Received Date:
16 June 2020 Revised Date:
11 September 2020 Available Online:
10 January 2021
Abstract:
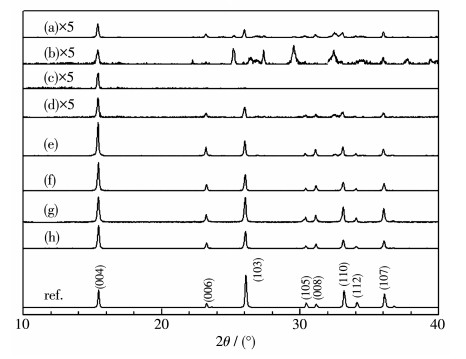
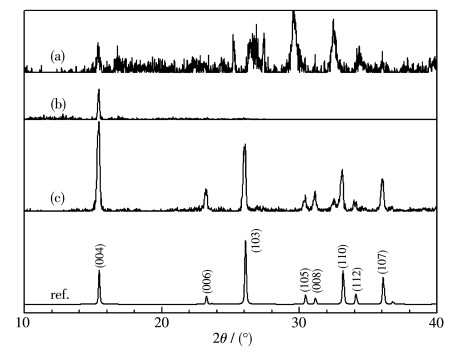
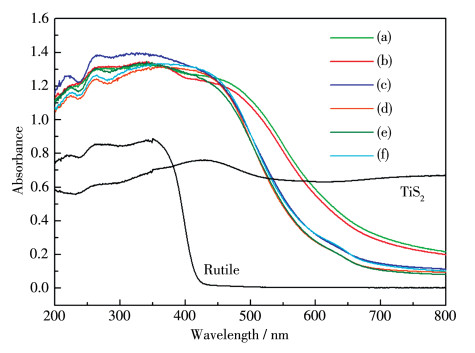
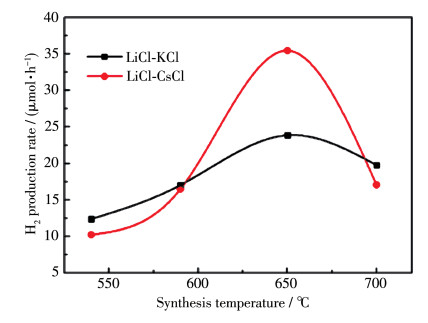
Sm2Ti2S2O5 (STSO) was prepared by a flux method using TiO2, TiS2 and Sm2O3 as reactants, and eutectic of LiCl and KCl (LiCl-KCl) or LiCl and CsCl (LiCl-CsCl) as flux. By analyzing X-ray diffraction patterns of the samples synthesized at different temperatures, it was firstly demonstrated that the threshold crystallization temperature of STSO was 520℃, much lower than the value of 650℃ that was reported previously as the lowest temperature for synthesizing STSO. Scanning electron microscope images showed that the synthesized STSO particles owned a platelike morphology. At the same temperature, LiCl-CsCl led to lower plate thickness than LiCl-KCl. The average photocatalytic H2 production rate showed volcano-like profile to synthesizing temperature, which was likely due to the effect of particle size and crystallinity on activity. Variation of hole sacrificial reagent showed that ascorbic acid produced much higher H2 evolution activity than Na2S-Na2SO3, triethylamine, triethanolamine and methanol. For the purpose of comparing with the literatures, Na2S-Na2SO3 was employed as sacrificial reagent in the following stability test. It was found that the as-prepared STSO exhibited stable H2 production over 20 h under visible light (Xe lamp, λ>420 nm) irradiation, and having nearly identical characterization results in XPS, XRD and TEM before and after photocatalytic reaction.
采用微波水热法担载IrO2。将0.25 g STSO分散在20 mL乙二醇中,加入0.5 mL IrCl3水溶液(相对STSO的质量分数为2%的IrO2),将该溶液放入微波反应器(Monowave 300,Anton Paar Company)后在150 ℃下反应0.5 h,反应液冷至室温后抽滤,并用乙醇及水洗涤数次,所得产物置于60 ℃烘箱中干燥。
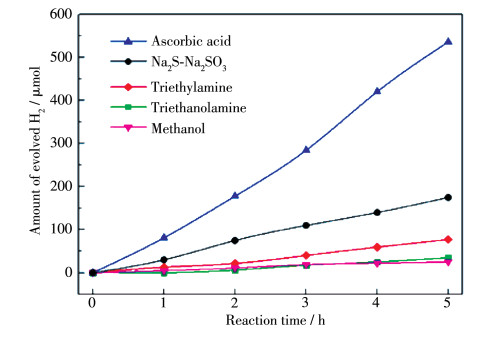
Figure 7.
Photocatalytic hydrogen production over STSO in solutions containing different sacrificial reagents
Amount of catalyst: 0.10 g Pt-IrO2/STSO (the STSO was synthesized at 650 ℃ using LiCl-CsCl as flux); Concentration of sacrificial reagent: 0.05 mol·L-1 aqueous solution for ascorbic acid and Na2S-Na2SO3, 10% volume fraction for methanol, triethylamine and triethanolamine; Light source: xenon lamp (300 W, λ>420 nm)
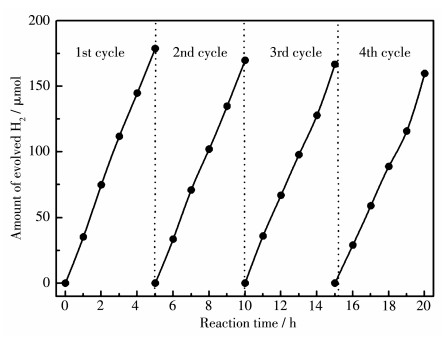
由于S2-离子的标准氧化电位较负(‒0.48 V vs RHE),硫氧化物半导体催化剂在反应过程中有可能发生自身的光氧化腐蚀反应,导致催化反应活性降低。我们选用LiCl-CsCl熔盐在650 ℃下所合成的STSO为催化剂,考察其光催化产氢反应稳定性,结果如图 8所示。由图可知,在20 h的反应过程中并未观察到催化剂活性的明显降低,这与高温下合成的STSO的稳定性结果相同[12, 20],表明本工作中在较低温度下合成的STSO同样具有持续的光催化产氢性能。此外,溶液中含有的Na2S-Na2SO3牺牲剂以及催化剂表面担载的Pt-IrO2助催化剂都有利于维持STSO表面光催化反应的稳定性。在这一反应体系中,该样品在(420±20) nm的单色光照射下光催化产氢的量子效率为0.3%。
图 8
图 8.
STSO的光催化稳定性
Figure 8.
Photocatalytic stability of STSO
Reaction condition: 0.10 g Pt-IrO2/STSO, 0.05 mol·L-1 Na2S-Na2SO3 aqueous solution, xenon lamp (300 W, λ>420 nm) as light source; System was re-evacuated after each cycle; STSO was synthesized at 650 ℃ using LiCl-CsCl as flux
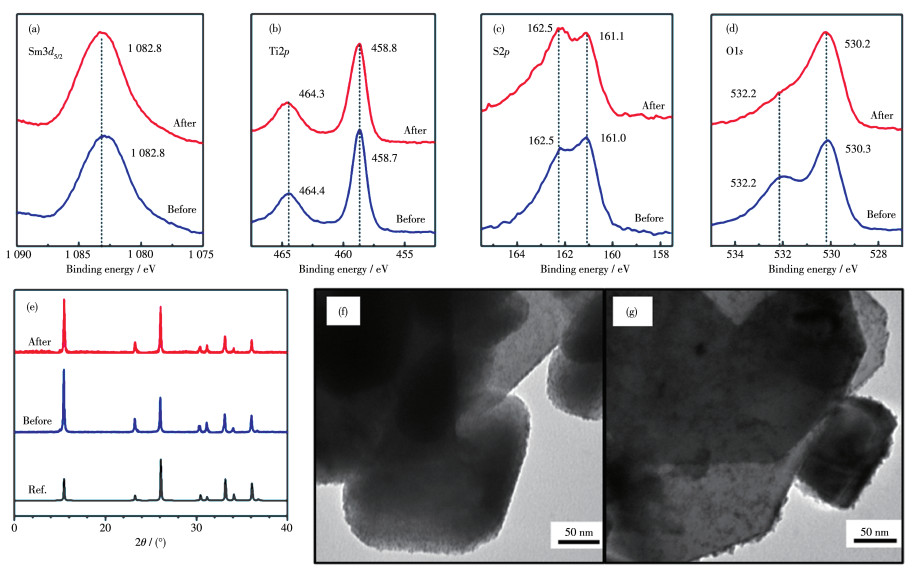
Figure 9.
XPS spectra, XRD patterns and TEM images of the STSO samples before and after stability test
(a~d) XPS spectra; (e) XRD pattern of IrO2/STSO powders before (blue line) and after (red line) stability test; (f, g) TEM images of IrO2/STSO samples before and after stability test, respectively
Takata T, Domen K. Dalton Trans., 2017, 46(32):10529-10544 doi: 10.1039/C7DT00867H
[3]
Michael G W, Emily L W, James R M, Shannon W B, Mi Q X, Eliza-beth A S, Nathan S L. Chem. Rev., 2010, 110:6446-6473 doi: 10.1021/cr1002326
[4]
Suzuki T, Hisatomi T, Teramura K, Shimodaira Y, Kobayashi H, Domen K. Phys. Chem. Chem. Phys., 2012, 14(44):15475-15481 doi: 10.1039/c2cp43132g
[5]
Ma G J, Suzuki Y, Singh R B, Iwanaga A, Moriya Y, Minegishi T, Liu J, Hisatomi T, Nishiyama H, Katayama M, Seki K, Furube A, Yama-da T, Domen K. Energy Environ. Sci., 2014, 7(7):2239-2242 doi: 10.1039/C4EE00091A
[6]
Ma G J, Suzuki Y, Singh R B, Iwanaga A, Moriya Y, Minegishi T, Liu J, Hisatomi T, Nishiyama H, Katayama M, Seki K, Furube A, Yama-da T, Domen K. Chem. Sci., 2015, 6(8):4513-4518 doi: 10.1039/C5SC01344E
[7]
Hisatomi T, Okamura S, Liu J Y, Shinohara Y, Ueda K, Higashi T, Katayama M, Minegishi T, Domen K. Energy Environ. Sci., 2015, 8(11):3354-3362 doi: 10.1039/C5EE02431E
[8]
Song Z M, Hisatomi T, Chen S S, Wang Q, Ma G, Li S, Zhu X, Sun S, Domen K. ChemSusChem, 2019, 12(9):1906-1910 doi: 10.1002/cssc.201802306
[9]
Wang Q, Nakabayashi M, Hisatomi T, Sun S, Akiyama S, Wang Z, Pan Z, Xiao X, Watanabe T, Yamada T, Shibata N, Takata T, Domen K. Nat. Mater., 2019, 18(8):827-832 doi: 10.1038/s41563-019-0399-z
[10]
Goto Y, Seo J, Kumamoto K, Hisatomi T, Mizuguchi Y, Kamihara Y, Katayama M, Minegishi T, Domen K. Inorg. Chem., 2016, 55(7):3674-3679 doi: 10.1021/acs.inorgchem.6b00247
[11]
Ishikawa A, Takata T, Junko N, Hara M, Kobayashi H, Domen K. J. Am. Chem. Soc., 2002, 124(45):13547-13553 doi: 10.1021/ja0269643
[12]
Ma G J, Chen S S, Kuang Y B, Akiyama S, Hisatomi T, Nakabayashi M, Shibata N, Katayama M, Minegishi T, Domen K. J. Phys. Chem. Lett., 2016, 7(19):3892-3896 doi: 10.1021/acs.jpclett.6b01802
[13]
Zhao W, Maeda K, Zhang F X, Hisatomi T, Domen K. Phys. Chem. Chem. Phys., 2014, 16(24):12051-12056 doi: 10.1039/c3cp54668c
[14]
Ishikawa A, Yamada Y, Ishikawa A, Kondo J N, Hara M, Kobayashi H, Domen K. Chem. Mater., 2003, 15(23):4442-4446 doi: 10.1021/cm034540h
Zhang F X, Maeda K, Takata T, Domen K. Catal. Today, 2012, 185(1):253-258
[19]
Zhang F X, Maeda K, Takata T, Domen K. Chem. Commun., 2010, 46(39):7313-7315 doi: 10.1039/c0cc02425b
[20]
Ma G J, Kuang Y B, Murthy D H K, Hisatomi T, Seo J, Chen S S, Matsuzaki H, Suzuki Y, Katayama M, Minegishi T, Seki K, Furube A, Domen K. J. Phys. Chem. C, 2018, 122(25):13492-13499 doi: 10.1021/acs.jpcc.7b12087
Figure 7
Photocatalytic hydrogen production over STSO in solutions containing different sacrificial reagents
Amount of catalyst: 0.10 g Pt-IrO2/STSO (the STSO was synthesized at 650 ℃ using LiCl-CsCl as flux); Concentration of sacrificial reagent: 0.05 mol·L-1 aqueous solution for ascorbic acid and Na2S-Na2SO3, 10% volume fraction for methanol, triethylamine and triethanolamine; Light source: xenon lamp (300 W, λ>420 nm)
Reaction condition: 0.10 g Pt-IrO2/STSO, 0.05 mol·L-1 Na2S-Na2SO3 aqueous solution, xenon lamp (300 W, λ>420 nm) as light source; System was re-evacuated after each cycle; STSO was synthesized at 650 ℃ using LiCl-CsCl as flux
Figure 9
XPS spectra, XRD patterns and TEM images of the STSO samples before and after stability test
(a~d) XPS spectra; (e) XRD pattern of IrO2/STSO powders before (blue line) and after (red line) stability test; (f, g) TEM images of IrO2/STSO samples before and after stability test, respectively

 下载:
下载:



 下载:
下载:










