-
[1]
Kumar V, Park S, Parida K, Parida K, Bhavanasi V, Lee P S. Mater. Today Energy, 2017, 4:41-57 doi: 10.1016/j.mtener.2017.03.004
-
[2]
Sekhar S C, Nagaraju G, Yu J S. Nano Energy, 2018, 48:81-92 doi: 10.1016/j.nanoen.2018.03.037
-
[3]
Armand M, Tarascon J M. Nature, 2008, 451(7179):652-657 doi: 10.1038/451652a
-
[4]
Simon P, Gogotsi Y. Nat. Mater., 2008, 7(11):845-854 doi: 10.1038/nmat2297
-
[5]
Gao M R, Xu Y F, Jiang J, Yu S H. Chem. Soc. Rev., 2013, 42:2986-3017
-
[6]
Lee Y W, Kim B S, Hong J, Choi H, Jang H S, Hou B, Pak S, Lee J, Lee S H, Morris S M, Whang D, Hong J P, Shin H S, Cha S N, Sohn J I, Kim J M. Nano Energy, 2017, 37:15-23 doi: 10.1016/j.nanoen.2017.05.006
-
[7]
Huo H H, Zhao Y Q, Xu C L. J. Mater. Chem. A, 2014, 2(36):15111-15117
-
[8]
Sun M, Tie J J, Cheng G, Lin T, Peng S M, Deng F Z, Ye F, Yu L. J. Mater. Chem. A, 2015, 3(4):1730-1736 doi: 10.1039/C4TA04833D
-
[9]
Chen D D, Yang L J, Li J F, Wu Q S. ChemistrySelect, 2019, 4(5):1586-1595 doi: 10.1002/slct.201803413
-
[10]
Liu T, Zhang L Y, You W, Yu J G. Small, 2018, 14(12):1702407 doi: 10.1002/smll.201702407
-
[11]
Chen S G, Wu B X, Qian H, Wu Z X, Liu P, Li F J, He H, Wu J H, Liu B. J. Power Sources, 2019, 438:227000 doi: 10.1016/j.jpowsour.2019.227000
-
[12]
Zhu Q C, Zhao D Y, Cheng M Y, Zhou J Q, Owusu K A, Mai L Q, Yu Y. Adv. Energy Mater., 2019, 9(36):1901081
-
[13]
Xu T H, Li G Y, Zhao L J. Chem. Eng. J., 2018, 336:602-611
-
[14]
Jia H N, Wang Z Y, Zheng X H, Lin J H, Liang H Y, Cai Y F, Qi J L, Cao J, Feng J C, Fei W D. Chem. Eng. J., 2018, 351:348-355
-
[15]
Ke Q Q, Guan C, Zhang X, Zheng M R, Zhang Y W, Cai Y Q, Zhang H, Wang J. Adv. Mater., 2017, 29(5):1604164
-
[16]
Liu Y, Huang M H, Lu M, Guan X H, Guan X, Wang G S, Jia B. Chem. Eng. J., 2019, 364:462-474
-
[17]
Jia H N, Wang Z Y, Zheng X H, Lin J H, Liang H Y Guan X H, Guan X, Wang G S, Jia B, Cai Y F, Qi J L, Cao J, Feng J C, Fei W D. Chem. Eng. J., 2018, 351:348-355
-
[18]
Wen Y X, Liu Y P, Dang S, Tian S H, Li H Q, Wang Z L, He D Y, Wu Z S, Cao G Z, Peng S L. J. Power Sources, 2019, 423:106-114
-
[19]
Sun S X, Luo J H, Qian Y, Jin Y, Liu Y, Qiu Y G, Li Y, Fang C, Han J T, Huang Y H. Adv. Energy Mater., 2018, 8(25):1801080
-
[20]
Xu R, Lin J M, Wu J H, Huang M L, Fan L Q, Chen H W, He X, Wang Y T, Xu Z D. Appl. Surf. Sci., 2018, 434:861-870
-
[21]
Duan B, Gao X, Yao X, Fang Y, Huang L, Zhou J, Zhang L. Nano Energy, 2016, 27:482-491
-
[22]
Wu B X, Qian H, Nie Z W, Luo Z P, Wu Z X, Liu P, He H, Wu J H, Chen S G, Zhang F F. J. Energy. Chem., 2020, 46:178-186
-
[23]
Li Z H, Li X, Xiang L, Xie X, Li X, Xiao D R, Shen J, Lu W Q, Lu L, Liu S Y. J. Mater. Chem. A, 2016, 4(47):18335-18341
-
[24]
Chen S G, Li Y H, Wu B X, Wu Z X, Li F J, Wu J H, Liu P, Liu P, Li H B. Electrochim. Acta, 2018, 275:40-49
-
[25]
Ye B R, Huang M L, Jiang S, Fan L Q, Lin J M, Wu J H. Mater. Chem. Phys., 2018, 211:389-398
-
[26]
Harish S, Nirmalesh-Naveen A, Abinaya R, Archana J, Ramesh R, Navaneethan M, Shimomura M, Hayakawa Y. Electrochim. Acta, 2018, 283:1053-1062
-
[27]
Hou B H, Wang Y Y, Guo J Z, Zhang Y, Ning Q L, Yang Y, Li W H, Zhang J P, Wang X L, Wu X L. ACS Appl. Mater. Interfaces, 2018, 10(4):3581-3589
-
[28]
Wang H Y, Liang M M, Duan D, Shi W Y, Song Y Y, Sun Z B. Chem. Eng. J., 2018, 350:523-533
-
[29]
Zhang Y, Yu L, Hu R D, Zhang J L, Wang Y N, Niu R C, Qian X Y, Zhu J W. J. Mater. Chem. A, 2018, 6(36):17417-17425
-
[30]
Zhang J L, Du C F, Dai Z F, Chen W, Zheng Y, Li B, Zong Y, Wang X, Zhu J W, Yan Q Y. ACS Nano, 2017, 11(10):10599-10607
-
[31]
Liu Y, Zhao D P, Liu H Q, Umar A, Wu X. Chin. Chem. Lett., 2019, 30(5):1105-1110
-
[32]
Yao D, Ouyang Y, Jiao X Y, Ye H T, Lei W, Xia X F, Lu L, Hao Q L. Ind. Eng. Chem. Res., 2018, 57(18):6246-6256
-
[33]
Wu B X, Zhang F F, Nie Z W, Qian H, Liu P, He H, Wu J H, Chen Z Y, Chen S G. Electrochim. Acta, 2021, 365:137325

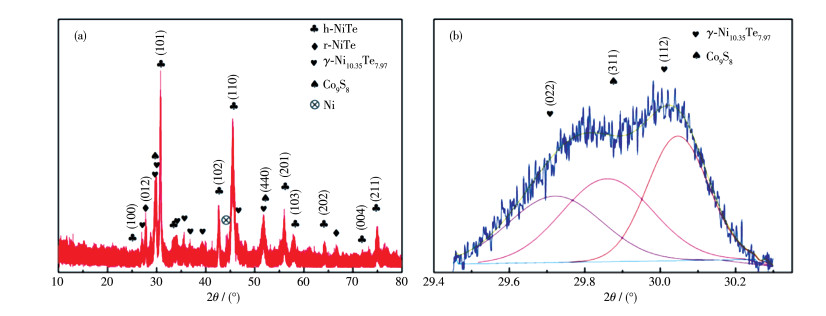
 下载:
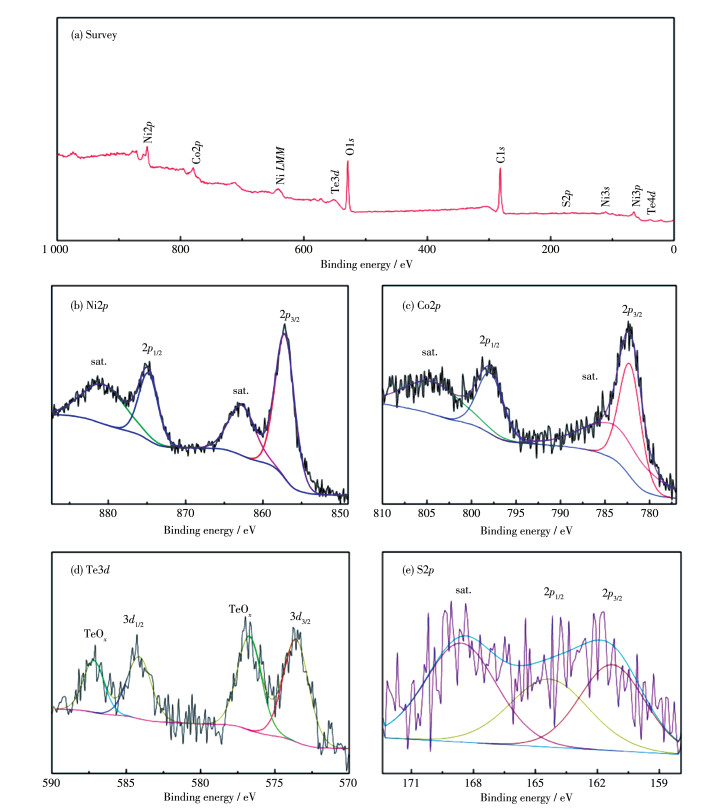
下载:
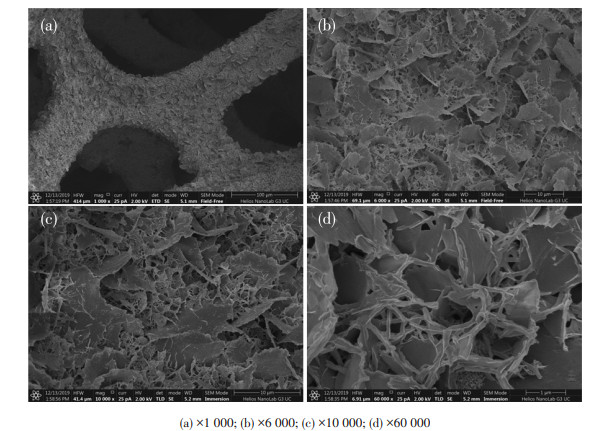


 下载:
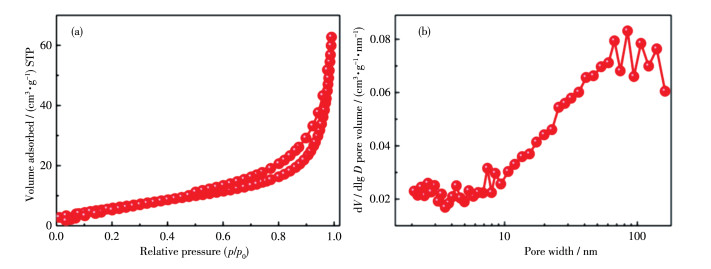
下载:










