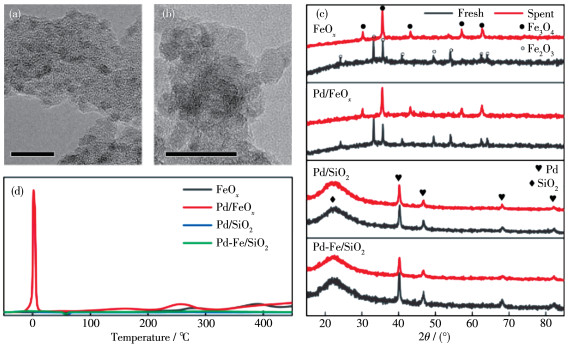
Figure 1.
TEM images of Pd-Fe/SiO2 (a) and Pd/FeOx (b), inserted scale bar is 10 nm; (c) XRD patterns of the fresh and spent catalysts; (d) H2-TPR results of Pd/FeOx, blank FeOx, Pd/SiO2, and Pd-Fe/SiO2 catalysts
Dynamic Formation of Pdδ+-Fe2+ Interface Promoting Reverse Water Gas Shift Reaction over Pd/FeOx Catalyst
Dian-Yu ZHANG , Fang LIU , Peng-Fei DU , Meng-Wei LI , Zhao-Xuan WU , Yi-Bing FENG , Yang ZHAO , Xiao-Yan XU , Xin-Xing ZHANG , Jun-Ling LU , Bing YANG

Carbon dioxide reduction has received tremendous attention for its potential in solving the energy and environmental crises[1-3]. The reverse water gas shift (RWGS) reaction is an effective route for syngas production via CO2 hydrogenation[4-6]. Numerous experimental studies have investigated the RWGS reaction, using Au/MoOx[7], Ru/Al2O3[8], Cu/FeOx[9], and other monometallic[10-12], bimetallic[13], and metal oxides[14] as catalysts. The reaction pathway can be summarized into two theories: the direct path and the indirect path. The direct path involves the direct CO2 dissociation into COad and Oad, and then the COad is desorbed as CO[15]. The indirect path involves the hydrogenation of CO2 via formate or carbonate species as intermediates, and the CO2 further decomposes into CO as the main product[3-4, 16-17]. The metal support interface also plays a vital role in the RWGS reaction[6-7, 18] and affects the selectivity[9]. To date, the reaction mechanism, including the active interface structure, is still under debate.
Iron oxide-supported palladium catalysts have been extensively studied for the RWGS reaction[19], water gas shift (WGS) reaction[20], and CO oxidation[21-23]. The Pd - FeO x interface has been found to be crucial in a variety of reactions, as it promotes oxygen vacancies and oxygen transportation[24-26]. The higher reducibility of the FeOx - supported Pd species, which feature active interface structures, has also been extensively reported[20, 26-28]. However, these works focus solely on the metal-support interaction from a static point of view, determined via ex situ characterization[5]. The dynamic evolution of the Pd-FeOx interface during the reaction has not yet been clarified owing to the lack of in situ techniques.
In this study, Pd/FeOx, Pd/SiO2, and Pd - Fe/SiO2 catalysts were systematically investigated for the RWGS reaction. The catalytic performance for the RWGS reaction was evaluated, and a promoting role of the Pd-FeOx interface was observed. Using a combination of ex situ and in situ characterizations, we identified a dynamic evolution of the Pd-FeOx interface during reaction, in which interfacial Pdδ+-Fe2+ species were formed as the active sites. The underlying reaction mechanism and the role of the Pd-FeOx interface were further investigated using temperature-programmed desorption/reduction/oxidation (TPD/TPR/TPO) and Fourier-transform infrared (FTIR) spectroscopy.
The FeOx sample was prepared by the precipitation of Fe(NO3)3·9H2O in a NaOH solution. The Pd/ FeOx catalyst was prepared by the impregnation method, with 28.8 mg Pd(acac)2 as the precursor and 200 mg FeOx and 60 mL toluene as the solvent in a flask[29]. Moreover, Pd/SiO2 was prepared by a wet impregnation method, using 148.5 mg Pd(NH3)4Cl2·H2O and 1 g SiO2. A GEMSTAR-6TM Benchtop atomic layer deposition (ALD) system was used to synthesize Pd-Fe/ SiO2[30]. Nitrogen (99.999%) was the carrier gas at 200 mL·min-1, and FeOx ALD was performed at 150 ℃ by exposing the as - prepared Pd/SiO2 to 20~25 cycles of ferrocene and oxygen (For more details, see in Supplementary Information).
Catalytic testing was performed in a continuous - flow fixed-bed reactor with 40 mg of catalyst under atmospheric pressure, with a feed gas of 24% CO2 and 72% H2 (volume fraction, unless otherwise specified, all other gas contents in the paper are volume fractions), balanced with Ar. The total flow was 24 mL· min-1, which resulted in a weight hourly space velocity of 36 000 mL·h-1·gcat-1. After the gas phase was allowed to stabilize for 30 min to reach the steady-state for each reaction temperature, the outlet gas composition was analyzed by an on - line gas chromatograph (Agilent 7820A) equipped with a thermal conductivity detector and a flame ionization detector, and He was the carrier gas.
CO2 conversion was calculated as follows:
|
$ {X_{{\rm{C}}{{\rm{O}}_{\rm{2}}}}} = \frac{{{n_0} - {n_1}}}{{{n_0}}} \times 100\% $ |
(1) |
Where n0 and n1 stand for the amounts of CO2 in feed gas and outlet gas.
