图 1.
YRO和YMRO的XRD图
Figure 1.
XRD patterns of YRO and YMRO

随着全球人口增长和工业化的发展,不可再生的能源储备有限,渐渐不能满足人类活动所需[1-2],因此,开发新的环保可再生能源是未来能源发展的主要趋势。太阳能、风能、地热能和潮汐能等新能源受到昼夜和季节性的影响限制了其发展,而氢能,由于其能量密度高,环境友好,逐渐成为最近的研究热点[3-4]。目前,利用光伏发电驱动电解水(PVE)制氢是最为可行的大规模可再生的能源制氢技术之一,而实现这一技术的前提是在酸性环境下采用质子交换膜(PEM)电解水。电解水制氢的电极总反应为2H2O→2H2+O2 (1),其电极电势会随酸碱环境的不同而发生改变[5-6]:
在碱性电解液中:
|
|
(2) |
|
|
(3) |
在酸性电解液中:
|
|
(4) |
|
|
(5) |
由以上方程式可以看出,酸性条件下的电极电势更高,这导致电极材料更容易发生分解,寿命下降[7-8]。阴极析出1 mol H2需转移2 mol电子,在阳极发生的析氧反应中,每生成1 mol O2会涉及4 mol电子的转移,相对析氢反应更加缓慢,因此,阳极反应决定了整体的反应进程[5]。故降低反应进行势垒,寻找合适的催化剂迫在眉睫。而金属氧化物具有一定的电导率,并且其多变的金属价态导致多样性的配位环境,调控了催化性能,因此吸引了广大研究学者的注意[9-13]。目前,Ru和Ir及其氧化物是已知在酸性条件下氧析出反应(oxygen evolution reaction,OER)催化活性最高的催化剂,Ru基氧化物催化剂相对于Ir基氧化物活性较好,但是稳定性更差[14]。在阳极极化下,RuO2在反应过程中会发生氧化转变成[RuO4]中间体而发生溶解,因此提高Ru基催化剂在OER过程中的稳定性至关重要[15-17]。而烧绿石结构(A2B2O7),类似于莹石结构,其阳离子形成面心立方堆积,阴离子占据其四面体间隙,从空间上看,[BO6]八面体沿立方晶胞的[110]方向以共角相连的方式向空间中伸展形成三维网络结构。但相比于莹石结构,其阴离子数量比莹石结构少1/8,因此在烧绿石结构中存在一定数量的氧空位,使其具有催化活性,并且其本身作为一种开放结构,可通过改变占据A、B位离子种类来调控氧空位浓度,合成Ru基烧绿石结构催化剂[18-19]。为了寻找稳定的催化剂,Yang等[20]制备出Ru基烧绿石型Y2Ru2O7-δ (YRO),相对于RuO2,烧绿石结构中的Ru氧化性较弱,导致吸附在其表面的自由基(OOH*、OH*、O*)更容易脱落,从而促进氧气生成,活性大大提高,并且经过循环测试后,YRO的表面结构几乎未发生变化,而RuO2的表面生成了小于2 nm的非晶层。Cho等[21]制备出Y2[Ru2-xYx]O7-y,通过X射线衍射吸收技术研究其金属离子价态变化,并发现在反应进行过程中,生成强化的[RuO6]结构,大大提高了催化剂的稳定性。Kuznetsov等[22]制备出部分替位的Y1.8M0.2RuO7-δ (M=Cu、Co、Ni、Fe),部分Cu、Co、Ni、Fe的替位影响了与OER活性有关的表面氧空位浓度,通过密度泛函理论证实替位削弱了金属表面与氧自由基的结合能,提高了氧析出效率。
在众多金属氧化物类催化剂中,烧绿石结构作为新兴的开放式结构吸引了广大研究学者的兴趣,然而其A位离子对OER性能的影响仍尚无定论,且提高Ru基催化剂的稳定性仍是解决当前电解水制氢的关键。因此,我们采用溶胶-凝胶法制备出稳定、高效的烧绿石型Y2-xMgxRu2O7-δ (YMRO,x=0.05、0.1、0.15),通过改变A位离子占位比,结合第一性原理进一步探究其对OER性能影响。其中,选择Mg替位Y主要是基于Mg为+2价离子,替位Y3+后可以产生氧空位,可以进一步提升催化剂的活性[23-25]。利用扫描电子显微镜(SEM)、透射电子显微镜(TEM)、X射线衍射(XRD)、能量散射X射线谱(EDS)、X射线光电子能谱(XPS)和热重(TG)等表征方法研究了YMRO催化剂的微观形貌结构并揭示了表面电子结构对其催化性能的影响。采用三电极系统,运用循环伏安(CV)法测试YMRO的催化性能,并使用交流阻抗技术探究其表面结构,证实所制备的YMRO-0.1催化剂的催化活性最高。同时,采用计时电位法对其进行稳定性测试发现,在酸性条件下YMRO-0.1催化剂较YRO更稳定。
采用简单高效的溶胶-凝胶法制备YMRO。按照化学计量比称取硝酸钇(YNO3·6H2O,99.9%)、乙酸镁(Mg(Ac)2·4H2O,99.98%)、三氯化钌(RuCl3· xH2O,Ru质量分数为37.44%)溶解于适量去离子水中,再称取相应含量的乙二胺四乙胺(EDTA,AR)和柠檬酸(C6H8O7·H2O,GR)(金属阳离子、EDTA和柠檬酸的物质的量之比为1:1:2)并加入氨水配成缓冲溶液,调节溶液pH=7,与上述金属离子水溶液混合,随后在水浴锅中85 ℃加热搅拌直到形成凝胶,然后转移至真空干燥箱内140 ℃加热12 h,获得干凝胶。待其冷却至室温后稍加研磨,放入坩埚中,在马弗炉以3 ℃·min-1的速率升温至600 ℃并保温6 h,随后以同样的速率升温至1 050 ℃并保温12 h。x= 0.05、0.1、0.15的YMRO样品分别命名为YMRO-0.05、YMRO-0.1、YMRO-0.15。作为对比的YRO和RuO2也采用相同方法制备。
采用由PANalytical公司生产的X射线衍射仪对上述合成的样品进行X射线衍射(XRD)分析,Cu Kα辐射,波长为0.15 nm,工作电压为40 kV,工作电流为40 mA,扫描步长为0.02°,2θ为5°~80°。样品表面微观形貌采用GeminiSEM 300场发射扫描电镜(SEM)分析,工作电压为5和50 kV。为进一步了解样品微观结构及粒径大小,对其进行高分辩透射电镜(HRTEM,JEM-2100F场发射透射电子显微镜,加速电压为200 kV)及选区电子衍射(SAED)分析,将样品分散在乙醇中超声分散得到均匀的悬浮液,随后滴加在碳膜上,干燥后测试。使用能谱(EDS)分析其组成。比表面积在TriStar П 3020比表面积与孔隙度分析仪上进行测试,测试前,样品先在120 ℃下脱气12 h,以去除表面杂质,然后在液氮温度下吸附氮气,最后随室温脱附并根据Brunauer-Emmet- Teller (BET)方程计算材料比表面积。为了鉴别样品表面化学组成以及元素价态,采用ESCALAB 250Xi X射线光电子能谱仪,以Al Kα为激发源,对其进行X射线光电子(XPS)分析,测试所得谱线均经过C1s标准污染峰(结合能284.8 eV)校对。采用STA-4495F5综合热分析仪分别在氮气和空气气氛中对YRO和YMRO进行TG分析。以10 ℃·min-1的速率从室温程序升温至1 000 ℃,通过NETZSCH Proteus Thermal Analysis软件分析在整个TG测试范围内催化剂在2种气氛下的质量变化,间接对氧空位进行定量分析。
为避免辅助电极发生极化,并且降低溶液欧姆降,所有的电化学测试均在室温下进行。以0.5 mol·L-1H2SO4溶液为电解质,在由硫酸亚汞电极、石墨棒电极和玻碳电极分别作为参比电极、辅助电极和工作电极的三电极系统内进行测试。准确称取0.005 mg催化剂粉末,0.001 mg的高导碳黑(ECP 600JD)作支撑载体以及导电介质,用移液枪分别移取400 μL异丙醇、600 μL去离子水于小号分装瓶中,再移取30 μL的Nafion作为粘结剂于上述混合溶液中。超声2 h以得到均匀的悬浮液。测试时移取4 μL上述悬浮液缓慢滴加于抛光后的玻碳电极上,载量为0.275 mg·cm-2,待异丙醇和Nafion挥发后可得到均匀的催化剂薄膜。采用上海华辰CHI 760e电化学工作站,使用旋转环盘电极仪(RRDE-3A)进行极化曲线测试。在测试之前,向电解液中通30 min氧气以使电解液处于氧饱和状态,电压范围为1.0~1.7 V,扫速为0.01 V·s-1,转速为1 600 r· min-1。为了研究电极表面状况对催化性能的影响,在1.5 V电位下进行交流阻抗测试,在不同频率(105~1 Hz)下对系统施加一个微小的5 mV交流电信号进行扰动,以探究电化学系统的结构和电极系统的动力学过程。采用计时电流法,对电化学系统施加1.5 V的恒定电位以测量电流随时间的变化关系。除特殊说明,所有的电位值均指相对于可逆氢电极。上述实验在测试之前均在1.0~1.5 V范围内,0.1 V·s-1的扫速下,使用CV法循环30圈以活化电极。由电解液引起的欧姆降均经过EiR-corrected=E-iR补偿,其中i是电流,R是欧姆电阻,由交流阻抗测得R=8 Ω。
为了进一步研究YMRO的空位及其电子结构,我们采用第一性原理进行计算。计算采用Vienna ab initio simulation package (VASP)软件,使用PBE泛函框架下的广义梯度近似(generalized gradient approximation,GGA)平面波赝势方法。由于GGA方法无法全面地考虑电子之间相互作用,通常不能合理地描述材料的电子结构。为了改善GGA方法的不足,通常采用GGA+U[26-27]方法修正Ru的d轨道位置(Ud),计算中使用的Ud=3.3 eV[28]。计算采用1×1×1的面心立方原胞,包含8个单元Y2Ru2O7,布里渊区K点选取为3×3×3和5×5×5,分别用于结构驰豫和电子结构计算,平面波截断能为400 eV;平面波能量的精度为1×10-5 eV·atom-1;迭代过程中作用在每个原子上的力不大于0.1 eV·nm-1。
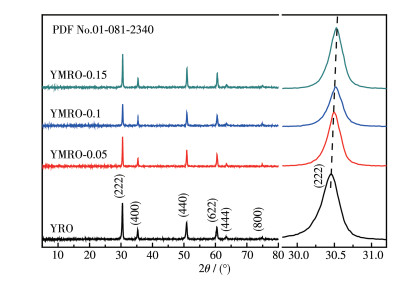
YRO和YMRO的XRD图如图 1所示,位于30.5°、35.4°、50.9°、60.6°、63.6°、74.9°的主要衍射峰分别对应于YRO晶体的(222)、(400)、(440)、(622)、(444)、(800)晶面,与烧绿石结构(PDF No.01-081-2340的特征衍射峰基本符合,属于Fd3m空间群。XRD图中显示其半峰宽较窄,说明合成样品的结晶性较好,同时,图中没有杂峰出现,表明采用溶胶-凝胶法合成的样品无杂相。图 1右侧为峰位在30.5°左右的放大图,可以看出,随着Mg掺杂量的增多,峰位整体右移,根据布拉格公式可知其晶格常数变小。这主要是由于Mg2+离子半径小于Y3+,发生替位后,离子的尺寸效应使晶格常数变小。
从YRO和YMRO的SEM图中可以发现(图 2),各样品颗粒之间均呈现一定程度的团聚,而颗粒本身呈现大小不一的不规则多面体形状,大颗粒周围环绕小颗粒,堆积形成孔道结构,这种结构有利于反应物质的输运。通过对样品BET比表面积测定可知,YMRO-0.05比表面积略大(YRO:5 m2·g-1,YMRO-0.05:6 m2·g-1,YMRO-0.1:5 m2·g-1,YMRO-0.15:4 m2·g-1),能为电极反应提供更多的活性位点,但通过电化学测试发现,其电荷迁移电阻较大,催化性能较差。而对于YMRO-0.15,可能由于其小颗粒增多,堵塞了孔道结构,导致比表面积略低。

(a, b) YRO; (c, d) YMRO-0.05; (e, f) YMRO-0.1; (g, h) YMRO-0.15
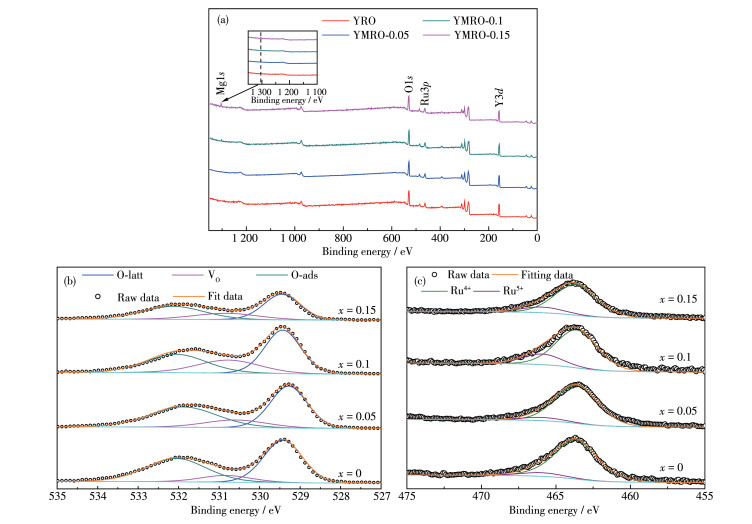
为了鉴别YRO和YMRO的表面化学组成以及元素价态,对其进行了XPS分析,通过全谱扫描探测到归属于Y、Mg、Ru、O等元素的特征峰信号,如图 3a所示。通过对O1s轨道窄谱分析发现,样品表面氧主要以晶格氧(Olatt)、氧空位(VO)以及羟基氧(O— H)形式存在[29-31],其结合能分别位于529.4、530.8和532.1 eV处,如图 3b所示。通过扣除背底,对峰面积积分发现,随Mg含量升高,Olatt含量降低,伴随VO浓度升高。YRO、YMRO-0.05、YMRO-0.1、YMRO-0.15的VO浓度占比分别为9.94%、13.8%、23.24%、18.94%。其中YMRO-0.15的部分Olatt转化为O—H,导致其VO浓度较YMRO-0.1有所下降。随VO浓度的升高,促使O—H键断开,溶液中更多含氧自由基吸附在表面,提高催化材料活性,促进反应进行[24-25]。为了评估Mg替位Y后Ru的价态变化,对YRO和YMRO中Ru窄谱双峰进行拟合分析,如图 3c所示。元素的价态因受所处化学环境和结合形式的影响而对应不同的结合能。当+2价Mg替位+3价Y时,为了保持电荷平衡,Ru会由+4变为+5价,查找文献可知其对应的结合能分别位于463.6和465.9 eV[32-35]。对于YRO,由于缺陷烧绿石结构本征VO的存在,使得其中Ru4+氧化为Ru5+以平衡价态。对于Mg替位的YRO,通过对其峰面积积分可以发现,Mg2+的引入使样品表面电荷密度分布发生改变,并且随着Mg2+含量升高,更多的Ru4+向Ru5+转变,与文献中报道一致[35]。由此可知,Mg的引入使得结构中VO浓度进一步升高,改变中心Ru的电子结构,从而影响其催化性能,其中YMRO-0.1催化剂的VO浓度最大,结合交流阻抗图发现有较多的Ru发生氧化,贡献电子到催化剂表面,促进表面电荷转移[36],加快OER反应进程。
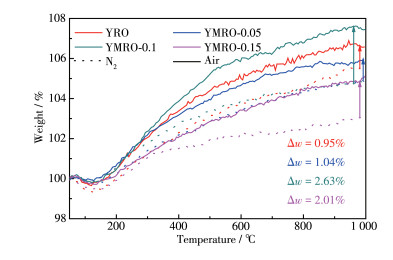
YRO和YMRO的TG曲线如图 4所示,图中虚线和实线分别表示在氮气和空气氛围中进行测试。在起始温度阶段,所有样品皆呈现小幅度增重后失重,可能是由于温度开始升高时,样品膨胀,随着温度继续升高,在样品表面物理吸附的杂质分解。相比于在氮气氛围下,样品质量均明显增加,这说明空气中的氧气与样品反应,填充到样品氧空位中,形成Olatt,使样品重量增加[37]。通过分析质量变化发现,YMRO-0.1在空气中有较大的增重量,这很有可能说明其氧空位含量最多,与XPS分析结果一致。
为了进一步了解YMRO-0.1的形貌与结构特点,将其与YRO对比。图 5a、5b分别为YMRO-0.1、YRO催化剂表面颗粒微观形态以及元素分布图,可以看出Mg均匀分布在基体的晶格中,且未发生明显偏析。图 5c为YMRO-0.1的HRTEM图,结合沿[220]晶带轴方向入射电子束得到的SAED分析可知,YMRO-0.1和YRO均具有较好的结晶态,同时仍保持面心立方结构。经测量可知,YMRO-0.1的(111)和(002)晶面间距分别为0.56和0.49 nm,均小于YRO的0.59和0.51 nm。这与前文XRD数据一致,再次表明Mg替位Y导致了晶格的收缩。

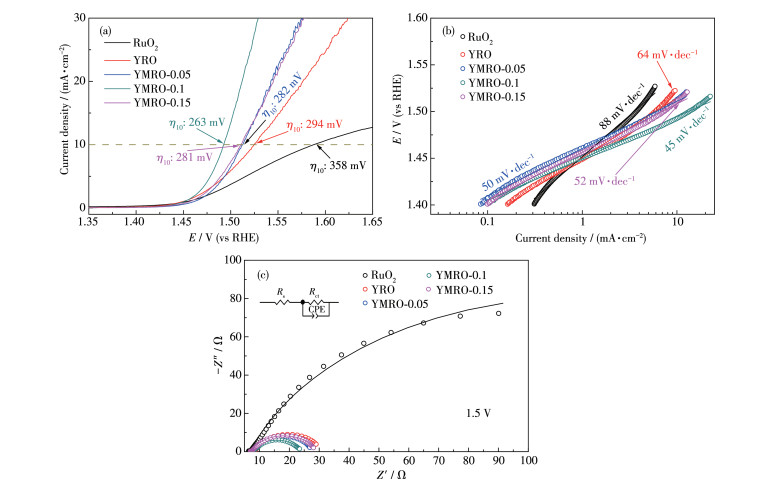
为了评价YMRO和YRO的OER催化性能,对其进行了电化学测试,同时选择RuO2作为对比。由图 6a可知,当电位在1.45 V左右时,就已经产生法拉第电流,开始析氧。相对于YRO,Mg的引入使得OER性能得到明显提高。尽管YMRO-0.1与YRO表现出相似的起始电位,但当流经电极表面的电流密度达到10 mA·cm-2时,YMRO-0.1所需电压仅1.49 V,过电位为263 mV,较YRO (294 mV)降低了10%,可与报道的烧绿石结构催化剂相媲美[35-36, 38]。为了进一步了解电极反应动力学过程,使用Tafel斜率表征电压响应效率,在相同的电流密度下,越低的Tafel斜率表明催动电极反应所需的电位越低,具有较快的动力学过程[39-40],电极反应进行得更加完全,催化活性更高。根据极化曲线并结合Tafel公式(6):η=a+blg|j|得到Tafel斜率[39],其中η、b、j分别为过电位、Tafel斜率以及电流密度,如图 6b所示。值得注意的是,YRO与YMRO-0.1的Tafel斜率(分别为64和45 mV·dec-1)进一步表明YMRO-0.1具有优异的OER性能。在电极反应过程中,电极催化层表面为反应粒子的电荷传递提供了场所。因此,了解电极催化表面的结构对探索反应机理至关重要。而交流阻抗技术是界面研究方法的重要手段,其通过对电极溶液界面施加电信号,获得溶液电极界面阻抗信息[41]。在1.5 V下测得RuO2、YRO和YMRO的交流阻抗图,如图 6c所示。由图可知,电极反应的决速步骤是由电荷转移控制,电化学等效电路如图 6c中插图所示,其中Rs指溶液电阻,常相位角元件CPE代指双电层电阻(纯电容元件过于理想化,考虑到实际催化剂表面的不均匀性及粗糙度),其由CPE-T以及CPE-P两部分组成,Rct指电荷传递电阻[17]。在高频区与横轴交点即为Rs,低频区与横轴交点即为Rct,较小的半圆直径代表较小的电荷传递阻力[42-43]。采用Zview软件进行拟合可知,RuO2、YRO、YMRO-0.05、YMRO-0.1、YMRO-0.15的Rct值依次为224、21.83、18.39、16.25、21.73 Ω。相对于YRO和YMRO催化剂,RuO2的Rct显著增加。Yang等[20]的工作表明,Rct受工作电压影响较大,微量的电压减小/增加,就能引起Rct指数级别的降低/升高。观察图 6a可知,在相同电压下,电极表面电流密度不同,并且在1.5 V下,RuO2电流密度远低于YRO及YMRO。由图 6b可知,RuO2具有最大的Tafel斜率,即电压效率最低,电极表面动力学过程较为缓慢。结合图 6a和6b,当RuO2与YRO和YMRO在相同电势下时,RuO2表现出更大的Rct,Palma-Goyes等[44]的工作也支持了这一结果。综上可知,Mg的占位确实提升了OER性能,并且在所合成的不同Mg含量催化剂中,YMRO-0.1具有最小的半圆直径,即其电荷传递所受阻力最小,从而大大促进了表面吸附物种(O*、OH*、OOH*)的脱落,YMRO-0.1的OER性能极佳。
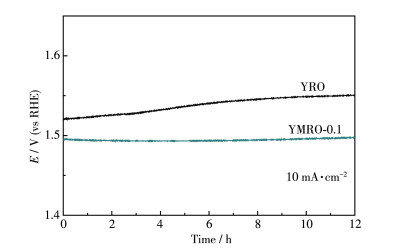
催化剂的稳定性也是评价其性能的关键因素,图 7为合成的YMRO-0.1和YRO催化剂在施加10 mA·cm-2的恒电流密度下,测试12 h的计时电位。对比YMRO-0.1和YRO的2条曲线可知,在相同时间内,为了维持在10 mA·cm-2电流密度,YRO所需的电位随着时间逐渐上升,在12 h后,其电位增加了30 mV;但YMRO-0.1所需的电位基本没有变化,表现出显著的稳定性。
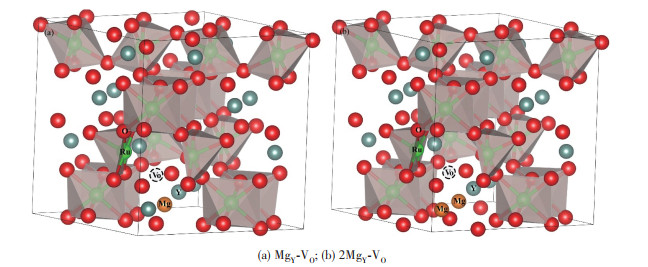
对于上述研究,我们尝试从第一性原理的角度解释Mg掺入对OER性能的影响。考虑到Mg掺杂后的释放应变,MgY与VO会产生相互作用,二者通常形成复合体,因此,我们构建出VO周围分别存在1个MgY (MgY-VO)和2个MgY (2MgY-VO)的2种理论模型,如图 8所示。这里主要讨论MgY-VO和2MgY-VO的性质,根据形成能Eform计算公式:
|
|
(7) |
|
|
(8) |
|
|
(9) |
其中,E(2MgY-VO)、E(MgY-VO)和E(host)分别为原胞中含有2MgY-VO和MgY-VO以及完整原胞的总能量,E(Y2O3)、E(MgO)和E(O2)分别为1个单元Y2O3、MgO和O2的总能量。计算结果如表 1所示,其中MgY-VO@YMRO与2MgY-VO@YMRO分别表示含有MgY-VO、2MgY-VO复合体的YMRO体系。由表 1可知,MgY-VO和2MgY-VO的形成能均比VO更低。这也说明MgY的确能降低VO的形成能,有利于VO浓度的增加。
 下载:
导出CSV
下载:
导出CSV
| Formation energy/eV | Band gap/eV | CTE/eV | |
| YRO | — | 1.03 | 4.71 |
| VO@YRO | 3.92 | — | 4.77 |
| MgY-VO@YMRO | 3.27 | — | 4.66 |
| 2MgY-VO@YMRO | 2.87 | 0.94 | 4.63 |
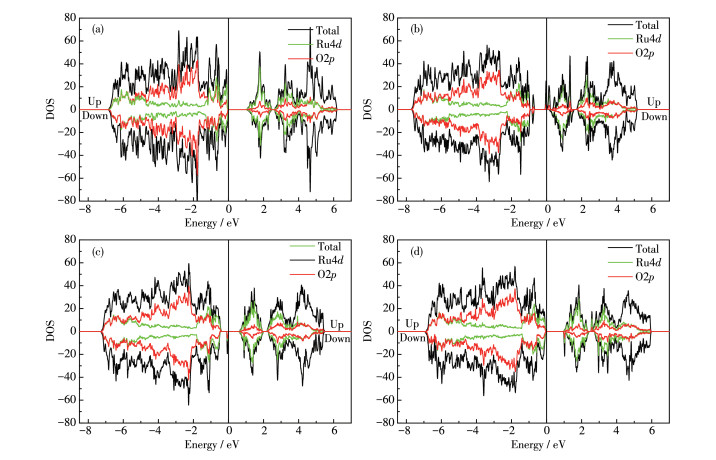
图 9为YRO以及含有MgY-VO和2MgY-VO复合体的YMRO的态密度(density of state,DOS)分析。可以看出YRO的禁带宽度为1.03 eV,低于实验值1.5 eV[45],但接近其他理论计算结果[28],通过禁带宽度可以判断其为半导体。而2MgY-VO复合体由于其电荷平衡,禁带中未出现杂质能级,但其带隙值降为0.94 eV。而YRO中的VO和YMRO中的MgY-VO复合体在禁带中引入了一个杂质能级,主要来源于VO旁Ru的4d轨道。杂质能级的存在促进了电子跃迁,也获得更好的导电性,这有利于催化活性的提高。这里需要指出的是,由于杂质能级的存在,无法计算正常的禁带宽度,因此在表 1中并未显示其值。此外,电荷迁移能(charge-transfer-energy,CTE)是过渡金属3d电子和O2p电子的相对能量,是关联体电子结构与表面性质OER活性的重要描述因子[46]。根据Hong的统计方法[46]计算可得,掺杂Mg后,2种复合体的CTE值均出现下降,这也说明Mg的确能提高催化活性。
实验表明,相对于YRO,YMRO催化剂降低了水裂解所需的过电位,具有较低的Tafel斜率,这主要是由于Mg2+替位部分Y3+,引入大量氧空位,促使表面O—H断开,同时,引起Ru氧化,向催化剂表面提供电子,促进表面电荷转移,改善材料本征活性。但随Mg进一步增多,相对于YMRO-0.1,YMRO-0.15的催化活性下降。由XPS可以看出,部分Olatt转变为羟基氧,VO含量并未进一步增多,导致催化性能有所下降。YMRO催化剂中,YMRO-0.1表现出最强的催化性能,稳定性测试发现其在酸性条件下比YRO更稳定。第一性原理研究进一步表明由于Mg的引入,降低了VO形成能,带隙减小,降低电荷迁移能,提高OER催化活性。
我们通过溶胶-凝胶法成功制备出YMRO催化剂,其中YMRO-0.1具有优异的OER催化性能。实验结果表明,在达到10 mA·cm-2电流密度时,所需电位仅为1.49 V,并且能在酸性条件下稳定工作。通过对其价态分析发现,采用小离子半径的Mg2+取代处于A位的部分Y3+,进一步增加了烧绿石结构中氧缺陷数量。第一性原理计算表明,Mg的引入可以降低氧空位形成能,同时Mg2+的存在影响了中心活性位点Ru4+的电子结构,降低了电荷迁移能,二者的协同作用大大增加了OER催化性能。
Montoya J H, Seitz L C, Chakthranont P, Vojvodic A, Jaramillo T F, Norskov J K. Nat. Mater., 2016, 16:70-81
Stamenkovic V R, Strmcnik D, Lopes P P, Markovic N M. Nat. Mater., 2016, 16:57-69
Khajehsaeidi Z, Sangpour P, Ghaffarinejad A. Int. J. Hydrogen Energy, 2019, 44:19816-19826 doi: 10.1016/j.ijhydene.2019.05.161
Zhao Z J, Zhao J, Wang H B, Li X L, Yang L Q, Zhao Z W, Liu X Y, Liu Y Z, Liu P, Cai Z Y. Int. J. Hydrogen Energy, 2020, 45:14199-14207 doi: 10.1016/j.ijhydene.2019.11.007
Kim J S, Kim B, Kim H, Kang K. Adv. Energy Mater., 2018, 8:1702774 doi: 10.1002/aenm.201702774
Xu H M, Ci S Q, Ding Y C, Wang G X, Wen Z H. J. Mater. Chem. A, 2019, 7:8006-8029 doi: 10.1039/C9TA00833K
Roy C, Rao R R, Stoerzinger K A, Hwang J, Rossmeisl J, Chorkendorff I, Shao-Horn Y, Stephens I E L. ACS. Energy Lett., 2018, 3:2045-2051 doi: 10.1021/acsenergylett.8b01178
Cherevko S, Zeradjanin A R, Topalov A A, Kulyk N, Katsounaros I, Mayrhofer K J J. ChemCatChem, 2014, 6:2219-2223 doi: 10.1002/cctc.201402194
Zhu Y L, Zhou W, Yu J, Chen Y B, Liu M L, Shao Z. Chem. Mater., 2016, 28:1691-1697 doi: 10.1021/acs.chemmater.5b04457
Suntivich J, May K J, Gasteiger H A, Goodenough J B, Shao-Horn Y. Science, 2011, 334:1383-1385 doi: 10.1126/science.1212858
Zhu J K, Gao Q M. Microporous Mesoporous Mater., 2009, 124:144-152 doi: 10.1016/j.micromeso.2009.05.003
Dresp S, Thanh T N, Klingenhof M, Brückner S, Hauke P, Strasser P. Energy Environ. Sci., 2020, 13:1725-1729 doi: 10.1039/D0EE01125H
Zhong H H, Liu T Y, Zhang S W, Li D Q, Tang P G, Alonso-Vante N, Feng Y J. J. Energy Chem., 2019, 33:130-137 doi: 10.1016/j.jechem.2018.09.005
Sardar K, Petrucco E, Hiley C I, Sharman J D, Wells P P, Russell A E, Kashtiban R J, Sloan J, Walton R I. Angew. Chem. Int. Ed., 2014, 53:10960-10964 doi: 10.1002/anie.201406668
Kötz R, Stucki S. Electrochim. Acta, 1986, 31:1311-1316 doi: 10.1016/0013-4686(86)80153-0
Hodnik N, Jovanovič P, Pavlišič A, Jozinović B, Zorko M, Bele M, Šelih V S, Šala M, Hočevar S, Gaberšček M. J. Phys. Chem. C, 2015, 119:10140-10147 doi: 10.1021/acs.jpcc.5b01832
Audichon T, Napporn T W, Canaff C, Morais C, Comminges C, Kokoh K B. J. Phys. Chem. C, 2016, 120:2562-2573 doi: 10.1021/acs.jpcc.5b11868
Talanov M V, Talanov V M. CrystEngComm, 2020, 22:1176-1187 doi: 10.1039/C9CE01635J
Fukina D G, Suleimanov E V, Fukin G K, Boryakov A V, Zubkov S Y, Istomin L A. J. Solid State Chem., 2020, 286:121267 doi: 10.1016/j.jssc.2020.121267
Kim J, Shih P C, Tsao K C, Pan Y T, Yin X, Sun C J, Yang H. J. Am. Chem. Soc., 2017, 139:12076-12083 doi: 10.1021/jacs.7b06808
Park J, Park M, Nam G, Kim M G, Cho J. Nano Lett., 2017, 17:3974-3981 doi: 10.1021/acs.nanolett.7b01812
Kuznetsov D A, Naeem M A, Kumar P V, Abdala P M, Fedorov A, Muller C R. J. Am. Chem. Soc., 2020, 142:7883-7888 doi: 10.1021/jacs.0c01135
Cheng F Y, Shen J, Peng B, Pan Y D, Tao Z L, Chen J. Nat. Chem., 2011, 3:79-84 doi: 10.1038/nchem.931
Li H, Shang J, Zhu H J, Yang Z P, Ai Z H, Zhang L Z. ACS Catal., 2016, 6:8276-8285 doi: 10.1021/acscatal.6b02613
Kim J, Yin X, Tsao K C, Fang S H, Yang H. J. Am. Chem. Soc., 2014, 136:14646-14649 doi: 10.1021/ja506254g
Sheetz R M, Ponomareva I, Richter E, Andriotis A N, Menon M. Phys. Rev. B, 2009, 80:195314 doi: 10.1103/PhysRevB.80.195314
Zhang S T, Li C M, Yan H, Wei M, Evans D G, Duan X. J. Phys. Chem. C, 2014, 118:3514-3522
Lan G Q, Song J, Yang Z. J. Alloys Compd., 2018, 749:909-925 doi: 10.1016/j.jallcom.2018.03.336
Banger K K, Yamashita Y, Mori K, Peterson R L, Leedham T, Rickard J, Sirringhaus H. Nat. Mater., 2011, 10:45-50 doi: 10.1038/nmat2914
Fan J C C, Goodenough B J. J. Appl. Phys., 1977, 48:3524-3531 doi: 10.1063/1.324149
Lu X F, Wu D J, Li R Z, Li Q, Ye S H, Tong Y X, Li G R. J. Mater. Chem. A, 2014, 2:4706-4713 doi: 10.1039/C3TA14930G
Chen S, Huang H, Jiang P, Yang K, Diao J F, Gong S P, Liu S, Huang M X, Wang H, Chen Q W. ACS Catal., 2019, 10:1152-1160
Patra A S, Gogoi G, Sahu R K, Qureshi M. Phys. Chem. Chem. Phys., 2017, 19:12167-12174 doi: 10.1039/C7CP01444A
Berti G, Sanna S, Castellano C, DuiJn J V, Ruiz-Bustos R, Bordonali L, Bussetti G, Calloni A, Demartin F, Duò L, Brambilla A. J. Phys. Chem. C, 2016, 120:11763-11768 doi: 10.1021/acs.jpcc.5b12411
Feng Q, Zhao Z L, Yuan X Z, Li H, Wang H J. Appl. Catal. B, 2020, 260:118176 doi: 10.1016/j.apcatb.2019.118176
Kim M, Ju H, Kim J. Chem. Eng. J., 2019, 358:11-19 doi: 10.1016/j.cej.2018.09.204
Huang Y, Li K, Li S, Lin Y, Liu H, Tong Y. ChemistrySelect, 2018, 3:7423-7428 doi: 10.1002/slct.201800908
Park J, Risch M, Nam G, Park M, Shin T J, Park S, Kim M G, Shao-Horn Y, Cho J. Energy Environ. Sci., 2017, 10:129-136 doi: 10.1039/C6EE03046G
Kim M, Ju H, Kim J. J. Mater. Chem. A, 2018, 6:8523-8530 doi: 10.1039/C8TA01374H
Kim M, Ju H, Kim J. Dalton Trans., 2018, 47:15217-15225 doi: 10.1039/C8DT03217C
Yuan X, Wang H, Colinsun J, Zhang J. Int. J. Hydrogen Energy, 2007, 32:4365-4380 doi: 10.1016/j.ijhydene.2007.05.036
Ehora G, Daviero-Minaud S, Steil M C, Gengembre L, Mentré O. Chem. Mater., 2008, 20:7425-7433 doi: 10.1021/cm801942c
Li X N, Zhang J, Feng Q, Pu C Y, Zhang L Z, Hu M M, Zhou X Y, Zhong X W, Yi W D, Tang J, Li Z W, Zhao X Z, Li H, Xu B M. J. Mater. Chem. A, 2018, 6:17288-17296 doi: 10.1039/C8TA05599H
Palma-Goyes R E, Vazquez-Arenas J, Romero-Ibarra I C, Ostos C. ChemistrySelect, 2018, 3:12937-12945 doi: 10.1002/slct.201802695
Subramanian M A, Aravamudan G, Rao G V S. Prog. Solid state. Chem., 1983, 15:55-143 doi: 10.1016/0079-6786(83)90001-8
Hong W T, Stoerzinger K A, Lee Y L, Giordano L, Grimaud A, Johnson A M, Hwang J, Crumlin E J, Yang W L, Shao-Horn Y. Energy Environ. Sci., 2017, 10:2190-2200 doi: 10.1039/C7EE02052J

图 2 不同分辨率下YRO和YMRO样品的SEM图
Figure 2 SEM images of YRO and YMRO samples at different magnifications
(a, b) YRO; (c, d) YMRO-0.05; (e, f) YMRO-0.1; (g, h) YMRO-0.15

图 3 YRO和YMRO样品的XPS谱图: (a)全谱图, 插图为结合能在1 300 eV附近的局部放大图; (b) O1s窄谱图; (c) Ru3p3/2窄谱图
Figure 3 XPS spectra of YRO and YMRO samples: (a) survey spectra, inset is the detail of binding energy at around 1 300 eV; (b) High-resolution spectra of O1s; (c) High-resolution spectra of Ru3p3/2

图 5 (a) YMRO-0.1样品的TEM图以及相应的元素分布图; (b) YRO样品的TEM图以及相应的元素分布图; (c) YMRO-0.1样品的HRTEM图; (d) YMRO-0.1样品沿[220]晶带轴的SADE花斑; (e) YRO样品的HRTEM图; (f) YRO样品沿[220]晶带轴的SADE花斑
Figure 5 (a) TEM image of YMRO-0.1 sample and corresponding element distribution diagram; (b) TEM image of YRO sample and corresponding element distribution diagram; (c) HRTEM image of YMRO-0.1 sample; (d) SADE pattern of YMRO-0.1 sample along the [220] zone axis; (e) HRTEM image of YRO sample; (f) SADE pattern of YRO sample along the [220] zone axis

图 6 RuO2、YRO和YMRO的电化学性能: (a)极化曲线; (b) Tafel曲线; (c)交流阻抗谱
Figure 6 Electrochemical performance of RuO2, YRO and YMRO: (a) polarization curves; (b) Tafel plots; (c) AC impedance spectra

图 7 YMRO-0.1和YRO在10 mA·cm-2恒电流密度下计时电位变化比较
Figure 7 Chronopotential changes of YMRO-0.1 and YRO under constant current density of 10 mA·cm-2

图 9 (a) YRO、(b) VO@YRO、(c) MgY-VO@YMRO和(d) 2MgY-VO@YMRO的DOS图, 费米能被设置为坐标零点
Figure 9 DOS spectra of (a) YRO, (b) VO@YRO, (c) MgY-VO@YMRO and (d) 2MgY-VO@YMRO, where Fermi energy was chosen to be zero in energy level
表 1 YRO、VO@YRO、MgY-VO@YMRO和2MgY-VO@YMRO的形成能、禁带宽度以及CTE
Table 1. Formation energy, band gap and CTE of YRO, VO@YRO, MgY-VO@YMRO and 2MgY-VO@YMR
| Formation energy/eV | Band gap/eV | CTE/eV | |
| YRO | — | 1.03 | 4.71 |
| VO@YRO | 3.92 | — | 4.77 |
| MgY-VO@YMRO | 3.27 | — | 4.66 |
| 2MgY-VO@YMRO | 2.87 | 0.94 | 4.63 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们