.7 | 0.47L → H | 4.9%dxz+4.7%dyz+88.7%π(ligand) → 95.8%π*(ligand) | 3MLCT/3LLCT | |
| 0.30L → H-1 | 98.5%π(ligand) → 95.8%π*(ligand) | ||||
| 1[14] | 626 |
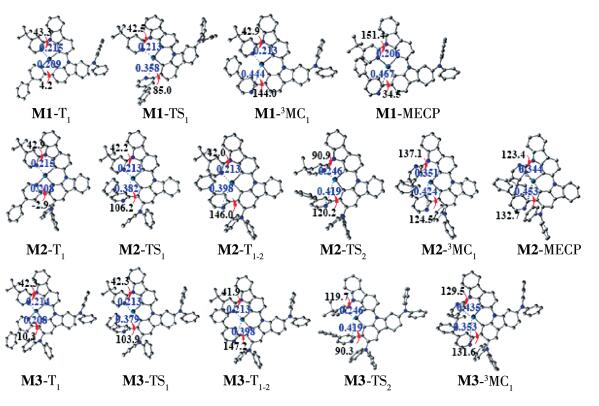
As described in the introduction, 3MC excited state acts as a very efficient activated non-radiative channel for the deactivation to S0 state, due to the presence of MECP between S0 and 3MC potential energy surface. In view of this, we attempt to optimize 3MC state by increasing the metal -ligand bond lengths. The optimized geometries of the TS state between T1 state and 3MC state as well as T1 state are also shown in Fig. 5. The calculated IRCs for T1 → 3MC pathways of studied complexes are shown in Fig. 6, together with the spin density distributions.
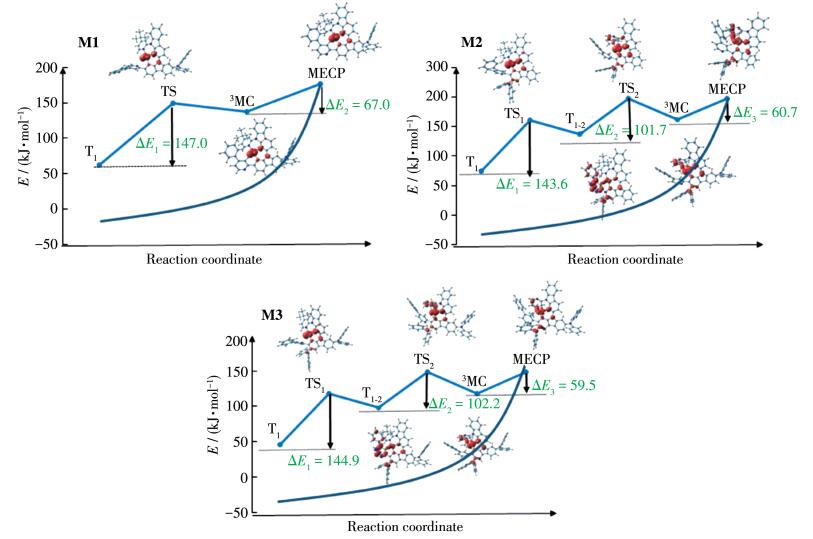
For M1, the metal-ligand bond length Pt—N1 of 3MC state was largely stretched (0.444 nm), which was much larger than those of complex 1 and 1a[14]. Coupled with the dissociation bond length, M1 showed large out of plane on the 4-phenylpyridine ligand. The intermolecular dihedral angle θ1 between the carbazole and biphenyl moiety increased from 4.2° (T1) to 144.0° (3MC1), as shown in Fig. 5. And the Pt—N1 bond increased by 0.151 nm from TS1 to T1 state. Coupled with the large geometry conformation, the energy barrier from T1 to 3MC state was 147.0 kJ·mol-1 for M1. And the MECP state of M1 was 67.0 kJ·mol-1 higher than that of 3MC, which was similar with that of 1[14]. The spin densities of TS, 3MC, and MECP state for these complexes with the typical feature of the large population at Pt atoms. Thus, the nonradiative deactivation pathway can be assigned to T1 (MLCT/π → π*) → TS (d-d) → 3MC (d-d) → MECP (d- d). Clearly, the introduction of diphenylamine moiety at R1 position plays small effect on tuning the non-radiative decay process.
For M2, because the large steric hindrance limited torsion of biphenyl moiety, the Pt—N1 bond length of T1-2 (MLCT/d-d) state was 0.398 nm, which was shortened ca. 0.05 nm relative to 3MC1 (d-d) state of M1. Therefore, the conjugation interaction between the metal center and ligand were still strong, and thus T1-2 state still belonged to 3MLCT character. The process of T1 (MLCT/π → π *) → TS1 (d-d) → T1-2 (MLCT/d-d) needed to overcome a large energy barrier of 143.6 kJ·mol-1. Next, starting from T1-2 state, the Pt—N2 bond was broken as well as the dihedral θ2 between pyridine and carbazole rotated from T1-2 to 3MC1. The TS2 connecting both T1-2 and 3MC1 were optimized. The metal-ligand bonds Pt—N1 in TS2 and 3MC1 became larger by 0.022 and 0.026 nm than those in T1-2 state of M2, respectively, while Pt—N2 of M2 in TS2 and 3MC1 became 0.033 and 0.138 nm relative to T1-2 state. The activation barrier of the reaction T1-2 → TS2 → 3MC1 of M2 was 101.7 kJ·mol-1. Besides, the MECP of M2 was 60.7 kJ·mol-1, higher than that of 3MC1 and thus the nonradiative deactivation channels of M2 became much difficult.
Similar with M2, the nonradiative deactivation channels of M3 followed two steps: one was T1 (MLCT/π → π*) → TS1 (d -d) → T1-2 (MLCT/d-d) with 144.9 kJ·mol-1 energy barrier; the other one was T1-2 (MLCT/d-d) → TS2 (d-d) → 3MC1(d-d) with 102.2 kJ·mol-1. For the first step, the Pt—N1 bond length of T1 state for M3 was elongated and the intermolecular dihedral angle θ1 was rotated. T1-2 Thus, T1 distortion opened up the first deactivation channel, which appeared for state of M3. T1-2 state was connected to 3MC1 state via a TS2 state. The MECP of M3 was found in 59.5 kJ·mol-1 above 3MC1 minimum. Furthermore, the energy gap between T1 and MECP of M2 and M3 were ca. 206.7 kJ·mol-1, which were much higher than that of M1 (192.0 kJ·mol-1). Thus, the high energy of MECP state of M2 and M3 makes its T1 state more stable; a deactivation pathway via the MECP state could be difficult. Moreover, the introduction of phenylamine moiety at R2 position could result in difficult nonradiative decay and possible increase of the quantum efficiency.
A detailed theoretical study on Pt(Ⅱ) cyclometalated complexes with diphenylamine group were carried out by DFT and TDDFT methods. The absorption spectra and emission spectra of these complexes are significantly affected by the diphenylamine substituents and their positions. The potential energy profiles for the nonradiative decay from T1 of studied complexes were explored to reveal the effect of nonradiative decay on phosphorescence. The calculated results show that the deactivation process is via MECP state. M2 and M3 have much higher energy of MECP state and larger energy barrier through the process of T1 (MLCT/π → π*) → TS1(d-d) → T1-2 (MLCT/d-d) → TS2 (d-d) → 3MC1 (d-d) than that of M1. Thus, a deactivation pathway of M2 and M3 via the MECP state could be difficult. So, the introduction of phenylamine moiety at R2 position could result in difficult nonradiative decay and possible increase of the quantum efficiency, which sheds light on a better understanding of the excited-state behavior of this type of Pt emitters.
Kang J S, Zaen R, Park K M, Lee K H, Lee J Y, Kang J Y. Cryst. Growth Des. , 2020, 20(9): 6129-6138 doi: 10.1021/acs.cgd.0c00838
Yang X L, Guo H R, Liu B, Zhao J, Zhou G J, Wu Z X, Wong W Y. Adv. Sci. , 2018, 5: 1701067-170107 doi: 10.1002/advs.201701067
秦梦娜, 王利东, 程美令, 刘露, 刘琦, 唐晓艳. 无机化学学报, 2020, 36(3): 529-535QIN M N, WANG L D, CHENG M L, LIU L, LIU Q, TANG X Y. Chinese J. Inorg. Chem. , 2020, 36(3): 529-535
武钰铃, 杜晓刚, 孙静, 李洁, 王华, 许并社. 无机化学学报, 2014, 30(11): 2516-2522WU Y L, DU X G, SUN J, LI J, WANG H, XU B S. Chinese J. Inorg. Chem. , 2014, 30(11): 2516-2522
Shafikov M Z, Daniels R, Pander P, Dias F B, Williams J A G, Kozhevnikov V N. ACS Appl. Mater. Interfaces, 2019, 11(8): 8182-8193 doi: 10.1021/acsami.8b18928
Li G J, Zhao X D, Fleetham T, Chen Q D, Zhan F, Zheng J B, Yang Y F, Lou W W, Yang Y N, Fang K, Shao Z Z, Zhang Q S, She Y B. Chem. Mater. , 2020, 32(1): 537-548 doi: 10.1021/acs.chemmater.9b04263
Liu B Q, Jabed M A, Guo J L, Xu W, Brown S L, Ugrinov A, Hobbie E K, Kilina S, Qin A J, Sun W F. Inorg. Chem. , 2019, 58(21): 1437714388
Fleetham T, Li G J, Li J. ACS Appl. Mater. Interfaces, 2015, 7(30): 16240-16246 doi: 10.1021/acsami.5b01596
Xu Y Y, Luo Y F, Li M, He R X, Shen W. J. Phys. Chem. A, 2016, 120(34): 6813-6821 doi: 10.1021/acs.jpca.6b03575
Liu Y Q, Gahungu G, Sun X B, Qu X C, Wu Z J. J. Phys. Chem. C, 2012, 116(50): 26496-26506 doi: 10.1021/jp3071019
Cui B B, Shao J Y, Zhong Y. Organometallics, 2014, 33(16): 42204229
Song C P, Tang J, Li J Q, Wang Z X, Li P, Zhang H Y. Inorg. Chem., 2018, 57: 12174-12186 doi: 10.1021/acs.inorgchem.8b01828
舒鑫, 周欣, 潘清江, 李明霞, 张红星, 孙家钟. 无机化学学报, 2008, 24(6): 971-976 doi: 10.3321/j.issn:1001-4861.2008.06.022SHU X, ZHOU X, PAN Q J, LI M X, ZHANG H X, SUN J Z. Chinese J. Inorg. Chem. , 2008, 24(6): 971-976 doi: 10.3321/j.issn:1001-4861.2008.06.022
Kang G J, Cheng H Y, Li K, Ma J F, Ren X F. Chem. Phys. Lett. , 2020, 740: 137077-137083 doi: 10.1016/j.cplett.2019.137077
Gaussian 09, Revision A 01, Gaussian, Inc., Wallingford, CT, 2009.
Ernzerhof M, Scuseria G E. J. Chem. Phys. , 1999, 110: 5029-5036 doi: 10.1063/1.478401
Adamo C, Barone V. J. Chem. Phys. , 1999, 110: 6158-6170 doi: 10.1063/1.478522
Häussermann U, Dolg M, Stoll H, Preuss H, Schwerdtfeger P, Pitzer R M. Mol. Phys. , 1993, 78: 1211-1224 doi: 10.1080/00268979300100801
Kuechle W, Dolg M, Stoll H, Preuss H. J. Chem. Phys. , 1994, 100: 7535-7542 doi: 10.1063/1.466847
Harvey J N, Aschi M. Phys. Chem. Chem. Phys. , 1999, 1: 5555-5563 doi: 10.1039/a907723e
Harvey J N, Aschi M, Schwarz H, Koch W. Theor. Chem. Acc. , 1998, 99: 95-99 doi: 10.1007/s002140050309

Figure 1 (a) Schematic structures with atomic labeling for metal-ligand bond of studied complexes M1~M3 and referenced complexes 1, 1a~1c[14]; (b) Optimized structures of M1~M3 in ground states

Figure 3 (a) Simulated absorption spectra for M1~M3 in CH2Cl2 solution; (b) Absorption spectrum (left) of synthesized compound 1 measured in CH2Cl2 at room temperature and emission spectra (right) measured in CH2Cl2 at room temperature (solid line) and in 2-methyl-THF at 77 K (dash-dot line)
Inset: T1 absorption transition, reproduced with permission from Ref.[8]

Figure 6 Energy levels of reaction path leading from triplet excited state T1 state to MECP state
Table 1. Selected optimized electron geometries (bond length (nm), bond angle (°), dihedral angles (°) of M1~M3 at S0 and T1 states
| M1 | M2 | M3 | ||||||
| S0 | T1 | S0 | T1 | S0 | T1 | |||
| Pt-N1 | 0.214 | 0.209 | 0.212 | 0.208 | 0.212 | 0.208 | ||
| Pt-C1 | 0.195 | 0.193 | 0.199 | 0.193 | 0.199 | 0.193 | ||
| Pt-N2 | 0.214 | 0.215 | 0.214 | 0.215 | 0.214 | 0.214 | ||
| Pt-C2 | 0.199 | 0.199 | 0.195 | 0.199 | 0.195 | 0.199 | ||
| N1-Pt-N2 | 100.35 | 98.93 | 100.18 | 99.33 | 100.27 | 98.89 | ||
| N1-Pt-C2 | 166.28 | 167.39 | 165.63 | 168.09 | 165.71 | 166.50 | ||
| N1-N2-C2-C1 | 14.95 | 13.55 | 14.28 | 12.82 | 14.22 | 14.74 | ||
 下载: 导出CSV
下载: 导出CSV
Table 2. Adsorption spectrum calculation results of M1~M3
| λ/nm | E/eV | f | Main configuration | Assignment | Contribution of MLCT /% | |
| M1 | ||||||
| S1 | 483.8 | 2.56 | 0.036 1 | H-1→L(0.68) | LLCT | 0 |
| S2 | 442.7 | 2.80 | 0.487 5 | H→L(0.68) | MLCT/LLCT | 8.6 |
| S19 | 303.4 | 4.09 | 0.252 4 | H→L+6(0.46) | MLCT/LLCT | 4.32 |
| H-1→L+5(0.37) | ||||||
| S28 | 281.4 | 4.41 | 0.299 5 | H-3→L+2(0.45) | MLCT/LLCT | 7.84 |
| H-1→L+7(-0.29) | ||||||
| H-5→L+2(-0.26) | ||||||
| M2 | ||||||
| S1 | 496.9 | 2.50 | 0.090 4 | H→L(0.69) | MLCT/LLCT | 5.86 |
| S17 | 305.3 | 4.06 | 0.323 2 | H-6→L(0.39) | MLCT/LLCT | 15.1 |
| H-2→L+3(0.36) | ||||||
| S29 | 277.8 | 4.46 | 0.341 4 | H→L+9(0.47) | MLCT/LLCT | 7.49 |
| M3 | ||||||
| S1 | 494.5 | 2.51 | 0.088 8 | H→L(0.64) | MLCT/LLCT | 6.28 |
| S2 | 449.8 | 2.76 | 0.311 2 | H-1→L(0.58) | LLCT | 0 |
| S18 | 312.5 | 3.97 | 0.203 9 | H-5→L(0.35) | MLCT/LLCT | 19.75 |
| H-3→L+2(0.25) | ||||||
| H-2→L+2(0.29) | ||||||
| H→L+5(0.24) | ||||||
| S22 | 305.4 | 4.06 | 0.219 2 | H-6→L(0.42) | MLCT/LLCT | 6.99 |
| H-3→L+3(-0.38) |
下载: 导出CSV
Table 3. Phosphorescence emission spectrum calculation results of M1~M3
| λ/nm | Main configuration | Orbital contribution | Assignment | ||
| M1 | 631.6 | 0.50L → H | 12.2%dxz+84.2%π(ligand) → 95.6%π*(ligand) | 3MLCT/3LLCT | |
| -0.30L → H-2 | 16.5%dyz+76.2%π(ligand) → 95.6%π*(ligand) | ||||
| M2 | 615.6 | 0.54L → H | 8.9%dyz+89.3%π(ligand) → 95.8%π*(ligand) | 3MLCT/3LLCT | |
| 0.24L → H-1 | 7.4%dyz+88.2%π(ligand) → 95.8%π*(ligand) | ||||
| -0.23L → H-2 | 15.3%dyz+82.9%π(ligand) → 95.8%π*(ligand) | ||||
| M3 | 602.7 | 0.47L → H | 4.9%dxz+4.7%dyz+88.7%π(ligand) → 95.8%π*(ligand) | 3MLCT/3LLCT | |
| 0.30L → H-1 | 98.5%π(ligand) → 95.8%π*(ligand) | ||||
| 1[14] | 626 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们