CO selectivity was calculated as follows:
|
$ {S_{{\rm{CO}}}} = \frac{{{n_{{\rm{CO}}}}}}{{{n_0} - {n_1}}} \times 100\% $ |
(2) |
Where nCO stands for the amount of CO in outlet gas.
X -ray diffraction (XRD, PAN analytical, X' Pert PRO X) was performed with a Cu Kα radiation source (λ =0.154 06 nm) in reflection mode. X - ray tube was operated at 40 kV and 40 mA. XRD diffraction range was set to 15° to 85°.
The catalysts were characterized via highresolution transmission electron microscopy (TEM), using a JEOL JEM - 2100 microscope at 200 kV. The sample was dispersed by ethanol under ultrasonication. The samples were also analyzed via energy-dispersive X-ray spectroscopy (EDS), using the Aztec X-Max 80T EDS system (Oxford instrument, UK).
Hydrogen temperature-programmed reduction (TPR) was performed on a Micromeritics AutoChem Ⅱ chemisorption analyzer. First, 50 mg catalyst was loaded into a U-shape quartz reactor and purged with helium at 120 ℃ for 2 h to remove physically adsorbed water and surface carbonates. After the catalyst was cooled to -50 ℃, the gas was switched to a flow of 10% (V/V) H2 balanced with Ar, and the catalyst was heated to 500 ℃ at a ramping rate of 10 ℃·min-1. The amount of H2 consumption was calculated with the H2 peak area and calibration curve of the 10% (V/V) H2 balanced with Ar standard gas.
Furthermore, CO2 temperature-programmed desorption (TPD) experiments were performed on the AutoChem Ⅱ 2920. The samples were placed in a quartz tube. Before the TPD experiment, all of the samples were pre-reduced by 10% (V/V) H2/Ar at 300 ℃ for 30 min and then cooled down to room temperature (RT). Then the gas was switched to 10% (V/V) CO2/Ar at 50 mL·min-1 for 1 h for the saturated adsorption of CO2. After N2 was purged for 30 min, the sample was heated from 50 to 800 ℃ at a ramping rate of 10 ℃ · min-1, during which TPD data were obtained.
Cyclic H2-TPR and CO2 temperature-programmed oxidation (TPO) experiments were performed on an OmniSTAR gas analysis system. First, 20 mg catalyst was loaded into a U-shape quartz reactor and purged with helium at 150 ℃ for 2 h to remove physically adsorbed water and surface carbonates. After the temperature was cooled to room temperature, the gas was switched to a flow of 5% (V/V) H2 balanced with He, and the catalyst was heated to 400 ℃ at a ramping rate of 10 ℃· min-1. Before the CO2-TPO experiment, the sample was purged again with He at 150 ℃ for 2 h. After the temperature was cooled to room temperature, the gas was switched to a flow of 5% (V/V) CO2 balanced with He, and the catalyst was heated to 400 ℃ at a ramping rate of 10 ℃·min-1.
The FTIR spectra of CO adsorption were recorded in transmission mode on a Bruker Tensor 27 model. CO was introduced for 30 min at room temperature to reach saturated adsorption, followed by purging with N2 to remove gaseous CO.
X-ray photoelectron spectroscopy (XPS) experiments were performed using ThermoFisher ESCALAB 250Xi. A semi-in situ experiment was conducted by transferring the catalyst directly for XPS measurements after a certain treatment without exposing the catalyst to air, as reported elsewhere[31]. The first set of experiments consisted of a reaction gas (24% CO2+72% H2+ 4% Ar, V/V) treatment for 30 min at room temperature and 400 ℃. The second set of experiments included three steps for each catalyst. First, the as-prepared catalyst was purged with N2 at room temperature. Then, the sample was heated to 400 ℃ under 10% (V/V) H2/ Ar for 30 min. Finally, the gas was switched to 10% (V/V) CO2/Ar for 30 min. The sample was transferred back to the analysis chamber for XPS measurements in between each treatment.
The TEM images of as-prepared Pd-Fe/SiO2 and the Pd/FeOx catalysts are displayed in Fig. 1a and 1b. The Pd-Fe/SiO2 showed uniformly dispersed Pd nanoparticles (NPs) of ~3 nm on amorphous SiO2. In contrast, Pd/FeOx displayed a highly polycrystalline FeOx, with no significant Pd particle present (see EDS mapping in Fig. S1). The EDS spectra identified the presence of Pd with a similar loading (Table S1) mea- sured by inductively coupled plasma-optical emission etry (Table S2). The results indicate that a system of highly dispersed Pd species, with a size likely below 2 nm, in as- prepared 3.6% (mass fraction) Pd/ FeOx catalysts was formed[32-33].
Fresh (as-prepared) and spent catalysts were examined via XRD, and the results are presented in Fig. 1c. The fresh FeOx and the 3.6% (mass fraction) Pd/ FeOx catalysts before reaction showed a typical XRD pattern of Fe2O3, which was reduced to Fe3O4 completely after the RWGS reaction. The absence of Pd and PdO XRD patterns in both the fresh and spent Pd/FeOx catalyst indicates the presence of small Pd/PdOx nanoparticles (<2 nm) or amorphous nature of Pd species in accordance with the TEM results[34]. In the XRD patterns of the Pd/SiO2 and Pd-Fe/SiO2, a broad band existed at 21.9°, which can be assigned to amorphous SiO2. The additional diffraction peaks at 40.4°, 46.7°, and 68.4° are ascribed to Pd NPs. Moreover, no FeOx or Fe diffraction peaks were observed in Pd - Fe/SiO2, suggesting a very thin layer of Fe species on the ALD- synthesized Pd NPs.
In the H2 - TPR results (Fig. 1d), the blank FeOx support showed two reduction peaks: at 280 and 390 ℃, which are characteristic of the reduction of Fe2O3 to Fe3O4[20]. In the case of the Pd/FeOx catalyst, the first peak (0 ℃) is ascribed to the reduction of PdO, and the latter two (150 and 250 ℃) are associated with the reduction of FeOx, which shift to lower temperatures. The presence of Pd thus facilitates the reduction of FeOx, likely via H2 spillover effect[20, 24]. Compared with the H2-TPR results of Pd/SiO2 and Pd-Fe/SiO2, the significantly enhanced H2 consumption of Pd/FeOx at 0 ℃ indicates a promoted reduction of the Pd - FeOx interface, since the H2 consumption was far more than the stoichiometric PdO amount (Table S3 and supplementary discussion). In addition, Pd-Fe/SiO2 also showed a small reduction peak at 320 ℃, which corre-sponds to the reduction of ALD - synthesized FeOx on Pd NPs.
The catalytic test was performed in a fixed - bed reactor under atmospheric pressure, with a feed gas of 24% CO2 and 72% H2 (V/V), balanced with Ar. As shown in Fig. 2a and 2b, the Pd/FeOx catalyst exhibited a remarkably high activity for the RWGS reaction, with a CO2 conversion of 10% at 300 ℃ and 29% at 400 ℃, and a CO selectivity of over 98% at all reaction temperatures. In contrast, both the blank FeOx and Pd/SiO2 catalysts were inactive, with CO2 conversion below 5% at temperatures up to 400 ℃ (see carbon balance data in Table S4). This strongly indicates that the Pd-FeOx interface is highly required for the RWGS reaction. To further prove this point, a thin layer of FeOx (mass fraction of 1.1%) was added onto the inactive Pd/SiO2 using ALD (the formed product was denoted as Pd-Fe/ SiO2 catalyst). The Pd-Fe/SiO2 catalyst showed a dramatically enhanced CO2 conversion, from 5% to 20%, at 400 ℃ (Fig. 2c). Only a slight deactivation (<4%) was observed, which was likely due to coking or mild sintering (Fig. S2 and S3). The enhanced activities of Pd/FeOx and Pd-Fe/SiO2 compared with those of their counterparts (FeOx and Pd/SiO2) reveal the promoting role of the Pd-FeOx interface as highly active sites for catalytic CO2 hydrogenation. The long-term stability test of the Pd/FeOx catalyst was performed at 400 ℃ with time on stream up to 30 h, as shown in Fig. 2d. The CO2 conversion and CO selectivity remain stable at ~29% and ~98% over the entire testing, suggesting an excellent stability of the catalyst. The water formation during reaction also shows a minor effect on the catalytic performance of Pd/FeOx catalyst, with no significant deactivation in both CO2 conversion and CO selectivity (Fig.S4).
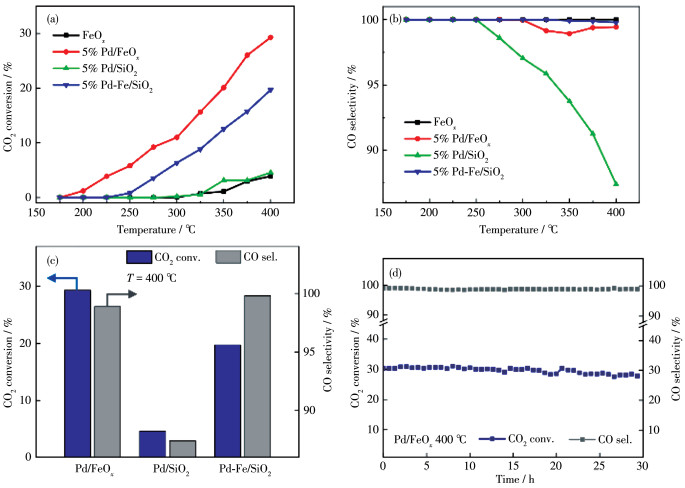
Furthermore, the Pd-FeOx interface also promoted CO selectivity. As shown in Fig. 2b, the CO selectivity on the Pd/SiO2 catalyst declined with increasing reaction temperature. The production of methane thus suggests a deep hydrogenation of CO on Pd/SiO2 at elevated temperatures. In comparison, both Pd/FeOx and Pd-Fe/SiO2 catalysts maintained a CO selectivity of over 98% at all temperatures, suggesting that the addition of FeOx could prevent the further hydrogenation of CO to methane. Table 1 summarizes the catalytic performance of other literature-reported RWGS catalysts. The 3.6% (mass fraction) Pd/FeOx and 4.4% (mass fraction) Pd-Fe/SiO2 catalysts in this work features a higher activity and CO selectivity than 1.5% (mass fraction) Rh/SrTiO3[35], 1.67% (mass fraction) Pt/TiO2[10], 3% (mass fraction) Pd/Fe3O4[19], and other metal carbide catalysts[13, 36].
 下载:
导出CSV
下载:
导出CSV
| Catalysta | VH2/VCO2 | T/℃ | Weight hourly space velocity/(L·h-1·g-1) | XCO2/ % | SCO/ % |
| 3.6% Pd/FeOx (this work) | 3 | 300 | 36 | 11 | 100 |
| 4.4% Pd-Fe/SiO2 (this work) | 3 | 300 | 36 | 6 | 100 |
| 3.6% Pd/FeOx (this work) | 3 | 400 | 36 | 29 | 100 |
| 4.4% Pd-Fe/SiO2 (this work) | 3 | 400 | 36 | 20 | 100 |
| Mo2C[36] | 3 | 300 | 36 | 8.7 | 93.9 |
| 7.5% Co/Mo2C[36] | 3 | 300 | 36 | 9.5 | 99 |
| 1.67% Pt/TiO2[10] | 1 | 300 | 351 | 4.5 | 99.1 |
| 1.67% Pt/SiO2[10] | 1 | 300 | 72.1 | 3.3 | 100 |
| 1.1% Rh/SrTiO3[35] | 1 | 300 | 120 | 7.9 | 95.4 |
| 5% Co/MCF-17[13] | 3 | 200~300 | 60 | ~5 | 90 |
| 5% Pt50Co50/MCF-17[13] | 3 | 200~300 | 60 | ~5 | 99 |
| 1% Pt/TiO2[11] | 3 | 600 | 12 | ~58 | ~95 |
| 3% Pd/Fe3O4 (dopPPh2)[19] | 4 | 400 | 60 | 8.8 | 94.6 |
| a All the percents are mass fraction. | |||||
To identify the dynamic evolution of surface active sites, semi-in situ XPS experiments were performed, which enabled transferring the catalyst directly for XPS measurements after certain treatment without exposing it to air (for details, see in supplementary methods). The Pd/FeOx, Pd/SiO2, and Pd-Fe/SiO2 catalysts were stepwise heated in 10% H2 and 10% CO2 at 400 ℃, and the Pd3d core-level evolution spectra are displayed in Fig. 3a. Three characteristic chemical states of Pd species could be identified from the evolu- tion spectra: 337.2 eV[37], for oxidized Pd2+ state; 335.7 eV[38], for partially oxidized Pdδ+ state; and 334.8 eV[38], for metallic Pd0 state (Fig.S5~S8). For Pd/SiO2, the pristine catalyst showed a Pd3d5/2 binding energy (BE) of 355.6 eV, corresponding to a Pdδ+ state, which likely originated from the native oxide on the Pd NPs. In previous studies, a similar Pdδ+ state was revealed by ambient - pressure XPS as a surface oxide and further assigned to Pd5O4 stoichiometry[39-41]. Reduction in H2 at 400 ℃ led to a prominent metallic Pd0 of 334.8 eV, which re-oxidized to a Pdδ+ state in the subsequent CO2 treatment (400 ℃). Considering the chemical inertness of the SiO2 support, we can conclude that Pd0 was oxidized to Pdδ+ by CO2 at 400 ℃, strongly indicating a direct dissociation of CO2 to CO on Pd NPs. The pristine Pd-Fe/SiO2 and Pd/FeOx catalysts showed a higher Pd3d5/2 BE of 336.8 and 337.2 eV respectively, characteristic of PdO oxides[37]. The similar redox properties of Pd - Fe/SiO2 and Pd/FeOx under H2 and CO2 treatments indicate the same reaction path of direct CO2 dissociation for the RWGS reaction. Moreover, the addition of FeOx significantly promoted the enrichment of Pdδ+ species in the reduced Pd/SiO2, Pd - Fe/SiO2, and Pd/FeOx catalysts after H2 treatment, as indicated by the gradual upshift of Pd3d5/2 BE from metallic Pd0 to Pdδ+ (334.8, 335.0, and 335.7 eV for Pd/SiO2, Pd-Fe/SiO2, and Pd/FeOx, respectively) with increasing FeOx content. The formation of Pdδ+ species during the RWGS reaction was further verified by the treatment of Pd/FeOx, Pd/SiO2, and Pd - Fe/SiO2 catalysts under the reaction gas of 24% CO2+72% H2+4% Ar (V/V) at 400 ℃. As shown in Fig. 3b, Fig.S9, and Table S5, the Pd/FeOx, Pd/SiO2, and Pd-Fe/SiO2 catalysts after reaction exhibited a prominent Pdδ++ state with a Pd3d5/2 peak of 335.7 eV, which further indicates the stability of the Pdδ+ surface oxides during the RWGS reaction.
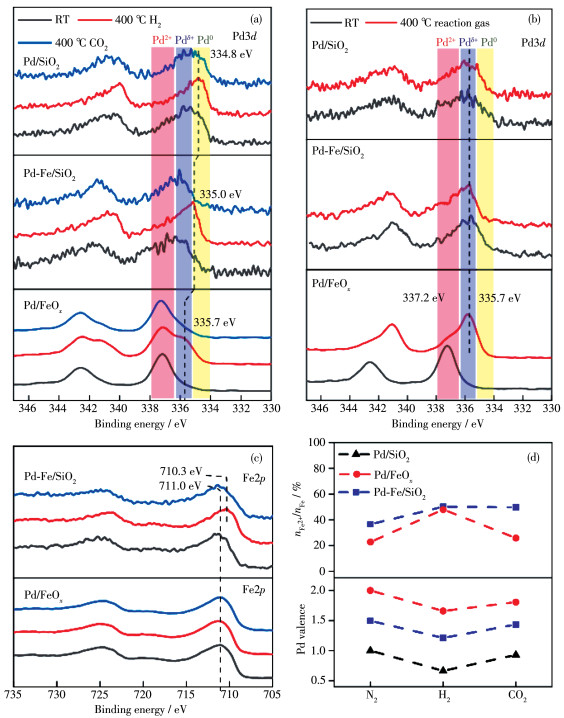
The Fe2p XPS core-level spectra are displayed in Fig. 3c and Fig.S10~S11. In the stepwise H2/CO2 heating experiments (Fig. 3c), an evolution of the chemical state of iron oxides in Pd-Fe/SiO2 occurred, resulting in a downshift of Fe2+p3/2 BE from 711.0 eV (Fe3+ state) to 710.3 eV (a reduced Fe2+ state) after H2 reduction and an upshift back to 711.0 eV after CO2 treatment. The dynamic formation of Fe2+ species was thus facilitated by H2 reduction, and the Fe2++ species were further re-oxidized to Fe3+ state by reacting with CO2. A similar oscillation of the Fe2+p3/2 core level was also observed on Pd/FeOx, but less prominent owing to the higher background signal from the bulk Fe2+O3 support in the Pd/FeOx catalyst. This further indicates the surfaceenrichment of Fe2+ species on the Pd/FeOx catalyst, formed via hydrogen spillover to the periphery of the Pd NPs[42-43]. After reaction gas treatment (Fig.S12), the Fe2+p3/2 of both Pd/FeOx and Pd - Fe/SiO2 underwent a downshift from 710.9 to 710.4 eV, and the characteristic satellite peak for Fe2+O3 (718 eV) was absent[44], suggesting a reduction in iron oxide support, which agrees with our XRD results. Further reduction of the surface/ bulk Fe species may require higher temperature or extended reaction time.
Fig. 3d displays the oscillation of Fe2+ concentra- tion and Pd valence state deduced from Fe2+p and Pd3d XPS results, as summarized in Tables S6 and S7. The enrichment of Pdδ+ - Fe2+ on both Pd - Fe/SiO2 and Pd/ FeOx was identified under dynamic reduction by H2, so that the Pdδ++ - Fe2++ further transformed into Pd2+ - Fe3+ after reacting with CO2. Based on the higher activities of Pd-Fe/SiO2 and Pd/FeOx compared with that of Pd/ SiO2, we can assign the dynamic Pdδ+-Fe2+ interface as a highly active site for CO2 dissociation during RWGS.
Fig. 4a displays the FTIR spectra of CO adsorption over spent catalysts. Two vibrational bands of CO stretching modes, at 2 086 and 1 965 cm-1, were observed on Pd/SiO2, which can be assigned to CO adsorption on multi-coordinated sites of Pd NPs[45-46]. Moreover, the CO adsorption intensity dramatically diminished on Pd-Fe/SiO2 and eventually became invisible on Pd/FeOx. The absence of CO adsorption on Pd/ FeOx can be ascribed to the higher oxidized Pdδ+/Pd2+ state, which may also lead to encapsulation via strong metal-support interaction[47]. The weakening of CO adsorption promoted by iron oxides facilitated the desorption of CO as a product and prevented the deep hydrogenation for methane formation.
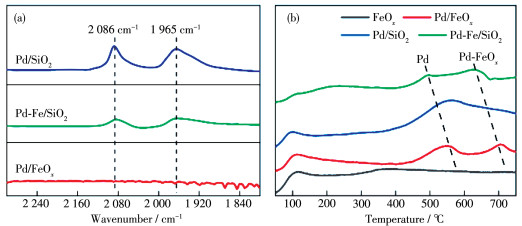
Fig. 4b presents the CO2 -TPD spectra of reduced FeOx, Pd/SiO2, Pd/FeOx, and Pd - Fe/SiO2 at temperatures up to 750 ℃. All of the tested catalysts were prereduced in 10% H2 at 300 ℃. FeOx presented only a subtle peak at 400 ℃, suggesting a very weak CO2 adsorption on Fe2+O3. In contrast, Pd/SiO2 showed a desorption peak at ~550 ℃, which can be ascribed to the molecular/reactive desorption of chemically adsorbed CO2 on Pd NPs in a mixed Pd0/Pdδ+ state, as identified by XPS. An additional desorption peak at higher temperatures (~670 ℃) was observed on both Pd/FeOx and Pd - Fe/SiO2, reflecting an enhanced CO2 adsorption owing to the Pdδ+-FeOx interface, which will lead to the formation of carbonate species or other adsorbed intermediates[48].
The semi-in situ XPS results demonstrated a dynamic evolution of Pd and Fe chemical states. To further reveal the reaction pathway, we performed cyclic H2 -TPR and CO2 -TPO on Pd/SiO2 and Pd/FeOx catalysts. In the case of the Pd/SiO2 catalyst (Fig. 5a), an indirect path was identified, as indicated by the pronounced CO production along the second H2-TPR cycle, via the hydrogenation of either adsorbed CO2 or carbonate species on the metallic Pd surfaces[17, 49]. Meanwhile, the formation of a little amount of CO was also detected in the CO2 - TPO cycle, suggesting the coexistence of direct CO2 dissociation, likely on the remanence of Pdδ+ species, as identified in the semi-in situ XPS experiments. Moreover, the Pd/FeOx (Fig. 5b) showed a remarkably enhanced CO formation in the CO2 - TPO cycle (direct path), while the hydrogenation path (indirect path) was significantly inhibited, as no CO signal was observed in the second H2-TPR cycle.

Based on the dynamic evolution of the Pd/Fe chemical state based on the semi- in situ XPS results, we can propose the overall reaction pathways and underlying mechanism for the RWGS reaction over Pd-Fe catalysts (Fig. 5c and 5d).
(1) On Pd/SiO2, the majority of CO2 is hydrogenated on the metallic Pd surface via an indirect path, leading to the formation of formate or bicarbonate intermediates, which are eventually decomposed to produce CO. Though less favorable, the direct path of CO2 dissociation also coexists on Pd/SiO2, which is likely facilitated by the remanence of Pdδ+ species near the SiO2 interface. The enhanced CO adsorption on metallic Pd0 favors the deep hydrogenation of CO, leading to undesired CH4 as a byproduct[50]. The low activity of Pd/SiO2 is caused by the weak CO2 adsorption, as indicated by the CO2-TPD experiments.
(2) On Pd/FeOx and Pd-Fe/SiO2, the Pd-FeOx interface significantly enhances the CO2 adsorption, resulting in a remarkably high CO2 conversion compared with that on Pd/SiO2. The dynamic formation of Pdδ+ - Fe2+ species promoted CO2 dissociation, likely following the Mars-van Krevelen mechanism, via active oxygen vacancies on the interface[8, 18, 43]:
|
$ {\rm{P}}{{\rm{d}}^{{\rm{2 + }}}}-{\rm{O}}-{\rm{F}}{{\rm{e}}^{{\rm{3 + }}}}{\rm{ + }}{{\rm{H}}_{\rm{2}}} \to {\rm{P}}{{\rm{d}}^{\delta + }}-{{\rm{O}}_{\rm{v}}}{\rm{ + F}}{{\rm{e}}^{{\rm{2 + }}}}{\rm{ + }}{{\rm{H}}_{\rm{2}}}{\rm{O}} $ |
(3) |
|
$ {\rm{P}}{{\rm{d}}^{\delta + }}-{{\rm{O}}_{\rm{v}}} + {\rm{F}}{{\rm{e}}^{2 + }} + {\rm{C}}{{\rm{O}}_2} \to {\rm{P}}{{\rm{d}}^{2 + }}-{\rm{O}} - {\rm{F}}{{\rm{e}}^{3 + }} + {\rm{CO}} $ |
(4) |
The weakening of CO adsorption on the oxidized Pdδ+/Pd2+ species enriched by iron oxide further enhances the CO selectivity (>98%) by preventing the deep hydrogenation for CH4 formation.
In this study, Pd/FeOx, Pd-Fe/SiO2, Pd/SiO2, and FeOx catalysts were systematically investigated for the RWGS reaction. The Pd/FeOx and Pd-Fe/SiO2 catalysts exhibited extraordinary activity for the RWGS reaction, with CO2 conversions of 29% and 20% at 400 ℃, respectively, and remarkably high CO selectivity (over 98%) at all considered reaction temperatures. Thus, the Pd-FeOx interface promoted the CO2 hydrogenation to CO. Moreover, semi - in situ XPS and cyclic TPR/ TPO experiments identified the dynamic formation of Pdδ+ -Fe2+ species during the RWGS reaction; the species acted as highly active sites for direct CO2 dissociation via oxygen vacancies enriched on the interface. The formation of Pdδ+ - FeOx species was confirmed to play a significant role in the CO2 adsorption and CO desorption, as they further enhanced the catalytic activity and selectivity of CO for the RWGS reaction. Our results provide new insights for the dynamic understanding of the metal- oxide interfaces during reactions and benefit the rational design of heterogeneous catalysts from a dynamic point of view.
Li W H, Wang H Z, Jiang X, Zhu J, Liu Z M, Guo X W, Song C S. RSC Adv., 2018, 8(14):7651-7669 doi: 10.1039/C7RA13546G
Dorner R W, Hardy D R, Williams F W, Willauer H D. Energy Environ. Sci., 2010, 3(7):884-890 doi: 10.1039/c001514h
Wang W, Wang S P, Ma X B, Gong J L. Chem. Soc. Rev., 2011, 40(7):3703-3727 doi: 10.1039/c1cs15008a
Daza Y A, Kuhn J N. RSC Adv., 2016, 6(55):49675-49691 doi: 10.1039/C6RA05414E
Parkinson G S. Surf. Sci. Rep., 2016, 71(1):272-365
Liu M, Yi Y H, Wang L, Guo H C, Bogaerts A. Catalysts, 2019, 9(3):275 doi: 10.3390/catal9030275
Carrasquillo-Flores R, Ro I, Kumbhalkar M D, Burt S, Carrero C A, Alba-Rubio A C, Miller J T, Hermans I, Huber G W, Dumesic J A. J. Am. Chem. Soc., 2015, 137(32):10317-10325 doi: 10.1021/jacs.5b05945
Yan Y, Wang Q J, Jiang C Y, Yao Y, Lu D, Zheng J W, Dai Y H, Wang H M, Yang Y H. J. Catal., 2018, 367:194-205 doi: 10.1016/j.jcat.2018.08.026
Halder A, Kilianová M, Yang B, Tyo E C, Seifert S, Prucek R, Panáček A, Suchomel P, Tomanec O, Gosztola D J, Milde D, Wang H H, Kvítek L, Zbořil R, Vajda S. Appl. Catal. B, 2018, 225:128-138 doi: 10.1016/j.apcatb.2017.11.047
Kattel S, Yan B H, Chen J G, Liu P. J. Catal., 2016, 343:115-126 doi: 10.1016/j.jcat.2015.12.019
Kim S S, Park K H, Hong S C. Fuel Process. Technol., 2013, 108:47-54 doi: 10.1016/j.fuproc.2012.04.003
Chen Y, Lin J, Li L, Qiao B T, Liu J Y, Su Y, Wang X D. ACS Catal., 2018, 8(2):859-868
Alayoglu S, Beaumont S K, Zheng F, Pushkarev V V, Zheng H M, Iablokov V, Liu Z, Guo J H, Kruse N, Somorjai G A. Top. Catal., 2011, 54(13/14/15):778-785
Zhang Y L, Fu D L, Liu X L, Zhang Z P, Zhang C, Shi B F, Xu J, Han Y F. ChemCatChem, 2018, 10(6):1272-1276 doi: 10.1002/cctc.201701779
Fujita S. J. Catal., 1992, 134(1):220-225
Goguet A, Meunier F C, Tibiletti D, Breen J P, Burch R. J. Phys. Chem. B, 2004, 108(52):20240-20246 doi: 10.1021/jp047242w
Boccuzzi F, Chiorino A, Manzoli M, Andreeva D, Tabakova T. J. Catal., 1999, 188(1):176-185
Bobadilla L F, Santos J L, Ivanova S, Odriozola J A, Urakawa A. ACS Catal., 2018, 8(8):7455-7467 doi: 10.1021/acscatal.8b02121
Caparrós F J, Soler L, Rossell M D, Angurell I, Piccolo L, Rossell O, Llorca J. ChemCatChem, 2018, 10(11):2365-2369 doi: 10.1002/cctc.201800362
Sun X C, Lin J, Zhou Y L, Li L, Su Y, Wang X D, Zhang T. AlChE J., 2017, 63(9):4022-4031 doi: 10.1002/aic.15759
Pavlova S N, Savchenko V I, Sadykov V A, Zaikovskii V I, Kalinkin A V. React. Kinet. Catal. Lett., 1996, 59(1):103-110 doi: 10.1007/BF02067998
Bi Y S, Dang G Y, Zhao X H, Meng X F, Lu H J, Jin J T. J. Hazard. Mater., 2012, 229-230:245-250 doi: 10.1016/j.jhazmat.2012.05.086
Liu L Q, Zhou F, Wang L G, Qi X J, Shi F, Deng Y Q. J. Catal., 2010, 274(1):1-10
Hensley A J R, Hong Y C, Zhang R Q, Zhang H, Sun J M, Wang Y, McEwen J S. ACS Catal., 2014, 4(10):3381-3392 doi: 10.1021/cs500565e
Cheng Z, Qin L, Guo M, Fan J A, Xu D, Fan L S. Phys. Chem. Chem. Phys., 2016, 18(24):16423-16435 doi: 10.1039/C6CP01287F
Liang J X, Yu Q, Yang X F, Zhang T, Li J. Nano Res., 2018, 11(3):1599-1611
Fu Q, Li W X, Yao Y, Liu H, Su H Y, Ma D, Gu X K, Chen L, Wang Z, Zhang H, Wang B, Bao X. Science, 2010, 328(5982):1141-1144 doi: 10.1126/science.1188267
Sun Y N, Giordano L, Goniakowski J, Lewandowski M, Qin Z H, Noguera C, Shaikhutdinov S, Pacchioni G, Freund H J. Angew. Chem. Int. Ed., 2010, 49(26):4418-4421 doi: 10.1002/anie.201000437
Xie Y H, Li B, Weng W Z, Zheng Y P, Zhu K T, Zhang N W, Huang C J, Wan H L. Appl. Catal. A, 2015, 504:179-186 doi: 10.1016/j.apcata.2014.12.008
Yi H, Xia Y J, Yan H, Lu J L. Chin. J. Catal., 2017, 38(9):1581-1587 doi: 10.1016/S1872-2067(17)62768-2
Hong Y C, Zhang H, Sun J M, Ayman K M, Hensley A J R, Gu M, Engelhard M H, McEwen J S, Wang Y. ACS Catal., 2014, 4(10):3335-3345 doi: 10.1021/cs500578g
Sun W Z, Li Q, Gao S A, Shang J K. Appl. Catal. B, 2012, 125:1-9 doi: 10.1016/j.apcatb.2012.05.014
Jiang T, Du S C, Jafari T, Zhong W, Sun Y, Song W Q, Luo Z, Hines W A, Suib S L. Appl. Catal. A, 2015, 502:105-113 doi: 10.1016/j.apcata.2015.05.013
Wang F G, Xu Y, Zhao K F, He D N. Nano-micro Lett., 2014, 6(3):233-241
Yan B H, Wu Q Y, Cen J J, Timoshenko J, Frenkel A I, Su D, Chen X Y, Parise J B, Stach E, Orlov A, Chen J G. Appl. Catal. B, 2018, 237:1003-1011 doi: 10.1016/j.apcatb.2018.06.074
Porosoff M D, Yang X F, Boscoboinik J A, Chen J G. Angew. Chem. Int. Ed., 2014, 53(26):6705-6709 doi: 10.1002/anie.201404109
Brun M, Berthet A, Bertolini J C. J. Electron. Spectrosc. Relat. Phenom., 1999, 104(1/2/3):55-60
Tura J M, Regull P, Victori L S, de Castellar M D. Surf. Interface Anal., 1988, 11(8):447-449 doi: 10.1002/sia.740110807
Klikovits J, Napetschnig E, Schmid M, Seriani N, Dubay O, Kresse G, Varga P. Phys. Rev. B, 2007, 76(4):045405
Duan Z Y, Henkelman G. ACS Catal., 2014, 4(10):3435-3443
Toyoshima R, Yoshida M, Monya Y, Kousa Y, Suzuki K, Abe H, Mun B S, Mase K, Amemiya K, Kondoh H. J. Phys. Chem. C, 2012, 116(35):18691-18697 doi: 10.1021/jp301636u
Jung K D, Bell A T. J. Catal., 2000, 193(2):207-223
Yang B, Yu X, Halder A, Zhang X B, Zhou X, Mannie G J A, Tyo E, Pellin M J, Seifert S, Su D S, Vajda S. ACS Sustainable Chem. Eng., 2019, 7(17):14435-14442
Grosvenor A P, Kobe B A, Biesinger M C, McIntyre N S. Surf. Inter-face Anal., 2004, 36(12):1564-1574
Sun K, Wilson A R, Thompson S T, Lamb H H. ACS Catal., 2015, 5(3):1939-1948
Fan Q N, He S, Hao L, Liu X, Zhu Y, Xu S L, Zhang F Z. Sci. Rep., 2017, 7(1):42172
d'Alnoncourt R N, Friedrich M, Kunkes E, Rosenthal D, Girgsdies F, Zhang B S, Shao L D, Schuster M, Behrens M, Schlögl R. J. Catal., 2014, 317:220-228
Jacobs G, Davis B H. Appl. Catal. A, 2005, 284(1/2):31-38
Chen C S, Wu J H, Lai T W. J. Phys. Chem. C, 2010, 114(35):15021-15028
Park J N, Mcfarland E W. J. Catal., 2009, 266(1):92-97

Figure 1 TEM images of Pd-Fe/SiO2 (a) and Pd/FeOx (b), inserted scale bar is 10 nm; (c) XRD patterns of the fresh and spent catalysts; (d) H2-TPR results of Pd/FeOx, blank FeOx, Pd/SiO2, and Pd-Fe/SiO2 catalysts

Figure 2 Catalytic performance of all of the catalysts tested for RWGS: (a) CO2 conversion; (b) CO selectivity; (c) Comparison of Pd/FeOx, Pd /SiO2, and Pd-Fe/SiO2 at 400 ℃; (d) Long term stability of Pd/FeOx catalyst at 400 ℃ with time on stream up to 30 h

Figure 3 Semi-in situ XPS spectra: Pd3d core-level spectra of Pd/FeOx, Pd/SiO2, and Pd-Fe/SiO2 catalysts during stepwise H2 and CO2 treatments (a) and after 400 ℃ treatment in the reaction gas (b); (c) Fe2p core-level spectra of Pd/FeOx, Pd/SiO2, and Pd-Fe/SiO2 catalysts during stepwise H2 and CO2 treatments; (d) Oscillation of Fe2+ concentration and Pd valence state, deduced from Fe2p and Pd3d XPS results

Figure 4 CO and CO2 adsorption: (a) FTIR spectra of CO adsorption over spent Pd/SiO2, Pd-Fe/SiO2, and Pd/FeOx catalysts (All IR spectra were acquired at room temperature after evacuating gaseous CO); (b) CO2-TPD spectra of reduced FeOx, Pd/SiO2, Pd/FeOx, and Pd-Fe/SiO2 at temperatures up to 750 ℃

Figure 5 Cyclic H2-TPR/CO2-TPO experiments on Pd/SiO2 (a) and Pd/FeOx (b) catalysts (Y-axis is the ion intensity of m/z=28 for CO); Schematic reaction path of CO2 hydrogenation on Pd/SiO2 (c) and Pd/FeOx (d) catalysts
Table 1. Summary of the conditions and reactivities of various catalysts in this work and the literature
| Catalysta | VH2/VCO2 | T/℃ | Weight hourly space velocity/(L·h-1·g-1) | XCO2/ % | SCO/ % |
| 3.6% Pd/FeOx (this work) | 3 | 300 | 36 | 11 | 100 |
| 4.4% Pd-Fe/SiO2 (this work) | 3 | 300 | 36 | 6 | 100 |
| 3.6% Pd/FeOx (this work) | 3 | 400 | 36 | 29 | 100 |
| 4.4% Pd-Fe/SiO2 (this work) | 3 | 400 | 36 | 20 | 100 |
| Mo2C[36] | 3 | 300 | 36 | 8.7 | 93.9 |
| 7.5% Co/Mo2C[36] | 3 | 300 | 36 | 9.5 | 99 |
| 1.67% Pt/TiO2[10] | 1 | 300 | 351 | 4.5 | 99.1 |
| 1.67% Pt/SiO2[10] | 1 | 300 | 72.1 | 3.3 | 100 |
| 1.1% Rh/SrTiO3[35] | 1 | 300 | 120 | 7.9 | 95.4 |
| 5% Co/MCF-17[13] | 3 | 200~300 | 60 | ~5 | 90 |
| 5% Pt50Co50/MCF-17[13] | 3 | 200~300 | 60 | ~5 | 99 |
| 1% Pt/TiO2[11] | 3 | 600 | 12 | ~58 | ~95 |
| 3% Pd/Fe3O4 (dopPPh2)[19] | 4 | 400 | 60 | 8.8 | 94.6 |
| a All the percents are mass fraction. | |||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们