0.
引言
纳米金属氟化物是一种多相酸催化剂,由于其具有独特的表面性质,有路易斯酸性[1 8 及很好的耐腐蚀性,尤其是能在HF酸体系中稳定存在,在氟氯交换、脱HF、氟氯烃的歧化、异构化等领域得到广泛的应用。固体金属氟化物的制备通常需经历高温煅烧以除去表面的结合水,以获得配位不饱和的金属位点,即路易斯酸位点。然而,这种由于高晶格能[9 引起的热处理结晶容易导致金属氟化物仅具有10~60 m2 ·g-1 的比表面积,使其具有较低的催化活性。
过去的几十年里,溶胶-凝胶(sol-gel)工艺已经被开发并应用于制备高比表面积催化剂。如在溶胶-凝胶方法制备金属氟化物的过程中,金属醇盐通常用作前驱体,在合适的有机溶剂中与无水HF反应,以合成金属氟化物[10 16 。Murthy等[17 利用溶胶-凝胶氟化的工艺成功地合成了具有150~350 m2 ·g-1 的高比表面积的无定形氟化镁(MgF2 )。Wuttke等[15 研究了溶胶-凝胶合成参数对MgF2 比表面积和结构的影响,证明溶胶-凝胶合成过程非常稳定,大多数操作参数的变化对它没有显著影响。由溶胶-凝胶法合成的高比表面积氟化镁(HS-MgF2 )由于其表面具有大量不饱和配位的Mg2+ 而表现出中等强度的路易斯酸性,然而,HS-MgF2 作为路易斯酸催化剂在应用时受到2个主要方面的限制:首先,HS-MgF2 在240 ℃加热时会发生结构坍塌,导致比表面急剧下降,酸度明显下降,最终导致催化剂失活[17 。其次,HS-MgF2 的路易斯酸度相对氟化铝的路易斯酸度仍太弱,因此不能提供足够的催化活性。为了克服这些问题,掺杂有催化活性的金属三氟化物(MF3 )[18 最近引起了极大的关注。当客体阳离子(即掺杂金属)的物质的量分数小于30%时,若2种阳离子具有相当的尺寸,则客体阳离子可以掺杂到主体的晶格中[19 20 。与未掺杂物质相比,其通常伴随着增强的路易斯酸度和催化活性[19 。据报道,Fe3+ 、Ga3+ 、V3+ 、In3+ 和Cr3+ 常作为MF3 /MgF2 的客体阳离子[7 9 21 。
催化脱HF反应是在路易斯酸催化剂的催化作用下,卤代烃按照E1消去机理[22 23 脱去HF,反应中C-F键在催化剂的路易斯酸酸性位上进行活化异裂[24 ,生成C+ 中间体。中等强度或较强的路易斯酸中心和F原子相互作用引发反应。我们采用溶胶-凝胶法,选择Fe作为掺杂金属,使用溶解在甲醇中的镁醇盐和溶解在乙醇中的Fe(OH)x 3 )3-x 作为金属离子的金属源,用质量分数为40%的HF溶液作为氟源制备Fe掺杂的具有高比表面积的FeF3 /MgF2 催化剂,并通过1,1-二氟乙烷(R152a,C2 H4 F2 )的脱HF催化活性来评价其活性和稳定性。
1.
实验部分
1.1
Fe掺杂MgF2 的制备
将Fe(NO3 )3 ·9H2 O置入65 ℃的减压烘箱中预处理2 h,制得Fe(OH)x 3 )3-x 备用。将Mg条置于体积分数为1%的稀盐酸中处理至银白色,用无水甲醇洗净表面残留的HCl后,干燥,称量4.86 g Mg条溶于200 mL的无水甲醇中,反应生成甲醇镁溶液。称量3.13 g的Fe(OH)x 3 )3-x 加入到无水乙醇中搅拌使之溶解。将2种溶液混合,搅拌0.5 h至混合均匀。在混合溶液中加入15 mL的40%HF水溶液氟化,直至溶液变为无色胶体溶液。反应结束后将胶体溶液老化14~16 h,然后在60 ℃的真空干燥箱中干燥过夜,得Fe-MgF2 干胶。然后将干凝胶置于300 ℃的马弗炉中空气气氛下焙烧4 h,得Fe 0.06Mg0.94F2。按Fe含量的不同,称取相应质量的Fe(OH)x 3 )3-x 和Mg条,采用相同方法制备Fex y 2 样品,其中x =0、0.06、0.15、0.20,相应的y =1、0.94、0.85、0.80。
1.2
催化剂的表征
在-196 ℃下使用Micromeritics ASAP 2000仪器上的N2 物理吸附测定催化剂的比表面积。在测量之前将样品在120 ℃下脱气6 h。选择吸附数据并使用Brunauer-Emmett-Teller(BET)方法计算比表面积。使用非局部密度泛函理论方法从吸附分支获得孔径分布。在相对压力p /p 0 为0.99时估算孔体积。XRD测试在X'pert PRO衍射仪上进行,Cu Kα 辐射(λ=0.154 056 nm),扫描范围10°~80°,电压40 kV,电流100 mA。NH3 程序升温脱附(NH3 -TPD)用于确定酸位点的强度及分布。首先将样品(约0.15 g)在300 ℃氩气中加热30 min进行脱气。冷却至室温后,将催化剂暴露于NH3 和He体积比为1:9的气体混合物中30 min。然后用N2 在100 ℃下冲洗过量的NH3 直至基线达到稳定状态,开始TPD程序(以10 ℃·min-1 的速率升至850 ℃)。通过在线质谱连续监测解吸NH3 的量。样品形貌测试在ZEISS扫描电镜(SEM)上进行,加速电压5 kV。能量散射X射线谱(EDS)测试在HITACHI 7700显微镜上进行,加速电压为100 kV。电子自旋共振(ESR)表征在室温下进行。X射线光电子能谱(XPS)表征在Kratos Axis Ultra仪器上进行。采用O2 程序升温氧化(O2 -TPO)测定反应前后的积碳,首先将约0.05 g样品在300 ℃氩气下吹扫1 h,冷却至室温后,将催化剂暴露于含有体积分数为5% O2 和95% Ar的混合气中以10 ℃· min-1 的升温速率升至700 ℃。通过TCD连续监测O2 的信号。
1.3
C2 H4 F2 脱HF评价反应
由于C2 H4 F2 在路易斯酸催化剂存在下会转化为氟乙烯(CH2 =CHF),使用其脱氢氟化物作为探针反应来测试FeF3 /MgF2 催化剂的催化活性。所涉及的反应如方程式(1 )所示。
实验中选用铬镍铁合金(直径9 mm)作为反应管,使用固定床反应装置。催化剂装填量为2 mL,反应前在N2 气氛下300 ℃预处理4 h。然后通C2 H4 F2 和N2 的混合气,流量控制在45 mL·min-1 ,而C2 H4 F2 占总流量的55.56%,反应温度为300 ℃,在标准大气压的条件下发生C2 H4 F2 的裂解反应。尾气通过色谱(捷岛GC1690,填充柱,TCD检测)分析。
2.
结果与讨论
2.1
Fe对MgF2 结构的影响
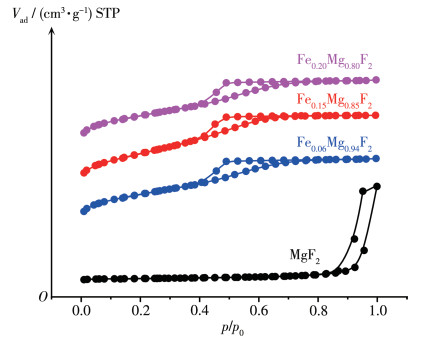
表 1 x y 2 催化剂的晶体结构与孔结构数据。由表 1 2 材料在300 ℃焙烧后比表面积小于50 m2 ·g-1 。掺杂Fe后,Fex y 2 的比表面积明显增加,并随着Fe掺杂量的增加而增加,当掺杂量为20%时,其比表面积最大,达到251 m2 ·g-1 。孔径分布也呈现相同的趋势:未掺杂样品的孔径较大,达12 nm,掺杂Fe后,样品孔径明显减小,并随掺杂量增加而变小。图 1 x y 2 的N2 吸附-脱附等温线。由图 1
表 1
图 1
根据谢乐公式对Fex y 2 的晶粒度进行计算,结果如表 1 2 的晶粒度大幅度减小,由原来的7.7 nm减小到2.6 nm。该现象可以用溶胶-凝胶法中Tanabe模型[25 中的2个前提来解释:(1)主体相以及客体相在形成金属氟化物固体后,依然各自保留其原有的配位数不变。(2)主体相不改变其原有的晶相。正如模型所预期的一样,实验证明Fe掺杂没有改变MgF2 原有的晶相,但是Fe元素的存在抑制了MgF2 晶粒的长大,且由于Fe3+ 离子半径及价态与Mg2+ 的差异,使MgF2 产生晶格畸变,晶胞结构被破坏,晶粒度减小。由图 2 x y 2 的SEM图可知,溶胶-凝胶法制备的MgF2 为比较均匀的球状颗粒。随着Fe掺杂量的增加,次级粒子的粒径从18 nm增加到21 nm。与表 1 2 的比表面积增加,这可能是晶粒度减小导致,与XRD的结果一致,即晶粒尺寸随Fe掺杂量增加而变小。
图 2
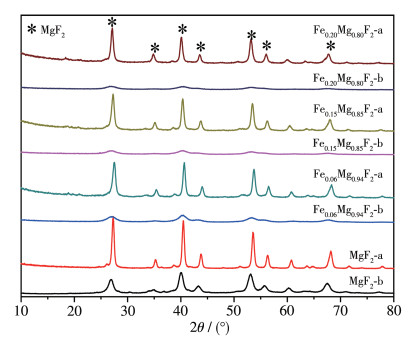
图 3 x y 2 样品的XRD图。无论是否掺杂Fe,Fex y 2 样品在300 ℃焙烧后,仅出现MgF2 的衍射峰,没有形成固溶体的峰和Fe的特征峰,且随着Fe含量的增加XRD峰逐渐减弱。结果表明,当Fe掺杂量小于20%时,溶胶-凝胶法制备的Fex y 2 中Fe3+ 遵循Tanabe模型,以晶格掺杂为主[26 。Fe3+ 金属掺杂不改变主体MgF2 ,只掺杂在MgF2 的晶格中,其引起的晶格畸变使MgF2 配位数减小从而使MgF2 晶粒度减小。图 2 图 3 表 1
图 3
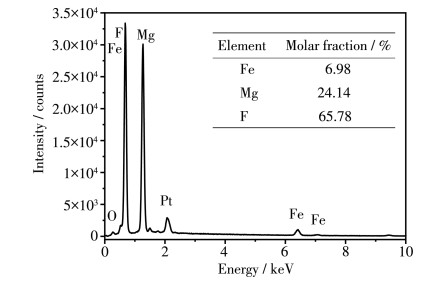
为了进一步明确样品中Fe的含量及分布,对Fe0.20 Mg0.80 F2 样品进行EDS和元素分布面扫描分析。由图 4 2 晶格中。由图 5 0.20 Mg0.80 F2 样品中,主体元素Fe、Mg、F均匀分布,说明Fe掺杂得较均匀。
图 4
图 5
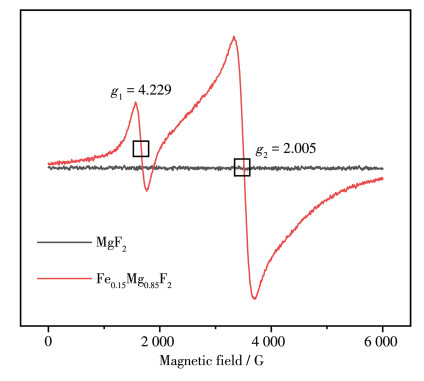
图 6 2 和Fe0.15 Mg0.85 F2 的ESR图,从图中可以看到MgF2 没有电子共振信号峰,掺杂Fe后的Fe0.15 Mg0.85 F2 有2组明显的信号峰,在g 1 =4.229处的峰是Fe3+ 的峰[27 ,而g 2 =2.005处的峰是F中心[28 的峰。Fe3+ 的掺杂使得MgF2 的晶格缺陷明显增加,形成了F中心,这与图 3 2 的峰变矮、结晶度变差相结合进一步说明Fe3+ 成功掺杂到MgF2 的晶格中。
图 6
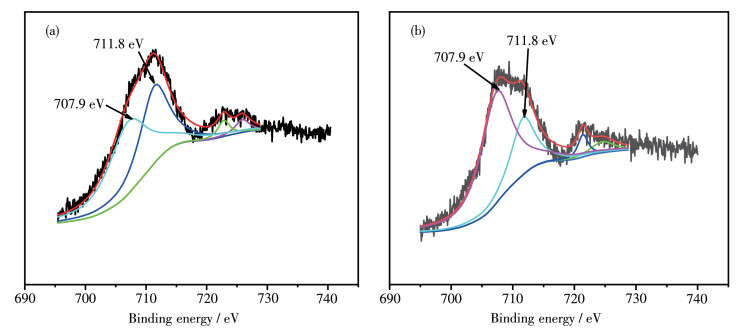
图 7 0.15 Mg0.85 F2 反应前后Fe2p 的XPS图,由图可知,Fe2p 3/2 在707.9和711.8 eV处各有一个分裂峰,分别归属为Fe3+ 峰和Fe2 O3 的特征峰[29 。因此Fe在MgF2 晶格中以Fe3+ 的形式存在,并且反应后也是以Fe3+ 形式存在,XPS结果与图 6
图 7
图 8 0.20 Mg0.80 F2 样品反应前后的SEM图。反应后Fe0.20 Mg0.80 F2 的团聚变少,但有更大的初级粒子产生,这与表 1
图 8
2.2
Fe对MgF2 酸性的影响
图 9 x y 2 的NH3 -TPD结果,对其进行分峰拟合得到表 2 x y 2 催化剂的酸强度及酸量数据。结合图 9 表 2 3 脱附峰对应的中等强度酸性的酸中心酸量减小,由1.09减小至几乎为0,而300 ℃以上对应较强酸性酸中心的NH3 脱附峰,其强度随着Fe含量的增加而增加,由2.79增至19.80,脱附温度也随之向高温迁移,说明随着Fe掺杂量的增加使催化剂酸性增强且酸量增大。因此Fe掺杂可以增强MgF2 酸性,增加MgF2 酸量。
图 9
表 2
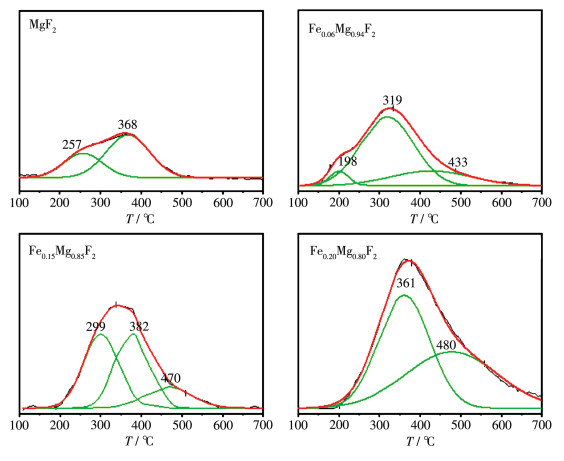
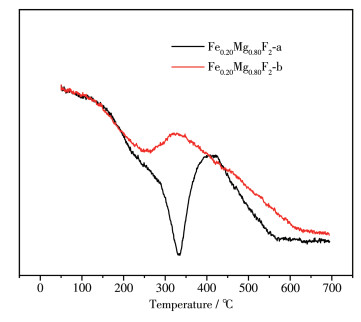
图 10 0.20 Mg0.80 F2 的O2 -TPO图。反应前在300 ℃的峰是催化剂本身碳的峰;而反应后在330 ℃左右对应的强峰是反应后产物氟乙烯发生聚合积碳的峰,说明Fe掺杂量为20%时催化剂在反应后发生了比较明显的积碳。
图 10
2.3
Fex y 2 催化C2 H4 F2 脱HF反应性能
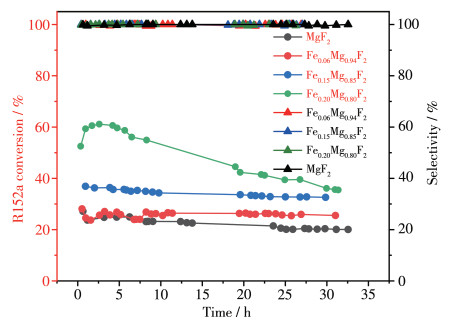
图 11 2 及Fex y 2 样品在300 ℃下催化C2 H4 F2 脱HF反应性能图。随着Fe掺杂量的增加,C2 H4 F2 的初始转化率随之提高,但当Fe掺杂量为20%时,催化剂失活明显。表 1 2 催化剂的晶粒度从7.7 nm增长至18.1 nm,增长了135%。Fex y 2 晶粒度变化很大,随着Fe掺杂量的增加,Fe0.06 Mg0.94 F2 、Fe0.15 Mg0.85 F2 和Fe0.20 Mg0.80 F2 催化剂在反应前后晶粒度分别增长了177%、411%和454%,Fe掺杂量越高,反应后晶粒度的增长率越高,但与不掺Fe3+ 的MgF2 晶粒度相比,掺Fe3+ 的MgF2 反应后晶粒度均小于不掺Fe3+ 的MgF2 ,与前期实验室研究的结果[30 相符,由此可得Fe掺杂量小于20%时,晶粒长大是催化剂失活主要原因。但在Fex y 2 催化剂中,Fe掺杂量为20%时,催化剂的酸量最大,且全部为强酸酸性位的酸量,据文献报道[31 ,这些强酸活性中心在催化C2 H4 F2 脱HF反应的同时也是积碳活性中心,酸性越强的催化剂越容易在脱HF的反应中产生积碳。结合O2 -TPO数据,当Fe掺杂量为20%时,催化剂有明显的积碳,而且催化剂活性明显下降,可能当Fe掺杂量过大时,并不是所有的Fe能以掺杂形式进入MgF2 晶格,部分Fe可能富集在催化剂表面,从而导致催化剂酸性增加,积碳明显,最终导致催化剂失活。故当Fe掺杂量小于20%时,晶粒长大是催化剂失活主要原因,但当Fe掺杂量为20%时,积碳最终导致催化剂失活。对于Fex y 2 而言,在C2 H4 F2 的空速为750 h-1 下的原料转化率可以达到60%,而有文献报道的AlF3 /SiC催化R152a脱HF转化率在相同空速下只有27%[32 ,掺Fe的MgF2 催化剂活性较之有较大提高。而由Fex y 2 在催化C2 H4 F2 脱HF反应中的选择性曲线可知,Fex y 2 的选择性均高于99%,几乎达到100%。
图 11
3.
结论
采用溶胶-凝胶法成功制备了一系列Fe掺杂的Fex y 2 催化剂,随着Fe掺杂量的增加,催化剂的比表面积、酸量和酸强度增加,晶粒度减小,催化C2 H4 F2 脱HF活性也显著增加。当Fe掺杂为20%时,催化剂明显失活,晶粒的长大和积碳可能是催化剂失活的主要原因。

 下载:
下载:




 下载:
下载:












